

Abstract
G protein βγ subunit-dependent signaling is important for chemoattractant-dependent leukocyte chemotaxis. Selective small molecule targeting of phosphoinositide 3-kinase (PI3-kinase) γ catalytic activity is a target of interest for anti-inflammatory pharmaceutical development. In this study, we examined whether small-molecule inhibition of Gβγ-dependent signaling, including Gβγ-dependent activation of PI3-kinase γ and Rac1, could inhibit chemoattractant-dependent neutrophil migration in vitro and inflammation in vivo. Small-molecule Gβγ inhibitors suppressed fMLP-stimulated Rac activation, superoxide production, and PI3-kinase activation in differentiated HL60 cells. These compounds also blocked fMLP-dependent chemotaxis in HL60 cells and primary human neutrophils. Systemic administration inhibited paw edema and neutrophil infiltration in a mouse carrageenan-induced paw edema model. Overall, the data demonstrate that targeting Gβγ-regulation may be an effective anti-inflammation strategy.
Chemoattractant-mediated recruitment of leukocytes is responsible for many of the deleterious effects of chronic inflammatory diseases. Many chemoattractants activate G protein-coupled receptors (GPCRs) coupled to the Gi family of heterotrimeric G proteins in leukocytes. Heterotrimeric G proteins are composed of Gα, Gβ, and Gγ subunits. Ligand binding to receptors catalyzes the exchange of tightly bound GDP for GTP on the Gα subunit, liberating it from the Gβγ subunits. Dissociation of the Gα and Gβγ subunits can allow each to directly bind to downstream effector proteins (Gilman, 1987; Oldham and Hamm, 2006). The free Gβγ subunits released from Gi heterotrimers upon chemoattractant receptor activation initiate critical signaling pathways to direct chemoattractant-dependent neutrophil functions including chemotaxis and superoxide production (Neptune and Bourne, 1997).
Key direct targets of Gβγ subunit binding and activation in neutrophils are phosphoinositide 3-kinase γ (PI3-kinase γ) (Stephens et al., 1994, 1997; Stoyanov et al., 1995), Phospholipase C β (PLCβ) (Wu et al., 2000), and P-Rex (Welch et al., 2002). PI3-kinase γ has been noted to be a central mediator of chemotaxis and plays a pivotal role in leukocyte recruitment to inflamed tissues (Hirsch et al., 2000; Li et al., 2000; Camps et al., 2005). PIP3, produced by PI3-kinase γ catalytic activity, is critical to the development of cell polarity, which is necessary for chemokine-mediated cell motility and directional sensing (Wu et al., 2000). PI3-kinase γ-deficient neutrophils have impaired responses to various chemoattractants, including diminished chemotaxis (Hirsch et al., 2000; Li et al., 2000) and respiratory burst (Li et al., 2000; Sasaki et al., 2000), in response to GPCR activation. Small-molecule inhibitors of PI3-kinase γ catalytic activity have been demonstrated to suppress joint inflammation in mouse models of inflammation (Barber et al., 2005; Camps et al., 2005). Critical to the success of a method that targets PI3-kinase γ activity as a therapeutic anti-inflammatory approach is development of selective inhibitors that do not target other PI3-kinase isoforms, because these enzymes are critically involved in multiple aspects of mammalian cell function (Rückle et al., 2006).
In this study, we demonstrate a novel strategy to inhibit chemoattractant-dependent chemotaxis and inflammation using recently identified compounds that block Gβγ-interactions with effectors, including PI3-kinase γ, by binding to a protein-protein interaction “hot spot” on the Gβ subunit (Bonacci et al., 2006). We show that these compounds block fMLP-dependent PI3-kinase γ activation, Rac1 activation, superoxide production, and neutrophil migration in vitro. Furthermore, when administered in vivo, neutrophil-dependent inflammation is inhibited, demonstrating that suppressing key Gβγ-dependent signaling functions with small molecules has significant anti-inflammatory potential.
Materials and Methods
Small Molecules
Compounds for this study were kindly provided by the National Cancer Institute repository except when indicated otherwise. Compound abbreviations are as follows: M119 (NSC119910) and M119B (NSC119892). Gallein (Acros Organics, Geel, Belgium), fluorescein, and wortmannin were obtained from Sigma Aldrich (St. Louis, MO).
Competition ELISA and Structure Activity Relationships
Binding of small molecules to Gβ1γ2 was assessed by competition with phage displaying the SIGK peptide as described previously (Scott et al., 2001; Bonacci et al., 2006). In brief, Gβ1γ2 (25 nM), with biotin incorporated via an N-terminal acceptor peptide on Gβ1, was immobilized in a 96-well plate coated with streptavidin. Compounds/DMSO and 0.1 × 1010 phage were added simultaneously and subsequently incubated for 1 h at room temperature. Plates were then washed with 1× Tris-buffered saline/0.5% Tween 20 and incubated with anti-M13, HRP-conjugated antibody (Amersham, Chalfont St. Giles, Buckinghamshire, UK). Phage binding was determined by monitoring A405 upon addition of 2,2′-azino-bis(3-ethylbenzothiazo-line-6-sulfonic acid) (Sigma Aldrich).
Surface Plasmon Resonance
Direct binding of “hot spot” binding small molecules was assessed using the Reichert SR7000 Surface Plasmon Resonance dual chamber instrument equipped with an autosampler (Reichert, Depew, NY). To activate the sensor chip surface, a mixture of 0.1 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride and 0.05 M N-hydroxysuccinimide was injected (flow rate 5 μl/min) over the sensor surface. Streptavidin (100 μg/ml; prepared in 20 mM sodium acetate buffer, pH 5.5) was coupled to the sensor chip surface followed by quenching the remaining activated carboxyl groups with 1 M ethanolamine, pH 8.5. bGβ1γ2 was conjugated to the streptavidin-coated chip in running buffer (50 mM HEPES, pH 7.6, 1 mM EDTA, 100 mM NaCl, 0.1% C12E10, and 1 mM dithiothreitol) to achieve 2000 micro-refractive index units. A second reference cell was treated similarly, but bGβ1γ2 was excluded. Direct binding of small molecules was tested out at room temperature with a flow rate of 75 μl/min. Compounds were prepared in running buffer and injected for 10 min followed by a dissociation phase of 10 min. The obtained sensorgrams were corrected for nonspecific background by running the same experiment in series over an identical sensor surface with immobilized streptavidin, blocked with biotin, without bGβγ. Binding rates and constants were independent of flow rate over a wide range and did not fit mass transport limited models, indicating that mass transport was not limiting association or dissociation rates. Best fit kinetic parameters were determined using kinetic titration model and ClampXP ver. 3.5, which accounts for incomplete dissociation between injections (Karlsson et al., 2006).
GFP-PH-Akt Translocation in Differentiated HL60 Cells
HL60 cells stably overexpressing GFP-PH-Akt (Servant et al., 2000) were maintained and differentiated as described previously (Bonacci et al., 2006). Cells (0.2 × 106 cells/ml) were differentiated by incubation with 1.2% DMSO for 5 days. Cells were washed with serum-free RPMI 1640 (Invitrogen, Carlsbad, CA). Cells (2 × 105; 100 μl) were transferred to a 1.8-ml Beckman ultracentrifuge tube. Cells were pretreated with DMSO or compounds for 10 min at room temperature and stimulated with 250 nM fMLP (Sigma Aldrich) for 2 min. at 37°C, snap-frozen in liquid nitrogen, and thawed in the presence of 100 μl of 2× lysis buffer (100 mM HEPES, pH 8.0, 6 mM MgCl2, 0.2 mM EDTA, 200 mM NaCl, 100 μM Na3VO4, and protease inhibitors). Membranes were harvested by centrifugation 100,000g for 20 min after four freeze-thaw cycles. All treatments contained the same final concentration of DMSO. Pellets were washed once with 100 μl of lysis buffer (50 mM HEPES, pH 8.0, 3 mM MgCl2, 0.1 mM EDTA, 100 mM NaCl, 50 μM Na3VO4, and protease inhibitors), pelleted as above, and boiled in 2× sample loading buffer. Samples were resolved by 12% SDS-PAGE, transferred to nitrocellulose, and probed with anti-GFP antibody (1:1000; Roche) followed by incubation with goat anti-mouse HRP-conjugated secondary antibody (1: 5000, Bio-Rad Laboratories, Hercules, CA). Chemiluminescence was analyzed using a charge-coupled device camera in a UVP Epi-Chem II Darkroom imaging system. All samples had background subtracted and were normalized to the fMLP-induced signal set at 100%.
Cell Motility Analysis
Chemotaxis was assayed using a Boyden chamber (Neuro Probe, Gaithersburg, MD) using 3-μm polyvinylpyrrolidone-free polycarbonate filters (Neuro Probe). HL60 cells (differentiated for 4 days) were washed with and resuspended in HBSS containing 1% bovine serum albumin to a final concentration of 1 × 106 cells/ml. Primary human neutrophils were washed and resus-pended in NaCl buffer (140 mM NaCl, 4 mM KCl, 10 mM D-glucose, 10 mM HEPES, pH 7.4, 1 mM MgCl2, and 1 mM CaCl2) to a final concentration of 1 × 106 cells/ml. Chemoattractant [1 μM granulocyte-macrophage–colony stimulating factor (GM-CSF), 10 nM IL-8, or 250 nM fMLP; Sigma Aldrich] was added to the bottom chamber in HBSS containing 1% bovine serum albumin. Cell suspensions (0.2 × 106 cells/well) were added to the top wells of the Boyden chamber and allowed to migrate for 1 h at 37°C. When applicable, cell suspensions were preincubated for 10 min with small-molecule inhibitors (in DMSO) at the indicated concentrations, and the bottom chamber was adjusted to the same concentration of small molecule. All treatments contained the same final concentration of DMSO. Filters were processed according to the manufacture's recommendations and stained using DifQuik (VWR Scientific, West Chester, PA). Chemotactic HL60 cells were scored by counting three microscope fields and subtracting the number of cells from fMLP wells as background. All samples had background subtracted and were normalized to the fMLP-induced signal set at 100%. For all the small molecules, effects of the compounds on chemokinesis was analyzed by measuring chemotaxis in the presence of 250 nM fMLP in both the upper and lower chambers of the Boyden Chamber and measuring changes in transwell migration. Unless otherwise indicated, none of the compounds had effects on chemokinesis.
Measurement of Superoxide Production
The nitro blue tetrazolium (NBT) method was used to assess the effects of Gβγ inhibitors on NADPH oxidase activity. HL60 cells (differentiated for 4 days) were washed with and resuspended in HBSS containing calcium and magnesium (Cellgro; Mediatech, Herndon VA) to a final concentration of 2 × 106 cells/ml. Cells (1 × 106/reaction) were pretreated with 10 μM Gβγ inhibitor compound (in DMSO) or 100 nM wortmannin (Sigma Aldrich) for 10 min at 37°C before the addition of NBT (25 μl of 10 mg/ml in methanol) and then incubated or an additional 5 min at 37°C. All treatments contained the same final concentration of DMSO. Cells were then activated with either 250 nM fMLP (Sigma Aldrich) or PMA (Sigma Aldrich) for 30 min in a 37°C water bath. Reactions were stopped by the addition of 500 μl of 1.2 N HCl, and cells were collected by centrifugation at 12,000g for 5 min. Cell pellets were then resuspended in 200 μl of DMSO, transferred to a 96-well plate, and absorbance was measured at 540 nM. All treatments and controls contained the same concentration of DMSO. All samples had background subtracted and were normalized to the fMLP-induced signal set at 100%.
Evaluation of Rac-1 Activation
HL60 cells (differentiated for 4 days) were washed with and resuspended in NaCl buffer to a final concentration of 20 × 106 cells/ml. Cells (10 × 106/reaction) were pretreated with 10 μM Gβγ inhibitor (in DMSO) for 10 min before challenge with 1 μM fMLP for 90 s in a 37°C water bath and then immediately transferred to an ice-water bath. Cells were recovered by centrifugation at 500g and washed two times with ice-cold Tris-buffered saline, pH 7.4. The Rac1 activation assay kit (Upstate Cell Signaling Solutions, Billerica, MA) was used to prepare cell extracts and evaluate Rac1 activation according to the manufacturer's instructions. Affinity-purified GTP-Rac1 was resolved by 15% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, probed with anti-Rac1 monoclonal antibody followed by detection with HRP-conjugated secondary antibody in accordance with the manufacturer's recommendations. Chemiluminescence was analyzed using a charge-coupled device camera in a UVP Epi-Chem II Darkroom imaging system. Results were expressed as percent fMLP-stimulated GTP Rac1 normalized to total Rac1 with the background subtracted.
Carrageenan-Induced Paw Edema and Neutrophil Abundance
Male Swiss-Webster mice (35–40 g b.wt.; Taconic Farms, Germantown, NY) were randomized and acclimated for 1 week in a 12-h light/dark cycle. Food and water were provided ad libitum. Animals were labeled with unique identifiers on their tails using indelible marker. One hour before challenge with carrageenan, mice were administered compounds (dissolved in PBS) or vehicle (PBS) either by intraperitoneal (300 μl with 27.5 gauge needle) injection or by oral gavage (100 μl with 1.5″, steel ball-tipped feeding needle; Popper and Sons, New Hyde Park, NY). Indomethacin (2.5 mg/kg) stock solution was prepared in methanol and diluted into PBS (Mediatech), small-molecule inhibitor (concentration as indicated in appropriate figure legend) was prepared in PBS. Mice were anesthetized by intraperitoneal injection with ketamine hydrochloride (175 mg/kg) and xylazine (7 mg/kg) using a 26 gauge needle. Mice were tested for pain reflex to ensure sedation and then injected subcutaneously into the plantar region of the hind paw with 25 μl of 2% carrageenan using a precision engineered 5/8″ 25 gauge Hamilton syringe (hypodermic needle; Hamilton Co., Reno, NV). Carrageenan (CarboMer, Inc., San Diego, CA) was suspended in PBS at 50°C for 10 min with stirring the night before experimentation. The contralateral paw was injected with vehicle as control. Mice were then transferred to bedded cages to minimize pain associated with the carrageenan injections. Dorsal-plantar swelling was measured using an electronic digital caliper (± 0.03 mm; VWR Scientific) at time 0 and every 2 h thereafter. To ensure measuring and injection consistency, the dorsal and plantar surface of the test paw and contralateral paws of each animal were marked with indelible marker. Each paw was measured two times at each time point and averaged. At the conclusion of the experiment, animals were euthanized in accordance with the University of Rochester and American Veterinary Medical Association standards by carbon dioxide narcosis and cervical dislocation. Paw edema was determined by subtracting the thickness of the contralateral paw from that of the carrageenan-injected paw at each time point.
Paws were amputated 2 h after carrageenan injection to determine the number of neutrophils present in the edematous fluid. Paws were transferred to prepared Eppendorf tubes, and the exudates were collected by centrifugation for 2 min. The paws were removed and the tubes re-weighed to determine the mass of the exudates, which was then converted to volume. The volume in the contralateral untreated control paw was subtracted from the cardgeenan-treated paw volume. To remove erythrocytes by hypertonic lysis, the exudate was resuspended in 250 μl of 1× PBS to which 50 μl of water was added. At the conclusion of a 30-s incubation, 75 μl of 4.5× PBS was added to return the solution to normal salt levels. The number of neutrophils present was determined by manual counting, and the small number of neutrophils in the control contralateral paw was subtracted.
Isolation of Primary Human Neutrophils
Human blood obtained by venous puncture from consenting, healthy adult donors, in accordance with University of Rochester standards, and collected in sterile vacutubes containing sodium heparin (BD Biosciences, San Jose, CA). Neutrophils were layered over PolymorphprepT11 (Accurate Chemical and Scientific Co., Westbury, NY) and isolated by centrifugation (470g for 50 min at room temperature). Trace erythrocytes were removed by hypertonic treatment followed by centrifugation. Isolated neutrophils were stored in HEPES-buffered saline solution (146 mM NaCl, 5 mM KCl, 5.5 mM d-glucose, 10 mM HEPES, 1 μM CaCl2, and 1 mM MgSO4) at pH 7.4.
Data Analysis
Ligand competition curves were determined by nonlinear regression using Prism software (GraphPad Software, Inc., San Diego, CA). Statistical significance was evaluated by one-way analysis of variance and Bonferroni's multiple comparison test. Statistical significance was defined as *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Results
Characterization of Novel Compound Binding to Gβγ
Previous studies identified multiple compounds that blocked effector binding to Gβγ. A lead compound, M119, inhibited Gβγ-dependent PI3-kinase γ and PLCβ activation in vitro and blocked chemoattractant/PI3-kinase-dependent GFP-PH-Akt translocation to membranes as well as Ca2+ release (Bonacci et al., 2006). We identified a related compound, gallein, that differs from M119 by the substitution of a benzene carboxylic acid for cyclohexane carboxylic acid at the 9 position of the core xanthene (Fig. 1A). Gallein is commercially available at high purity as a single isomer and in quantities necessary for in vivo analysis, so the Gβγ binding properties of gallein were compared with M119. Gallein effectively competed for binding of SIGK peptide to Gβγ in a phage ELISA assay with an IC50 comparable with that of M119 (Fig. 1B). We have shown previously that compounds that block peptide binding in this assay are effective competitors of many Gβγ protein-protein interactions.
Fig. 1.
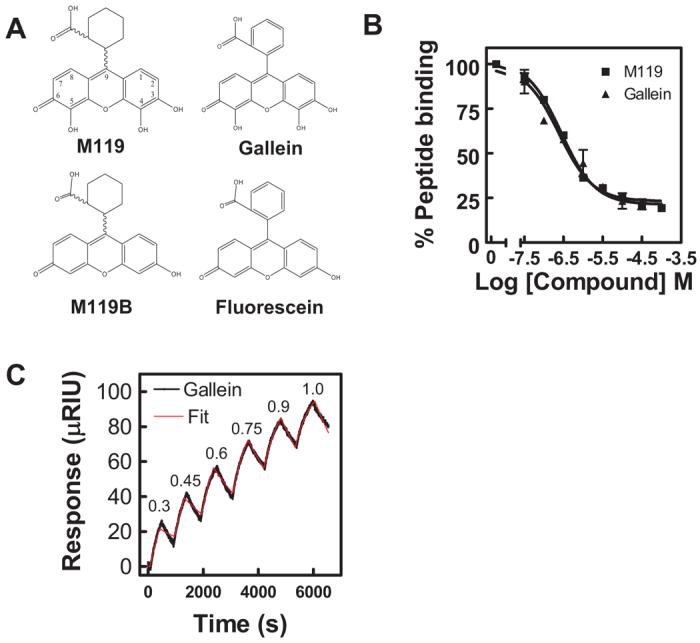
Small molecule binding profiles. A, structures of M119, M119B, gallein, and fluorescein are shown. B, M119 and gallein bind with comparable affinities in the competition phage ELISA. M119 and gallein were tested for their ability to inhibit binding of a phage displaying the peptide SIGK to the Gβγ “hot spot” as described previously (Bonacci et al., 2006). Data shown is representative of three independent experiments, each in duplicate, ± S.D. C, direct binding analysis of gallein bind to Gβγ by SPR. A representative experiment for gallein binding to bGβ1γ2. Gallein binding was tested at sequentially higher micromolar concentrations indicated at the peak of each association followed by a dissociation phase with the compound removed between each addition. All data were fit with a kinetic titration model (Karlsson et al., 2006) to give ka and kd values. In the experiment shown, the fits resulted in ka = 1130 ± 17 M−1 s−1 and k = 4.3 ± 0.04 × 10−4 s−1. Pooled data from three separate experiments are given in Table 1.
To determine direct binding equilibrium and kinetic constants for gallein binding to Gβγ, surface plasmon resonance (SPR) measurements were performed with streptavidin-immobilized biotinylated Gβ1γ2. Binding and dissociation of gallein was monitored as a function of concentration and time. Data were fit to one site association and dissociation models to calculate association and dissociation rate constants from which affinity constants were derived (Fig. 1C, Table 1). Based on the SPR analysis, gallein bound to Gβ1γ2 (Fig. 1C) with a Kd value of approximately 400 nM (Table 1), in relatively close agreement with the IC50 of 200 nM observed in the competition ELISA assay. The control compound, fluorescein, which did compete for SIGK binding in the competition ELISA, did not have detectable binding by SPR (Table 1). It is noteworthy that binding and dissociation rates for gallein were relatively slow (Fig. 1C). These data confirm that gallein binds directly to Gβγ, and resulting effects on competition for effector binding are likely to result from direct binding to Gβγ with high affinity. Based on these data, gallein would be predicted to have similar effects to M119 when tested in in vitro and in vivo assays.
TABLE 1.
Equilibrium and kinetic binding constants for small molecule binding to β1γ2 calculated from surface plasmon resonance analysis and ELISA competitionValues are mean ± S.E. from three separate experiments.
| Kd | kon | koff | ELISA IC50 | |
|---|---|---|---|---|
| nM | ×103 M−1 s−1 | ×104 M−1 s−1 | nM | |
| Gallein | 422 ± 49 | 0.99 ± 0.11 | 4.09 ± 0.09 | 241 ± 24 |
| Fluorescein | N.B. |
N.B., no binding detected.
Inhibition of G Protein-Dependent Chemotactic Peptide Signaling with Small Molecules
A key molecule involved in chemotactic peptide signaling is PI3-kinase γ. To determine whether gallein, like M119, modulates the receptor-dependent activation of PI3-kinase, HL60 cells expressing GFP-PH-Akt were pretreated with compound and challenged with fMLP. Gallein inhibited fMLP-dependent GFP-PH-Akt translocation in differentiated HL60 cells with an efficacy comparable with that of M119 (Fig. 2A).
Fig. 2.

“Hot spot” binding small molecules modulate key leukocyte functions. A, M119 and gallein inhibit GFP-PH-Akt translocation. Differentiated HL60 cells stably expressing GFP-PH-Akt were challenged with 250 nM fMLP in the presence and absence of 10 μM concentrations of the indicated compounds. Translocation of GFP-PH-Akt to the plasma membrane was evaluated by Western blot. Quantification shown below. ***, P < 0.001 analysis of variance (ANOVA) is statistically different from control (PBS + vehicle). Western blot shown is representative of three independent experiments and quantitation contains data pooled from three independent experiments ± S.E.M. B, M119 and gallein block activation of Rac1. Differentiated HL60 cells were challenged with 1 μM fMLP in the presence and absence of 10 μM concentrations of the indicated compounds. Rac1 activation was assessed by Western blots of affinity-precipitated GTP-Rac1 from HL60 cell lysates. Western blot shown is representative of three independent experiments and quantitation contains data pooled from three independent experiments ± S.E.M. *P < 0.05 ANOVA is statistically different from control (DMSO only). n = 3. C and D, M119 and gallein inhibit superoxide production in fMLP-challenged HL60 cells. Differentiated HL60 cells were challenged with 250 nM fMLP or 250 nM PMA in the presence and absence of 10 μM compounds or 100 nM Wortmannin (Wtmn). NADPH oxidase activity was determined after reaction with NBT by absorbance at 540 nm. Data shown is contains data pooled from three independent experiments (each in duplicate) ± S.E.M. *, P < 0.05 ANOVA is statistically different from control (DMSO only).
Activation of Rac in neutrophils is critical for fMLP-dependent activation of NADPH-oxidase and subsequent superoxide production and is dependent on Gβγ- and PIP3-dependent activation of the Rac-specific exchange factor P-Rex. Other previous work showed that M119 inhibited P-Rex1 activation by fMLP suggesting the compounds could inhibit Rac activation (Zhao et al., 2007). We determined whether Gβγ-binding compounds would inhibit receptor-dependent activation of Rac1. Rac1GTP levels were measured in cytosolic extracts of HL60 cells that had been pretreated with compounds before stimulation with fMLP. M119 and gallein inhibited fMLP-induced Rac1 activation in differentiated HL60 cells, whereas the negative control compound M119B had little effect (Fig. 2B).
Gβγ Inhibitors Blocked fMLP-Dependent Superoxide Production and Neutrophil Chemotaxis
The ability of these small molecules to block fMLP-induced chemotaxis and superoxide production was assessed to determine whether the compounds could block relevant cellular functions downstream of PI3-kinase γ and Rac. Both of these functions are critical to the inflammatory process and inhibition of both processes could contribute to anti-inflammatory effects of Gβγ inhibitors.
M119 and gallein both significantly inhibited fMLP-dependent superoxide production. Control compounds M119B and fluorescein did not significantly affect this response (Fig. 2C). Wortmannin also blocked fMLP-dependent superoxide production, indicating that the pathway was a PI3-kinase-dependent pathway. We also examined PMA-dependent superoxide production, a process not dependent on Gβγ. PMA-dependent superoxide production was not inhibited by any of the Gβγ binding compounds or controls (Fig. 2D), indicating that the compounds specifically inhibited the Gβγ-dependent pathway to superoxide production.
fMLP-dependent chemotaxis was assessed in a Boyden Chamber. M119 and gallein inhibited fMLP-induced chemotaxis in differentiated HL60 cells with similar efficacy (Fig. 3A). Neither M119 nor gallein had any effects on chemokinesis measured in the transwell assay with fMLP in both the upper and lower chambers (data not shown). To support the idea that the mode of action of these compounds is dependent on Gβγ heterodimers liberated from Gαi-coupled chemokine receptors, cells were challenged with GM-CSF. Chemotaxis induced by GM-CSF is partly dependent on PI3-kinase activity in human neutrophils but is independent of Gβγ-stimulation of PI3-kinase (Gomez-Cambronero et al., 2003). If the small molecules were acting directly on PI3-kinase or by a nonspecific mechanism, they would be expected to block GM-CSF-induced chemotaxis. Neither M119 nor gallein blocked GM-CSF-induced chemotaxis in differentiated HL60 cells (Fig. 3B). These data demonstrate two key points: 1) the general chemotaxis machinery in HL60 cells is not affected by these compounds and 2) the compounds selectively inhibited GPCR-dependent chemotaxis, consistent with inhibition of Gβγ signaling.
Fig. 3.

“Hot spot” binding small molecules inhibit GPCR-coupled chemoattract-dependent chemotaxis. A, M119 and gallein inhibit fMLP-induced chemotaxis in differentiated HL60 cells. Differentiated HL60 cells (200k) were pretreated with 10 μM concentrations of the indicated compound, challenged with 250 nM fMLP, and assayed for chemotaxis in a Boyden chamber for 1 h at 37°C. Chemotaxis was quantified by counting Diffquik-stained cells in three random microscope fields, subtracting out background cells (0–10 cells) in the absence of chemoattractant to obtain total transmigrated cells (∼125 cells fMLP + vehicle), and represented as the percentage of fMLP-treated control cells. ***, P < 0.001 ANOVA is statistically different from control. Data shown pooled from three independent experiments, each in duplicate, ± S.E.M. B, neither M119 nor gallein blocks 1 μM GM-CSF–induced chemotaxis in a Boyden chamber. Chemotaxis was quantified as above by subtracting out background cells (0–10 cells) in the absence of chemoattractant to obtain total transmigrated cells (∼100 cells GM-CSF + vehicle) and represented as the percentage of GM-CSF–treated control cells. No statistically significant difference from control was seen by ANOVA. Data shown are pooled from two independent experiments, each performed in duplicate, ± S.E.M. C, M119 and gallein inhibit fMLP- and IL-8-induced chemotaxis in human neutrophils in a Boyden chamber. Primary human neutrophils were isolated from whole blood to ≥80% purity. Neutrophils (2 × 105) were pretreated with gallein (10 μM), M119 (10 μM), M119B (10 μM), or wortmannin (wtmn.) (1 μM) and then challenged with 250 nM fMLP or 10 nM IL-8 to evaluate chemotaxis in a Boyden chamber for 1 h at 37°C. Chemotaxis was quantified as above by subtracting out background cells (0–10 cells) to obtain total transmigrated cells (fMLP ∼100 cells and IL-8 ∼175 cells) and represented as the percentage of chemoattractant-treated control cells. ***, P < 0.001 ANOVA is statistically different from control. NS, not statistically different from control. Data are mean ± S.E.M. Data shown are pooled from three independent experiments, each in duplicate, ± S.E.M. D, gallein dose-dependently inhibits human neutrophil chemotaxis in a Boyden chamber. Primary human neutrophils were isolated and treated (250 nM fMLP ± gallein) as described above. Chemotaxis was quantified as above by subtracting out background cells to obtain total transmigrated cells and represented as the percentage of fMLP-treated control cells. Data shown are pooled from two independent experiments, each in duplicate, ± S.E.M.
The inhibitory properties of these compounds in potentially more clinically relevant isolated primary human neutrophils were also evaluated. Again, both M119 and gallein significantly inhibited fMLP-induced chemotaxis (Fig. 3C). Wortmannin, a general PI3-kinase inhibitor (Okada et al., 1994), also blocked chemotaxis (Fig. 3C). Conversely, M119B binds only weakly to the Gβγ “hot spot” (Table 1 in Bonacci et al., 2006) and had no affect on cell motility (Fig. 3C), suggesting that the observed effects on chemotaxis are dependent on small-molecule-binding to the Gβγ “hot spot.” IL-8 regulated chemotaxis was also blocked by M119 and gallein (Fig. 3C) extending our findings to other Gi coupled chemoattractants and supporting the idea that the mechanism of action of these compounds is to inhibit Gβγ subunit signaling downstream of chemoattractant/chemokine receptors.
To assess the potency of gallein for inhibition of chemotaxis, primary human neutrophils were treated with a range of concentrations and assayed for fMLP-dependent chemotaxis. Gallein blocked fMLP-dependent chemotaxis with an IC50 of approximately 5 μM (Fig. 3D). Thus, small molecules that bind to Gβγ and block Gβγ-dependent PI3-kinase γ regulation in vitro and in HL60 cells potently inhibit GPCR-dependent neutrophil chemotaxis with an IC50 comparable with what has been published for direct PI3-kinase catalytic inhibitors on chemokine-dependent monocyte chemotaxis (Camps et al., 2005).
Gallein Attenuates Inflammation and Neutrophil Recruitment in Vivo
Inhibition of chemoattractant-dependent chemotaxis of neutrophils would be predicted to inhibit neutrophil-dependent inflammation based on data from PI3-kinase γ knock-out mice (Hirsch et al., 2000; Li et al., 2000). To assess the in vivo efficacy of Gβγ-binding small molecules in blocking neutrophil chemotaxis we tested gallein in the carrageenan paw edema model. Carrageenan, when injected into the glabrous tissue of the hind paw, leads to rapid acute inflammation characterized by infiltration of neutrophils (Siqueira-Junior et al., 2003). Gallein or vehicle control was delivered by intraperitoneal injection 1 h before injection of carrageenan into the paw. Peak paw edema was observed 2 h after carrageenan injection (Fig. 4A). Indomethacin, a nonselective nonsteroidal anti-inflammatory drug that inhibits cyclooxygenases 1 and 2 (Siqueira-Junior et al., 2003), is the drug of choice for comparative in vivo efficacy studies. Pretreatment with indomethacin sharply reduced paw edema. It is noteworthy that prophylactic administration of 100 mg/kg gallein reduced paw edema to levels comparable with that of indomethacin. Injection with gallein in the absence of carrageenan had no effect on paw thickness (data not shown). Additional experimentation determined that the ED50 value of gallein for inhibiting paw edema to be approximately 20 mg/kg (Fig. 4B). Acute phase inflammation, as seen in the carrageenan-induced paw edema model, is characterized by neutrophil infiltration (Siqueira-Junior et al., 2003; Posadas et al., 2004). To confirm that neutrophil infiltration was inhibited, the number of neutrophils in paw exudates was quantified. Pretreatment with 100 mg/kg gallein reduced the number of neutrophils in the edematous fluid by approximately half to levels comparable with that seen with indomethacin (Fig. 4C). These data correlated well with the actual volume of exudate fluid. Pretreatment with indomethacin and gallein also reduced exudate volume by approximately 75% (Fig. 4D).
Fig. 4.

Gβγ “hot spot” binding small molecules inhibit neutrophil recruitment and acute phase inflammation in vivo. A, gallein inhibition of carrageenan-induced paw edema. Male mice (35–40 g) were injected i.p. with 100 mg/kg gallein or 2.5 mg/kg indomethacin in PBS 1 h before subplantar injection of 50 μl of 2% carrageenan (Cg) into the test paw. The contralateral paw was injected with saline as control. Each paw was measured 3 times every 2 h. Change in paw thickness was quantified by subtracting the average thickness of the contralateral paw from the average thickness of the test paw. Each point represents the average paw thickness of four mice, each measurement done in duplicate. Data shown is representative of more than three independent experiments. Data are mean ± S.E.M. B, gallein inhibits carrageenan-induced paw edema in a dose-dependent manner. Mice (four mice per plotted point) were treated and quantified as described above. Data are mean ± S.E.M. C, neutrophil recruitment is attenuated by gallein. Mice (four mice per plotted point) were treated as described above. Two hours after carrageenan injection, paws were severed and the number of neutrophils contained within edematous fluid was determined. Data are mean ± S.E.M. ***, P < 0.001 and **, P < 0.01 ANOVA are statistically different from control. D, paw swelling is reduced by gallein. Mice (four mice per plotted point) were treated as described above. Two hours after carrageenan injection, paws were severed, and the volume of edematous fluid was determined. ***P < 0.001 ANOVA is statistically different from control.
Oral administration of gallein (30 mg/kg) in mice 1 h before carrageenan challenge also significantly reduced paw swelling by 40% comparable with that of indomethacin (Fig. 5). These data demonstrate that gallein is absorbed into systemic circulation, is bioavailable, and systemically impairs neutrophil recruitment. To support the conclusion that the observed reduction in paw swelling was Gβγ-dependent, we tested the M119B-like compound fluorescein under identical experimental conditions. Fluorescein differs from M119B only by the substitution of an aromatic benzene ring for a cyclohexane at the 9 position of the core xanthene (Fig. 1A) and binds Gβγ very weakly if at all (Table 1). Fluorescein did not reduce paw swelling under identical experimental conditions (Fig. 5), which indicates that the observed reduction in paw swelling and neutrophil infiltration was correlated with the ability of the small molecules to bind to Gβγ.
Fig. 5.
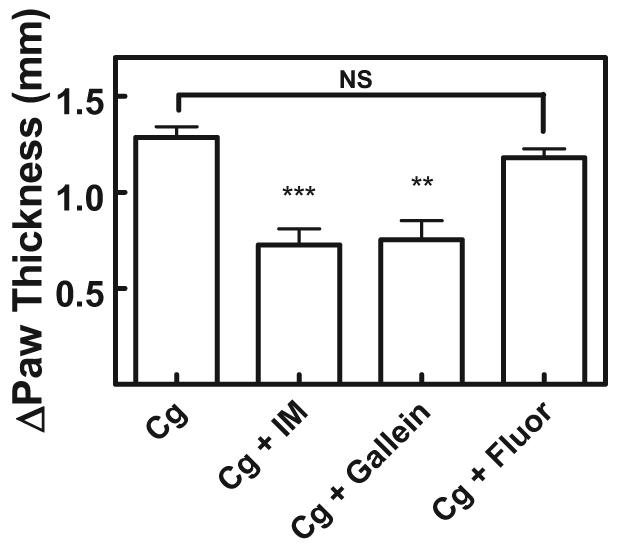
Gallein is effective with oral administration. Male mice (35–40 g) were dosed by oral gavage with 30 mg/kg gallein 1 h before challenge with 2% carrageenan. Methods are as described in Fig. 4. Each bar represents the average paw thickness of four mice at 3 h after carrageenan injection, each measurement done in duplicate. Data are mean ± S.E.M. ***, P < 0.001 and **, P < 0.01 ANOVA are statistically different from vehicle. NS, not statistically different from control. Data shown are representative of two independent experiments.
Discussion
Herein we present a novel strategy for treatment of inflammation that targets interactions between G protein βγ subunits and effectors that are critical for neutrophil migration in response to activation of chemoattractant receptors. Gβγ-responsive PI3-kinase γ production of PIP3 is critical to neutrophil functions, including superoxide production, chemotaxis, and cellular polarization (Hirsch et al., 2000; Li et al., 2000). We have shown that Gβγ-binding small molecules inhibit interactions between Gβγ and PI3-kinase γ. These same molecules block PI3-kinase and Rac1 activation in HL60 cells, chemoattractant-dependent superoxide production and chemotaxis in differentiated HL60 cells. These findings were extended to inhibition of chemoattractant-dependent chemotaxis in primary human neutrophils and ultimately neutrophil-dependent inflammation in vivo.
PI3-kinases play diverse roles in normal cellular physiology, including cell motility and survival (Cantley, 2002). Camps and colleagues identified and characterized small-molecule inhibitors of PI3-kinase γ that are competitive with ATP (Camps et al., 2005). These small molecules were efficacious in mouse models of rheumatoid arthritis and systemic lupus, providing further rationale for pharmacological targeting of the pathway as therapeutic strategy (Barber et al., 2005; Camps et al., 2005). However, a concern with inhibitors of this class is that because many kinases of the same family have significant homology, there may be difficulty in developing inhibitors that are selective for PI3-kinase γ, although some progress has been made in this area (Rückle et al., 2006). The strategy presented here selects for a single isoform of the PI3-kinase family because PI3-kinase γ is the only PI3-kinase isoform that uses Giβγ-dependent activation as a major mechanism for regulation (Stephens et al., 1994, 1997; Hirsch et al., 2000).
Compounds such as M119 and gallein also inhibit interactions between Gβγ and other targets that are critical for chemoattractant-dependent directed migration or reactive oxygen species production by neutrophils or other monocytes. For example, M119 blocks membrane translocation of P-Rex, a PIP3- and Gβγ-regulated Rac2 exchange factor, in human neutrophils, which could be due in part to directly blocking P-Rex binding to Gβγ in addition to blocking PIP3 production by PI3-kinase (Zhao et al., 2007). It has recently been shown that Ras is required for full PI3-kinase γ activation in neutrophils (Suire et al., 2006). It is possible that the inhibitors block Gβγ-dependent activation of Ras and in part act to block PI3-kinase γ activation in cells indirectly through inhibition of Ras activation. In Dictyostelium discoideum, both PI3-kinase and phospholipase A2 activities downstream of G protein activation are important for chemotaxis (Chen et al., 2007). Other studies have shown chemoattractant-dependent chemotaxis becomes PI3-kinase–independent under certain conditions (Ferguson et al., 2007). Thus, a broad-spectrum Gβγ inhibitor could be more effective, in some cases, than selective PI3-kinase γ inhibitors because they can block other Gβγ interactions besides PI3-kinase-γ, a property that could contribute to therapeutic efficacy. Nevertheless, because of the wide roles of Gβγ in regulation of cell physiology, development of Gβγ binding small molecules that are more selective for PI3-kinase γ inhibition relative to other Gβγ-depen-dent pathways is an important direction.
Inflammation is a central mediator of many human conditions, including atherosclerosis, allergic reactions, psoriasis, virus-induced myocarditis, ischemia-reperfusion injury, and rheumatoid arthritis. The inflammatory process involves complex signaling cascades partly coordinated by chemokines, which recruit leukocytes, including large numbers of neutrophils, to sites of inflammation (Wu et al., 2000). Targeting chemokines with antibodies or binding proteins as well as targeting chemokine receptors has been attempted as a therapeutic strategy (Gong et al., 1997; Ogata et al., 1997; Plater-Zyberk et al., 1997; Barnes et al., 1998; Halloran et al., 1999; Matthys et al., 2001; Podolin et al., 2002; Yang et al., 2002) However, the overwhelming complexity of these signaling molecules (multiple chemokines, chemokine receptors, and redundancy) is a significant hurdle. Polychemokine (Carter, 2002) or combinations of different chemokine (al Mughales et al., 1996) antagonists have been suggested, but there may be chemokines that act as an agonist at one receptor and an antagonist at another (Xanthou et al., 2003). Despite this complexity, these chemokine receptors and ligands represent a tantalizing therapeutic target because of their integral role in the inflammatory process. Targeting PI3-kinase γ has been suggested as strategy for blocking a common signal downstream of chemokine receptors (Camps et al., 2005; Rückle et al., 2006). The data presented here indicate that targeting Gβγ could also circumvent the redundancy of the chemokine system and may offer some advantages to direct targeting of PI3-kinase γ.
Acknowledgments
We thank the Developmental Therapeutics Program at the National Cancer Institute/National Institutes of Health for providing small molecules used in this study, H. Bourne (San Francisco, CA) for providing HL60 cells stably overexpressing GFP-PH-Akt, R. Waugh for purifying human neutrophils, and J. Bidlack and B. Fulton for valuable discussions.
Supported by National Institutes of Health grants GM60286 (A.V.S.) and Postdoctoral Training grant in Cancer Biology (D.M.L.) the Johnson and Johnson Discovery Fund (A.V.S.), and an Arthritis Foundation Postdoctoral Fellowship (D.M.L.).
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- PI3
phosphoinositide 3
- PLC
phospholipase C
- P-Rex
phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchange factor
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- bGβγ
N terminally biotinylated Gβγ
- fMLP
formyl-Met-Leu-Phe
- ELISA
enzyme-linked immunosorbent assay
- DMSO
dimethyl sulfoxide
- HRP
horseradish peroxidase
- HBSS
Hanks' balanced saline solution
- GM-CSF
granulocyte-macrophage–colony stimulating factor
- IL
interleukin
- NBT
nitro blue tetrazolium
- PMA
phorbol 12-myristate 13-acetate
- GFP-PH-Akt
Green fluorescent protein fusion with the pleckstrin homology domain from Akt protein kinase
- ANOVA
analysis of variance
References
- al Mughales J, Blyth TH, Hunter JA, Wilkinson PC. The chemoattractant activity of rheumatoid synovial fluid for human lymphocytes is due to multiple cytokines. Clin Exp Immunol. 1996;106:230–236. doi: 10.1046/j.1365-2249.1996.d01-836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DF, Bartolome A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, Camps M, Rückle T, Schwarz MK, Rodriguez S, et al. PI3Kγ inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005;11:933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- Barnes DA, Tse J, Kaufhold M, Owen M, Hesselgesser J, Strieter R, Horuk R, Daniel Perez H. Polyclonal antibody directed against human RANTES ameliorates disease in the Lewis rat adjuvant-induced arthritis model. J Clin Invest. 1998;101:2910–2919. doi: 10.1172/JCI2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Differential targeting of Gβγ-subunit signaling with small molecules. Science. 2006;312:443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- Camps M, Rückle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, et al. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Carter PH. Chemokine receptor antagonism as an approach to anti-inflammatory therapy: ‘just right’ or plain wrong? Curr Opin Chem Biol. 2002;6:510–525. doi: 10.1016/s1367-5931(02)00351-4. [DOI] [PubMed] [Google Scholar]
- Chen L, Iijima M, Tang M, Landree MA, Huang YE, Xiong Y, Iglesias PA, Devreotes PN. PLA2 and PI3K/PTEN pathways act in parallel to mediate chemotaxis. Developmental Cell. 2007;12:603–614. doi: 10.1016/j.devcel.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GJ, Milne L, Kulkarni S, Sasaki T, Walker S, Andrews S, Crabbe T, Finan P, Jones G, Jackson S, et al. PI(3)Kγ has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol. 2007;9:86–91. doi: 10.1038/ncb1517. [DOI] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Gomez-Cambronero J, Horn J, Paul CC, Baumann MA. Granulocyte-macrophage colony-stimulating factor is a chemoattractant cytokine for human neutrophils: involvement of the ribosomal P70 S6 kinase signaling pathway. J Immunol. 2003;171:6846–6855. doi: 10.4049/jimmunol.171.12.6846. [DOI] [PubMed] [Google Scholar]
- Gong JH, Ratkay LG, Waterfield JD, Clark-Lewis I. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-Lpr mouse model. J Exp Med. 1997;186:131–137. doi: 10.1084/jem.186.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halloran MM, Woods JM, Strieter RM, Szekanecz Z, Volin MV, Hosaka S, Haines GK, III, Kunkel SL, Burdick MD, et al. The role of an epithelial neutrophil-activating peptide-78-like protein in rat adjuvant-induced arthritis. J Immunol. 1999;162:7492–7500. [PubMed] [Google Scholar]
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Karlsson R, Katsamba PS, Nordin H, Pol E, Myszka DG. Analyzing a kinetic titration series using affinity biosensors. Anal Biochem. 2006;349:136–147. doi: 10.1016/j.ab.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLCβ-2 and β-3 and PI3K γ in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- Matthys P, Hatse S, Vermeire K, Wuyts A, Bridger G, Henson GW, De Clercq E, Billiau A, Schols D. AMD3100, a potent and specific antagonist of the stromal cell-derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-γ receptor-deficient mice. J Immunol. 2001;167:4686–4692. doi: 10.4049/jimmunol.167.8.4686. [DOI] [PubMed] [Google Scholar]
- Neptune ER, Bourne HR. Receptors induce chemotaxis by releasing the βγ subunit of Gi, not by activating Gq or Gs. Proc Natl Acad Sci U S A. 1997;94:14489–14494. doi: 10.1073/pnas.94.26.14489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Takeya M, Yoshimura T, Takagi K, Takahashi K. The role of monocyte chemoattractant protein-1 (MCP-1) in the pathogenesis of collagen-induced arthritis in rats. J Pathol. 1997;182:106–114. doi: 10.1002/(SICI)1096-9896(199705)182:1<106::AID-PATH816>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Okada T, Sakuma L, Fukui Y, Hazeki O, Ui M. Blockage of chemotactic peptide-induced stimulation of neutrophils by wortmannin as a result of selective inhibition of phosphatidyl inositol 3-kinase. J Biol Chem. 1994;269:3563–3567. [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Structural basis of function in heterotrimeric G proteins. Q Rev Biophys. 2006;39:117–166. doi: 10.1017/S0033583506004306. [DOI] [PubMed] [Google Scholar]
- Plater-Zyberk C, Hoogewerf AJ, Proudfoot AEI, Power CA, Wells TNC. Effect of a CC chemokine receptor antagonist on collagen induced arthritis in DBA/1 mice. Immunol Lett. 1997;57:117–120. doi: 10.1016/s0165-2478(97)00075-8. [DOI] [PubMed] [Google Scholar]
- Podolin PL, Bolognese BJ, Foley JJ, Schmidt DB, Buckley PT, Widdowson KL, Jin Q, White JR, Lee JM, Goodman RB, et al. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit. J Immunol. 2002;169:6435–6444. doi: 10.4049/jimmunol.169.11.6435. [DOI] [PubMed] [Google Scholar]
- Posadas I, Bucci M, Roviezzo F, Rossi A, Parente L, Sautebin L, Cirino G. Carrageenan-induced mouse paw oedema is biphasic, age-weight dependent and displays differential nitric oxide cyclooxygenase-2 expression. Br J Pharmacol. 2004;142:331–338. doi: 10.1038/sj.bjp.0705650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rückle T, Schwarz MK, Rommel C. PI3Kγ inhibition: towards an ‘aspirin of the 21st century’? Nat Rev Drug Discov. 2006;5:903–918. doi: 10.1038/nrd2145. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, et al. Function of PI3K in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- Scott JK, Huang SF, Gangadhar BP, Samoriski GM, Clapp P, Gross RA, Taussig R, Smrcka AV. Evidence that a protein-protein interaction ‘hot spot’ on heterotrimeric G protein βγ subunits is used for recognition of a subclass of effectors. EMBO J. 2001;20:767–776. doi: 10.1093/emboj/20.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant G, Weiner OD, Hezmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siqueira-Junior JM, Peters RR, Brum-Fernandes AJ, Ribeiro-do-Valle RM. Effects of valeryl salicylate, a COX-1 inhibitor, on models of acute inflammation in mice. Pharmacol Res. 2003;48:437–443. doi: 10.1016/s1043-6618(03)00188-9. [DOI] [PubMed] [Google Scholar]
- Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT. A novel, phosphoinositide 3-kinase activity in myeloid-derived cells is activated by G-protein βγ-subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- Stephens LR, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AV, Thelen M, Cadwallader K, Tempst P, Hawkins PT. The Gβγ sensitivity of a PI3K is dependent upon a tightly associated adaptor, P101. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Stoyanov B, Volinia S, Hanck T, Rubio I, Loubchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nurnberg B, et al. Cloning and characterization of g protein-activated human phosphoinositide-3 kinase. Science. 1995;269:690–693. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- Suire S, Condliffe AM, Ferguson GJ, Ellson CD, Guillou H, Davidson K, Welch H, Coadwell J, Turner M, Chilvers ER, et al. Gβγs and the ras binding domain of P110γ are both important regulators of PI3Kγ Signalling in Neutrophils. Nat Cell Biol. 2006;8:1303–1309. doi: 10.1038/ncb1494. [DOI] [PubMed] [Google Scholar]
- Welch HCE, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR. P-Rex1, a PtdIns(3,4,5)P3- and Gβγ-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- Wu D, Huang CK, Jiang H. Roles of phospholipid signaling in chemoattractant-induced responses. J Cell Sci. 2000;113:2935–2940. doi: 10.1242/jcs.113.17.2935. [DOI] [PubMed] [Google Scholar]
- Xanthou G, Duchesnes CE, Williams TJ, Pease JE. CCR3 functional responses are regulated by both CXCR3 and its ligands CXCL9, CXCL10 and CXCL11. Eur J Immunol. 2003;33:2241–2250. doi: 10.1002/eji.200323787. [DOI] [PubMed] [Google Scholar]
- Yang YF, Mukai T, Gao P, Yamaguchi N, Ono S, Iwaki H, Obika S, Imanishi T, Tsujimura T, Hamaoka T, et al. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur J Immunol. 2002;32:2124–2132. doi: 10.1002/1521-4141(200208)32:8<2124::AID-IMMU2124>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Zhao T, Nalbant P, Hoshino M, Dong X, Wu D, Bokoch GM. Signaling requirements for translocation of P-Rex1, a key Rac2 exchange factor involved in chemoattractant-stimulated human neutrophil function. J Leukoc Biol. 2007;81:1127–1136. doi: 10.1189/jlb.0406251. [DOI] [PubMed] [Google Scholar]