

Abstract
Activated protein C (APC) is a serine protease with anticoagulant and direct cytoprotective activities. Early postischemic APC application activates the cellular protein C pathway in brain endothelium and neurons, which is neuroprotective. Whether late APC administration after a transient ischemic attack is neuroprotective and whether APC influences brain repair is not known. Here, we determined safety and efficacy of late APC and tissue-plasminogen activator (tPA) administrations in a mouse model of transient brain ischemia. tPA given at 6 h after onset of ischemia killed all mice within 2 d, whereas APC given at 6 or 24 h after ischemia onset improved significantly functional outcome and reduced spread of the ischemic lesion. At 7 d postischemia, APC multiple dosing (0.8 mg/kg, i.p.) at 6–72 or 72–144 h enhanced comparably cerebral perfusion in the ischemic border by ∼40% as shown by in vivo lectin-FITC angiography, blocked blood–brain barrier leakage of serum proteins, and increased the number of endothelial replicating cells by 4.5- to 4.7-fold. APC multidosing at 6–72 h or 72–144 h increased proliferation of neuronal progenitor cells in the subventricular zone (SVZ) by 40–50% and migration of newly formed neuroblasts from the SVZ toward the ischemic border by approximately twofold. The effects of APC on neovascularization and neurogenesis were mediated by protease-activated receptor 1 and were independent of the reduction by APC of infarction volume. Our data show that delayed APC administration is neuroprotective and mediates brain repair (i.e., neovascularization and neurogenesis), suggesting a significant extension of the therapeutic window for APC intervention in postischemic brain.
Keywords: activated protein C, serine protease, transient cerebral ischemia, neuroprotection, neurogenesis, angiogenesis
Introduction
Activated protein C (APC) is a serine protease with systemic anticoagulant activity, which is mediated by irreversible proteolytic inactivation of factors Va and VIIIa with contributions by various cofactors (Mosnier et al., 2007). Independent of its anticoagulant activity, APC exerts direct cellular effects that are mediated by the protein C cellular pathway resulting in the following: (1) cytoprotective alteration of gene expression profiles, (2) antiinflammatory activities, (3) antiapoptotic activity, and (4) protection of endothelial barriers (Joyce et al., 2001; Riewald et al., 2002; Cheng et al., 2003; Dömötör et al., 2003; Mosnier and Griffin, 2003; Feistritzer and Riewald, 2005; Finigan et al., 2005). APC substrates for anticoagulant actions are factors Va and VIIIa, and, for cytoprotective actions, protease-activated receptor 1 (PAR1) (Mosnier et al., 2007).
Early postischemic application of APC during cerebral arterial occlusion is neuroprotective in rodent models of transient brain ischemia (Shibata et al., 2001; Cheng et al., 2003) or embolic stroke (Zlokovic et al., 2005). APC inhibits the death of neurons from NMDA, tissue-plasminogen activator (tPA), or staurosporine toxicity in vitro and in vivo (Guo et al., 2004; Liu et al., 2004), blocks p53-mediated apoptosis in brain endothelium (Cheng et al., 2003), and reduces tPA-mediated neurotoxicity and blood–brain barrier (BBB) breakdown (Liu et al., 2004; Cheng et al., 2006). APC also exhibits angiogenic activity in vitro and in vivo (Uchiba et al., 2004). Recent studies suggest that APC protects against diabetic endothelial and glomerular injury (Isermann et al., 2007) and its variant is protective in a mouse model of multiple sclerosis (Han et al., 2008).
In addition to reducing organ damage in different animal models of sepsis, thrombosis, and ischemic injury (Griffin et al., 2002; Mosnier et al., 2007), APC substantially reduces mortality in patients with severe sepsis (Bernard et al., 2001). Currently, the safety and feasibility of intravenous APC is being newly studied in patients with acute ischemic stroke who are given APC within a relatively narrow therapeutic window [APCAST (Activated Protein C in Acute Stroke Trial); http://clinicaltrials.gov/ct2/show/NCT00533546?term=apc&rank=25]. A key question in brain ischemia therapy is how late after an ischemic insult can patients still benefit from a given treatment? Whether late administration of APC after a transient ischemic brain attack is neuroprotective and whether APC affects positively or negatively brain repair mechanisms and regeneration of tissue damaged by ischemia is not known. Here, we use a transient ischemia model in mice to address these questions and compare late interventions with APC versus tPA, the only Food and Drug Administration (FDA)-approved agent for stroke, in postischemic brain.
Materials and Methods
Reagents and antibodies.
Recombinant mouse APC was prepared from culture medium of a stably transfected K293 cell line expressing protein C, as described previously (Fernández et al., 2003). Recombinant mouse tPA was purchased from Innovative Research. FITC-labeled Lycopersicon esculentum (tomato) lectin (FL-1171) was purchased from Vector Laboratories. For antibodies, see supplemental methods (available at www.jneurosci.org as supplemental material).
Experimental animals.
We used 3- to 4-month-old C57BL/6 male mice (The Jackson Laboratory) and 3- to 4-month-old male PAR1-null mice on 97% C57BL/6 background obtained from Dr. S. Coughlin, University of California, San Francisco, San Francisco, CA (Connolly et al., 1996). We used a total of 124 mice.
Stroke model.
We used a 1 h transient middle cerebral artery occlusion (MCAO) suture model, as we reported previously (Wang et al., 1997; Cheng et al., 2003, 2006; Liu et al., 2004), followed by 3–7 d reperfusion. All procedures were conducted according to the National Institutes of Health guidelines and approved by the Animal Care Committee at the University of Rochester. Mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine.
Physiological parameters.
See supplemental methods (available at www.jneurosci.org as supplemental material).
Treatment schedules.
The first group of mice received 6 h after ischemia either mouse recombinant APC (0.2 mg/kg via the femoral vein 50% as a bolus and 50% as a 30 min infusion) or mouse recombinant tPA (1 mg/kg, i.v.; 10% as a bolus followed by 90% infusion over 30 min) or vehicle. In the second group, mice received 24 h after ischemia APC at two dose levels (i.e., 0.8 mg/kg, i.p.; 1.6 mg/kg, i.p.). It is of note that the plasma APC pharmacokinetic profile after an intravenous administration of 0.2 mg/kg APC (50% bolus:50% infusion over 30 min) was comparable with the APC plasma profile after an intraperitoneal administration of 0.8 mg/kg APC (see Results). In the third group, mice were treated with a multiple-dose APC (M6–72) (0.8 mg/kg, i.p.) at 6, 24, 48, and 72 h after ischemia. In the fourth group, mice were treated with a multiple-dose APC (M72–144) (0.8 mg/kg, i.p.) at 72, 96, 120, and 144 h after ischemia. In the fifth group, PAR1-null mice were treated with vehicle or a multiple-dose APC (M6–72) (0.8 mg/kg, i.p.) at 6, 24, 48, and 72 h after ischemia.
Behavioral studies.
We used a motor neurological deficit score and rotarod test to evaluate motor function, the beam balance test to evaluate sensorimotor coordination and forelimb and hindlimb functionality, and the wire grip test to evaluate the vestibulomotor reflex, as reported previously (Aronowski et al., 1996; Chen et al., 2003a; Zlokovic et al., 2005). See supplemental methods (available at www.jneurosci.org as supplemental material).
Neuropathological analysis.
Coronal brain sections (1 mm thick) were cut and stained with 2% TTC (2,3,5-triphenyltetrazolium chloride) solution in PBS for 5 min at 37°C, as reported previously (Shibata et al., 2001; Cheng et al., 2003). The injury volume (in cubic millimeters) was calculated by multiplying the surfaces of all injured areas in square millimeters by the thickness of brain sections (i.e., 1 mm). Brain swelling volume was calculated by subtracting the volume of the contralateral nonischemic hemisphere from the volume of the ipsilateral ischemic hemisphere, as described previously (Wang et al., 1997).
Hemoglobin assay.
To determine whether tPA or APC treatment produced microhemorrhages in brain tissue, we used a spectrophotometric hemoglobin assay, as we reported previously (Shibata et al., 2001; Cheng et al., 2006). See supplemental methods (available at www.jneurosci.org as supplemental material).
APC plasma levels.
In separate experiments, plasma APC levels were determined after the intravenous or intraperitoneal APC administration using a mouse APC-specific ELISA (Fernández et al., 2006). See supplemental methods (available at www.jneurosci.org as supplemental material).
In vivo lectin-FITC angiography.
In vivo lectin-FITC angiography. FITC-labeled L. esculentum (tomato) lectin at 10 mg/kg (Beck et al., 2000) was administered via the femoral vein in anesthetized mice 5 min before killing at day 7 of reperfusion. See supplemental methods (available at www.jneurosci.org as supplemental material).
5-Bromo-2′-deoxyuridine labeling in vivo.
5-Bromo-2′-deoxyuridine (BrdU) was used to measure proliferation of neuronal progenitor cells and migration of neuroblasts in combination with other markers of neurogenesis [i.e., nestin and doublecortin (Dcx) and/or βIII-tubulin (TuJ1), respectively], as reported previously (Z. G. Zhang et al., 2000; R. Zhang et al., 2002; Chen et al., 2003b; Sun et al., 2003; Wang et al., 2004; Yamashita et al., 2006; Breunig et al., 2007; R. L. Zhang et al., 2007). See supplemental methods (available at www.jneurosci.org as supplemental material).
Immunohistochemistry.
Coronal cryostat sections (20 μm) were mounted on slides, postfixed with acetone, and stored at −20°C until immunostaining.
For the analysis of blood vessels, sections from brains that were used for in vivo lectin-FITC angiography were incubated with an antibody to CD31 or CD105 overnight at 4°C followed by incubation with the secondary antibodies [e.g., anti-rat IgG-Cy5 to detect CD31+ endothelial structures (Cheng et al., 2006) or an anti-mouse IgG-Cy3 to detect CD105+ endothelial structures or mouse serum IgG leakage across the BBB]. In some experiments, sections were incubated with an antibody to Ki-67, a marker for cells undergoing mitosis (Breunig et al., 2007) and an antibody to CD31 followed by incubation with anti-mouse IgG-FITC and anti-rat IgG-Cy3, respectively. All secondary antibodies were incubated for 1 h at 37°C.
For the analysis of neurogenesis, brain sections were initially prepared for BrdU staining as explained above, and incubated with primary antibodies to nestin, BrdU, and Dcx followed by incubation with their respective secondary antibodies (e.g., anti-mouse IgG-Cy3 to detect nestin, anti-rat IgG-Cy5 or IgG-Cy3 to detect BrdU, and anti-rabbit IgG-FITC to detect Dcx). All secondary antibodies were incubated for 1 h at 37°C. See supplemental methods (available at www.jneurosci.org as supplemental material).
Image analysis and quantification.
Confocal laser-scanning three-color microscopy (LSM510 META; Carl Zeiss MicroImaging) was used to acquire images from immunostained sections. Argon laser (excitation, 488 nm; emission, 500–550 nm), helium–neon laser (excitation, 543 nm; emission, 560–615 nm), and helium–neon laser (excitation, 633 nm; emission, 650–710 nm) were used to excite FITC, Cy3, and Cy5, respectively. In studies focused on analysis of blood vessels, we used the following pseudocolors: CD31-Cy5, blue; lectin-FITC, green; IgG-Cy3, red. In studies focused on analysis of neuronal progenitor cells and neuroblasts, the following pseudocolors were used: nestin-Cy3, red; BrdU-Cy5, blue; Dcx-FITC, green. See supplemental methods (available at www.jneurosci.org as supplemental material).
Statistics.
Data are presented as mean ± SEM. To compare differences in the infarction and edema volumes between different treatments, we used Student's t test. The differences in motor neurological deficit score and beam balance score were determined by Kruskal–Wallis nonparametric test. The ANOVA followed by Tukey's post hoc test was used for all other comparisons. Differences were considered statistically significant if p < 0.05.
Results
In all studies, we used mouse rather than human recombinant proteins to avoid potentially confounding effects of species specificities.
Physiological variables
Physiological variables including PaO2, PaCO2, pH, hematocrit, mean arterial blood pressure, and rectal temperature remained within normal limits during the MCAO surgical procedure compared with basal values (data not shown), as reported previously (Shibata et al., 2001; Cheng et al., 2006). MCAO resulted in ∼80% reduction in the cerebral blood flow (CBF) compared with the preocclusion baseline values. During reperfusion, the CBF recovered within the first 2 h to 85–87% of the baseline values (data not shown), as reported previously (Shibata et al., 2001).
APC single-dose neuroprotective therapy at 6 h after transient MCAO
First, we compared the safety and efficacy of mouse recombinant tPA alone (1 mg/kg, i.v.) at a dose equivalent to an FDA-approved tPA dose in humans (National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995) and mouse recombinant APC alone (0.2 mg/kg, i.v.) at a dose that was highly neuroprotective in mice after an early application (<1 h) after transient ischemia (Cheng et al., 2003, 2006; Liu et al., 2004). tPA and APC were both administered 6 h after ischemia onset. tPA killed all mice within 2 d of reperfusion and increased by ∼50% the infarct volume (Fig. 1A–E), consistent with its reported neurotoxicity seen previously in models of transient brain ischemia (Wang et al., 1998; Nagai et al., 1999; Nicole et al., 2001; Liu et al., 2004; Cheng et al., 2006). However, all animals treated with APC survived. APC-treated mice did not exhibit macroscopic bleeding or tissue microhemorrhages as demonstrated by complete absence of extravasated red blood cells on brain tissue sections analysis (data not shown). In contrast to a marked increase in hemoglobin tissue levels after tPA treatment, mice treated with APC did not have signs of occult bleeding, as shown by negligible brain tissue levels of hemoglobin (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

APC is neuroprotective when given at 6 h after transient brain ischemia in mice. A–E, Motor neurological score (A), wire grip latency (B), beam balance score (C), rotarod latency (D), and infarct volume and brain swelling (edema) (E) in mice subjected to 1 h MCAO and 3 d reperfusion, and treated intravenously with either vehicle, mouse recombinant APC, or mouse recombinant tPA. Vehicle (blue), APC (0.2 mg/kg; 50% bolus:50% infusion over 30 min; red), or tPA (1 mg/kg; 10% bolus:90% infusion over 30 min; pink) were administered 6 h after an ischemic insult. Shown are mean ± SEM (n = 6 mice per group). F, Incidence and topography of infarct at level of optic chiasm in vehicle-treated and APC-treated mice. Shown are mean ± SEM (n = 5–6 mice per group). G, Plasma levels of APC after intravenous administration of APC (0.2 mg/kg; 50% bolus:50% infusion over 30 min) were determined in a separate group of control animals as described previously (Fernández et al., 2006) Shown are mean ± SEM (n = 6 mice per group). ap < 0.5; bp < 0.01.
All APC-treated mice had significant improvements in motor and sensorimotor activities, motor coordination, and the vestibular motor reflex, as shown by the corresponding behavioral tests including motor neurological score, wire grip latency, beam balance score, and rotarod latency (Aronowski et al., 1996; Chen et al., 2003a; Zlokovic et al., 2005) (Fig. 1A–D). The first group of APC-treated mice was killed for neuropathological analysis after 3 d. This analysis showed that APC significantly reduced the infarct volume by >45% as well as the edema volume (Fig. 1E). The infarction topography indicated that all vehicle-treated mice had significant injury in the ipsilateral cortex and lateral striatum and ≥50% of the mice had changes in the medial striatum, whereas <50% mice exhibited changes in the dorsomedial and ventromedial cortex (Fig. 1F). Late APC treatment localized injury to a smaller area in the lateral cortex and lateral striatum and reduced significantly injury in other regions.
Measurements of APC circulating in plasma (Fernández et al., 2006) indicated that the used intravenous administration protocol for APC resulted in a transient rise in APC levels with a plateau of ∼6 nm over 30 min, which was about two orders of magnitude greater than the basal levels of endogenous circulating APC in mice (Fig. 1G).
APC single-dose and multiple-dose therapies at 6–72 and 72–144 h after transient ischemia
Next, we developed an APC intraperitoneal administration protocol suitable for treatment at multiple later time points after stroke with minimal discomfort to mice. The pilot studies suggested that administration of 0.8 mg/kg intraperitoneal APC resulted in an APC plasma pharmacokinetic profile that resembled that obtained with 0.2 mg/kg intravenous APC (Fig. 2A). However, 0.8 mg/kg single-dose APC given 24 h after 1 h of ischemia gave only marginal neuroprotection (data not shown). In contrast, doubling the dose to 1.6 mg/kg APC intraperitoneal given at 24 h after ischemia onset produced within 7 d significant improvements in body weight, motor neurological score, beam balance score, and wire grip latency (Fig. 2B–E), and reduced by ∼35% the infarct volume (Fig. 2F). Thus, APC late administration had a dose-dependent neuroprotective effect, similar to that as seen with early (≤1 h) APC postischemic applications (Shibata et al., 2001; Cheng et al., 2003, 2006).
Figure 2.

Neuroprotection is provided by single-dose or delayed multiple-dose APC therapies at 6–72 and 72–144 h after transient ischemia in mice. A, Plasma levels of APC after intraperitoneal administration of mouse recombinant APC (0.8 mg/kg) were determined in a separate group of animals. Values are mean ± SEM (n = 6 mice per group). B–F, Body weight (B), motor neurological score (C), beam balance score (D), wire grip latency (E), and infarct volume (F) in mice subjected to 1 h MCAO and 7 d reperfusion, and treated intraperitoneally with either vehicle (blue) or APC that was administered in a single-dose (1.6 mg/kg, i.p.; S, green) at 24 h after the MCAO or as multiple doses M6–72 (0.8 mg/kg, i.p., at 6, 24, 48, and 72 h post-MCAO; red) and M72–144 (0.8 mg/kg, i.p., at 72, 96, 120, and 144 h post-MCAO; brown). Shown are mean ± SEM (n = 6 mice per group). G, Incidence and topography of infarct at level of optic chiasm in mice treated with vehicle or multiple-dose APC M6–72 (0.8 mg/kg, i.p.) after ischemia onset. Areas 3 and 4, and 1, 2, and 3 were used for the analysis in Figures 3 and 4, and 5 and 6, respectively. ap < 0.5; bp < 0.01; ns, nonsignificant.
Multiple-dose APC M6–72 therapy (i.e., 0.8 mg/kg, i.p., APC at 6, 24, 48, and 72 h) after ischemia onset resulted within 7 d in striking neuroprotection compared with single-dose therapy both in terms of functional recovery (Fig. 2B–E) and in a remarkable 50% reduction in the infarct volume (Fig. 2F). The infarction topography in vehicle-treated animals at 7 d confirmed similar injury distribution as seen at 3 d, but the total area affected by injury was somewhat smaller at 7 d, consistent with the smaller infarction volume (Fig. 2G), as reported previously (Sun et al., 2003). Multiple-dose M6–72 APC treatment localized after 7 d the infarction mainly to a smaller area in the lateral cortex and lateral striatum and reduced significantly injury to other regions.
We also studied the effect of M72–144 very late multidose APC therapy (0.8 mg/kg, i.p.) administered at 72, 96, 120, and 144 h after ischemia onset. Our data show that APC M72–144 multidose treatment beginning 3 d after ischemia onset did not reduce the infarct volume (Fig. 2F), which is consistent with the concept that the infarcted lesion is likely to mature by 3 d after transient MCAO. However, M72–144 multidose APC therapy had a significant effect on functional recovery at day 7 (Fig. 2B–E), suggesting the beneficial effects of APC may extend beyond its effect of reducing the size of the ischemic lesion.
APC mediates neovascularization after transient ischemia
The analysis of blood vessels in the ischemic border by simultaneous three-color confocal laser-scanning microscopy indicated that, compared with vehicle, APC multidose M6–72 therapy after ischemia onset, increased at 7 d the total capillary length of CD31+ capillary endothelial structures in the ischemic border in the cortex and striatum by 32 and 40%, respectively (Fig. 3A–C, white bars). These results were confirmed by measuring the total length of CD105+ brain microvessels in the periinfarct areas of the same respective regions (data not shown). To determine whether APC-mediated increase in total capillary length reflects an increase in cerebral perfusion and represents functional vessels, we performed in vivo L. esculentum lectin-FITC (molecular weight, 100 kDa) angiography (Beck et al., 2000), as described in Materials and Methods. This experiment showed that the total length of brain capillaries perfused in vivo with lectin-FITC was increased in the ischemic border in the cortex and striatum by a comparable 35 and 41%, respectively (Fig. 3A–C, black bars).
Figure 3.

APC therapies at 6–72 and 72–144 h after transient ischemia enhance neovascularization in the ischemic border in mice via PAR1. A, B, Expression of CD31 (brain endothelial marker), in vivo systemic lectin-FITC angiography, and serum IgG leakage studied simultaneously by three-color confocal laser-scanning microscopy in the ischemic border in the striatum 7 d after 1 h transient MCAO in vehicle-treated mice (A) or APC-treated mice (APC at 0.8 mg/kg, i.p., at 6, 24, 48, and 72 h after ischemia) (B). Inset was taken from the merged image. Scale bar, 50 μm. CD31, blue; lectin-FITC, green; IgG, red. C, Total capillary length of CD31-positive structures (open bars) and of in vivo lectin-perfused vessels (closed bars) in the cortex and striatum in the periinfarct regions 7 d after 1 h transient MCAO in mice treated with vehicle and APC multiple doses M6–72 (0.8 mg/kg, i.p., at 6, 24, 48, and 72 h post-MCAO) and M72–144 (0.8 mg/kg, i.p., at 72, 96, 120, and 144 h post-MCAO), and in PAR1-null mice (PAR1−/−) treated with vehicle or APC multiple-dose M6–72. D, Quantification of the IgG signal intensity in the striatum 7 d after 1 h transient MCAO in mice treated with vehicle, APC multiple-dose M6–72, or APC single dose (1.6 mg/kg, i.p.; S) 24 h post-MCAO, compared with sham-operated controls and in PAR1−/− mice treated with vehicle or APC multiple-dose M6–72. Shown are mean ± SEM (n = 5–6 mice per group). For sham-operated control mice, n = 3. ap < 0.5; bp < 0.01; ns, nonsignificant.
APC M72–144 multidose therapy produced a significant increase in total capillary length at 7 d after ischemia onset that was comparable with that seen with APC M6–72 multidose therapy after ischemia onset (Fig. 3C). Single-dose APC (1.6 mg/kg, i.p.) given at 24 h after ischemia onset increased also cerebral perfusion in the periinfarct regions as shown by in vivo lectin-FITC angiography, but the effects were less pronounced compared with APC M6–72 and M72–144 multidose therapies (data not shown). These results are consistent with previous findings demonstrating that APC increases angiogenesis from systemic endothelial cells both in vitro and in vivo (Uchiba et al., 2004). The length of vessels in the periinfarct areas in mice challenged with ischemia was increased by ∼30% compared with controls (Fig. 3C) consistent with previous reports (Lin et al., 2002; Hayashi et al., 2003; Tomita et al., 2005; Liu et al., 2007).
Single-dose APC therapy given at 24 h after ischemia onset and multiple-dose APC therapy (as above) reduced substantially postischemic BBB disruption as shown by blockade of postischemic leakage of serum proteins (e.g., IgG) by 63 and 80%, respectively, corrected for the residual vascular IgG immunoreactivity and background values obtained in sham-operated controls (Fig. 3A,B,D). These results are consistent with findings that APC enhances the endothelial barriers in vitro (Feistritzer and Riewald, 2005; Finigan et al., 2005) and in vivo, as for example after tPA-mediated BBB breakdown (Cheng et al., 2006). The intensity of IgG staining 7 d after stroke was increased by approximately fivefold, indicating severe BBB disruption (Fig. 3D), as reported previously (Z. G. Zhang et al., 2002; Yu et al., 2007).
Single-dose APC given at 24 h after ischemia onset and multiple-dose APC therapies given at 6–72 and 72–144 h after ischemia onset increased by 2.5-, 4.5-, and 3.8-fold, respectively, the numbers of Ki-67+/CD31+ endothelial cells (Fig. 4A–C). Ki-67 is a marker for cells undergoing mitosis (Breunig et al., 2007), suggesting APC increased the number of endothelial replicating cells. Similar results were obtained by double Ki-67 and CD105 staining (data not shown).
Figure 4.
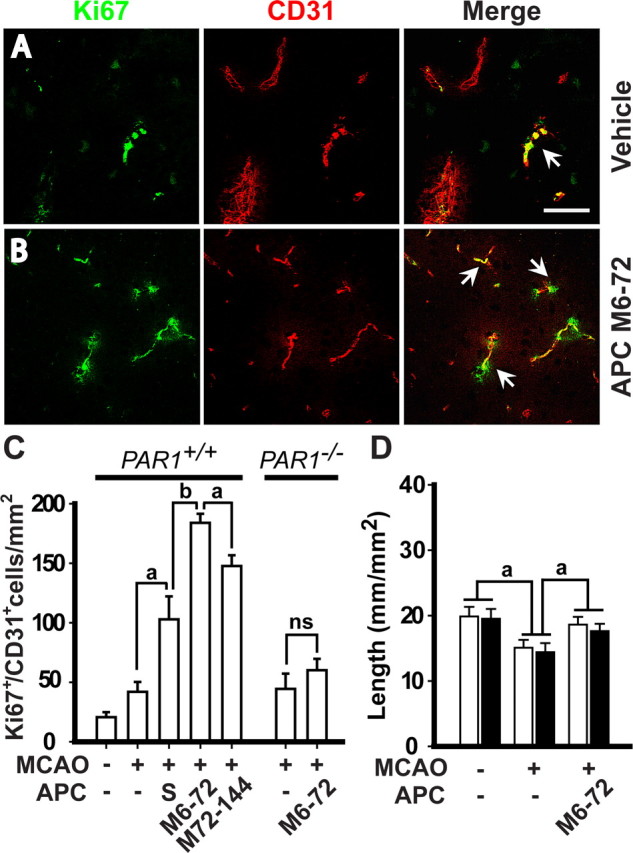
APC therapies at 6–72 and 72–144 h after transient ischemia increase the number of newly replicating endothelial cells in the ischemic border. A, B, Ki-67 and CD31 colocalization in the ischemic border in the striatum in a mouse treated with vehicle or with multiple-dose APC M6–72 (0.8 mg/kg, i.p., at 6, 24, 48, and 72 h post-MCAO, respectively). Tissues were analyzed at 7 d after the transient MCAO. Scale bar, 25 μm. C, Ki-67+/CD31+ cells in the striatum in mice treated with vehicle, single-dose of APC (1.6 mg/kg, i.p., 24 h after ischemia onset; S) or multiple-doses of APC M6–72 and M72–144 (0.8 mg/kg, i.p., at 72, 96, 120, and 144 h post-MCAO) and in PAR1−/− mice treated with vehicle or APC multiple-dose M6–72, 7 d after 1 h transient MCAO, compared with controls. Shown are mean ± SEM (n = 6 mice per group). D, Total capillary length of CD31-positive structures (open bars) and of in vivo lectin-perfused vessels (closed bars) in the cortex in periinfarct region 3 d after 1 h transient MCAO in mice treated with vehicle or multiple-dose APC M6–72. Shown are mean ± SEM (n = 6 mice per group). ap < 0.5; bp < 0.01; ns, nonsignificant.
To address whether the increased vessel length in the periinfarct region with APC treatment represents only the generation of new vessels as opposed to protection of some vessels that are otherwise lost because of ischemia at 2–3 d after MCAO, we studied how APC treatment affects the vessel length at 3 d after ischemia onset before significant appearance of Ki67-positive vascular cells. In vehicle-treated MCAO mice at 3 d after ischemia onset, there was a modest loss of ∼22% of total vessel length (Fig. 4D) consistent with the reported 12% loss of brain vessels because of 45 min ischemia at day 1 (Liu et al., 2004). APC completely prevents a modest loss of vessels to ischemia at 24 h after ischemia onset (Liu et al., 2004), and here we show that the length of vessels at 3 d postischemia in APC-treated group was similar to the length of vessels in sham-operated controls (Fig. 4D), suggesting complete protection. Quantitatively, it appears that the effect of APC on promoting generation of new vessels and on decreasing loss of existing vessels contributed to 82 and 18%, respectively, to the overall effects of APC on increasing total length of vessels at day 7 after ischemia onset (Fig. 3C).
APC promotes neurogenesis after transient MCAO
As shown by three-color confocal laser-scanning microscopy, APC multidose M6–72 and M72–144 therapies increased in the ipsilateral subventricular zone (SVZ) within 7 d after ischemia onset the number of Nestin+/BrdU+/Dcx− neuronal progenitor cells by 40–45% and the number of Nestin+/BrdU+/Dcx+ forming neuroblasts by ∼50% (Fig. 5A–C). The number of Nestin+/BrdU+/Dcx+ neuroblasts migrating toward the periinfarct areas along the corpus callosum was increased by APC multiple-dose M6–72 and M72–144 therapies by 2- and 1.8-fold, respectively (Fig. 6A–C). The number of Dcx+/Ki-67+ neuroblasts accumulated in the clusters within the ischemic boundary in the striatum and cortex and dispersing into the periinfarct tissue was increased by approximately twofold by APC multidose therapy (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Consistent with the concept that multiple markers should be used in search for newborn cells in the brain (Breunig et al., 2007), the Dcx+ neuroblasts were also TuJ1+ (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Compared with multidose treatment, single-dose APC treatment given at 24 h after ischemia onset produced a significant, although more moderate, beneficial effect on postischemic neurogenesis (Figs. 5C, 6C). Data for control vehicle-treated animals are consistent with reports showing a peak in postischemic neurogenesis in rodents 1–3 weeks after stroke (Yamashita et al., 2006; R. L. Zhang et al., 2007).
Figure 5.

APC therapies at 6–72 and 72–144 h after transient ischemia increase the number of neuronal progenitor cells and neuroblasts in the SVZ in mice via PAR1. A, B, Immunostaining for nestin, BrdU, and Dcx by three-color confocal laser-scanning microscopy in the SVZ in the ischemic side 7 d after 1 h transient MCAO in mice treated with either vehicle (A) or APC M6–72 (0.8 mg/kg, i.p., at 6, 24, 48, and 72 h after ischemia) (B). Nestin, red; BrdU, blue; Dcx, green. Scale bar, 50 μm. C, Nestin+/BrdU+/Dcx− neuronal progenitor cells and nestin+/BrdU+/Dcx+ neuroblasts in the SVZ after a single dose of APC (1.6 mg/kg, i.p., given at 24 h after ischemia onset; S) or APC multiple doses M6–72 and M72–144 (0.8 mg/kg, i.p., at 72, 96, 120, and 144 h post-MCAO), and in PAR1−/− mice treated with vehicle or APC multiple-dose M6–72, in the ischemic side 7 d after the transient MCAO compared with sham-operated controls. Shown are mean ± SEM (n = 6 mice per group). For sham-operated control mice, n = 3. ap < 0.5; ns, nonsignificant.
Figure 6.

APC therapies at 6–72 h and 72–144 h after transient ischemia increase the number of migrating neuroblasts toward the periinfarct areas along the corpus callosum. A, B, Migration of Dcx+/BrdU+ neuroblasts along the corpus callosum toward the ischemic penumbra 7 d after the transient MCAO in mice treated with vehicle or multiple-dose APC M6–72 (0.8 mg/kg, i.p., at 6, 24, 48, and 72 h after ischemia onset), respectively. Dcx, green; BrdU, red. C, Graph shows the effects of APC single dose (1.6 mg/kg, i.p., at 24 h after ischemia onset; S) and APC multiple doses M6–72 and M72–144 (0.8 mg/kg, i.p., at 72, 96, 120, and 144 h post-MCAO) on the number of Dcx+/BrdU+ neuroblasts migrating toward the ischemic border across the corpus callosum. The number of Dcx+/BrdU+ migrating neuroblasts along the corpus callosum in PAR1−/− mice treated with vehicle or APC multiple-dose M6–72. Scale bar, 50 μm. Shown are mean ± SEM (n = 6 mice per group). For sham-operated control mice, n = 3. ap < 0.5; bp < 0.01; ns, nonsignificant.
PAR1 mediates effects of APC on postischemic neovascularization and neurogenesis
The length of perfused vessels in the periinfarct area in cortex and striatum in PAR1 null mice subjected to 1 h MCAO and treated with APC multidose M6–72 therapy was similar to the respective lengths observed in nontreated PAR1 null mice, but was ∼40% lower compared with the values seen in normal treated PAR1+/+ mice (Fig. 3C). APC therapy failed to block IgG leakage in PAR1−/− mice (Fig. 3D). The number of Ki-67+/CD31+ replicating endothelial cells in PAR1−/− mice treated with APC multidose therapy was lower by 4.7-fold than in control PAR1+/+ mice treated with APC multidose therapy, but was not different from values obtained for nontreated PAR1−/− mice (Fig. 4C). The number of neuronal progenitor cells and neuroblasts in the SVZ zone and Dcx+/BrdU+ migrating neuroblasts along the corpus callosum in nontreated and multidose APC-treated PAR1−/− mice were comparable and lower by 50–55% and twofold, respectively, than in PAR1+/+ mice treated with multidose APC (Fig. 6C). These results suggest that PAR1 is required for the effects of APC on neovascularization and neurogenesis. Deletion of the PAR1 gene also resulted in loss of the beneficial effect of APC of reducing infarction volume (data not shown), consistent with previous findings (Cheng et al., 2003, 2006).
APC effects on neovascularization and neurogenesis are not attributable to the reduced infarct size
Our study with late APC multidose M72–144 therapy given beginning at 72 h after 1 h MCAO showed no effect on the infarct volume (Fig. 2F). However, this APC therapy mediated significant increases in neovascularization and neurogenesis compared with untreated control group (Figs. 4–6), suggesting the effects of APC on brain repair mechanisms were independent of its effect on infarct size. To provide additional evidence that APC effects on neovascularization and neurogenesis obtained after an earlier initiated postischemic therapy are not secondary to the reduced infarct size, we compared the postischemic vascular and neurogenesis responses after 45 min MCAO with no treatment versus 1 h MCAO with APC multidose M6–72 h treatment. The 45 min MCAO produced an infarct lesion at 7 d that was by 41% smaller than that produced by 1 h MCAO (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The infarct size produced by 45 min MCAO without APC treatment was comparable with that obtained with APC multidose M6–72 neuroprotective therapy after 1 h MCAO (Fig. 2F). Despite the similar size of the infarct volume for these two protocols, the protocol using APC multidose therapy compared with that without APC therapy was associated with ∼35–40% significantly greater total length of perfused capillaries and a twofold increase in the number of migrating neuroblasts at 7 d, respectively (supplemental Fig. 6, available at www.jneurosci.org as supplemental material).
Discussion
Our major finding is that late administration of APC to mice undergoing transient focal cerebral ischemia improves functional outcome, reduces cerebral infarction, and enhances repair of damaged brain tissue by promoting postischemic cerebral angiogenesis and neurogenesis. As found previously and here, APC has multiple and unique properties in being able to protect ischemic brain tissue after a delayed administration and after an early postischemic application (Shibata et al., 2001; Cheng et al., 2003; Zlokovic et al., 2005). The properties of APC are especially notable when compared with intravenous tPA, which is protective in humans if administered within 3 h after stroke (National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995) and in rodent models of cerebral embolism if administered early (Zivin et al., 1985; Overgaard et al., 1992; Jiang et al., 2000), or compared with repair therapy with vascular endothelial growth factor (VEGF), which is protective after a delayed postischemic administration (Sun et al., 2003), but not if administered early after ischemia because it promotes hemorrhagic transformation of the infarct (Z. G. Zhang et al., 2000).
Several agents have important effects on postischemic neurogenesis and/or angiogenesis. For example, delayed administration of sildenafil has a robust effect on neurogenesis and promotes postischemic functional recovery, although it apparently does not reduce the cerebral infarction (R. Zhang et al., 2002) in contrast to APC or VEGF administration (Sun et al., 2003). Delayed therapies with statins (Chen et al., 2003b) and erythropoietin (Wang et al., 2004) can importantly enhance postischemic angiogenesis and neurogenesis and improve functional recovery in rodents, but apparently they do not reduce the infarct volume.
Although reduction of cerebral infarction after single systemic administration of APC given at 24 h after ischemia is unusual, similar findings have been reported with late administration of VEGF (Sun et al., 2003), magnesium (Heath and Vink, 1999), and cyclooxygenase-2 inhibitors (Candelario-Jalil et al., 2002). These findings suggest that APC prevents delayed neuronal cell death and spread of ischemic lesion in addition to its beneficial effects that minimize acute excitotoxic injury in both in vivo and in vitro models of NMDA excitotoxicity (Shibata et al., 2001; Cheng et al., 2003; Guo et al., 2004). The results here are also consistent with reports showing that APC protects neuronal and brain endothelial cells from mitochondria-mediated and death receptor-mediated delayed apoptosis (Guo et al., 2004; Liu et al., 2004) and that APC is protective in a mouse model of familial amyotrophic lateral sclerosis (Z. Zhong, L. Hallagan, N. Paquette, M. Thiyagarajan, R. Deane, J. A. Fernandez, S. M. Lane, T. Liu, J. H. Griffin, N. Chow, K. Stojanovic, D. W. Cleveland, and B. V. Zlokovic, unpublished observations) and multiple sclerosis (Han et al., 2008).
The substantial increase in cerebral perfusion and in the number of functional nonleaky blood vessels in the ischemic border by APC, as shown by in vivo systemic lectin-FITC angiography, could be attributable to APC-mediated postischemic survival of the existing vessels (Liu et al., 2004) and/or attributable to its angiogenic effect on newly formed brain vessels. Our analysis indicates that APC prevents almost completely ischemia-induced loss of vessels measured at day 3 after ischemia onset before the appearance of Ki67-positive vascular cells and that the increased vessel length observed at day 7 after multiple-dose APC therapy is primarily (>80%) attributable to new vessel formation. A 4.5-fold increase in the endothelial cell replication rate with APC most likely reflects an enhancement of sprouting angiogenesis, although one cannot rule out completely the possibility that APC may also enhance vasculogenesis or endothelial progenitor cells-mediated vascular formation (Arenillas et al., 2007). Future work should determine the respective contributions of sprouting angiogenesis and vasculogenesis to the observed increase in APC-mediated postischemic neovascularization. The present findings are consistent with reported APC angiogenic effects demonstrated in models of HUVEC-mediated angiogenesis in vitro and in vivo in the cornea assay (Uchiba et al., 2004). APC also stimulates brain endothelial cells-mediated angiogenesis in three-dimensional Matrigel in vitro assays (I. Singh and B. V. Zlokovic, unpublished observations). Enhanced angiogenesis by APC may play an important role in tissue recovery and functional outcome after ischemia (Arenillas et al., 2007) and would enable macrophage infiltration of necrotic brain tissue to promote clean-up tasks (Manoonkitiwongsa et al., 2001).
APC promoted both postischemic proliferation of neuronal progenitor cells in the SVZ as well as migration of newly formed neuroblasts from the SVZ along the corpus callosum to the periinfarct areas. Several experimental brain ischemia studies have reported that enhanced neurogenesis correlates well with the improvement in functional recovery (R. Zhang et al., 2002; Chen et al., 2003b; Wang et al., 2004). Moreover, a high rate of neurogenesis, as for example in the dentate gyrus, may translate into an enhanced performance on a hippocampal-dependent tasks, whereas a decreasing rate of neurogenesis may be correlated with impairment of these tasks (van Praag et al., 1999a,b; Shors et al., 2001).
Our findings suggest that the effects of APC on neovascularization and neurogenesis may occur independently of its neuroprotective effects controlling the spread of infarct lesion. Namely, mice treated with multidose APC therapy (M72–144) beginning after 3 d of ischemia onset did not show a reduction in infarct size compared with vehicle-treated mice, but still displayed significant increases in neovascularization and neurogenesis, which correlated with an improvement in functional recovery as seen at day 7 after ischemia onset. Therefore, enhancement of neurogenesis that is accompanied by enhanced migration of neuroblasts to the periinfarct regions and neovascularization may contribute to functional recovery seen after treatment with APC.
The present transient ischemia mouse model produces a submaximal injury of 50–60 mm3 compared with that of a permanent proximal MCAO model. It remains to be explored whether positive effects of APC on neovascularization and neurogenesis that were obtained in the present model would occur in a model with more severe ischemic insult corresponding to large hemispheric strokes in humans that are typically lethal. In this regard, human APC therapy prevented stroke-related premature death in a mouse model with large hemispheric stroke in >90% animals (Shibata et al., 2001).
With respect to the mechanism of action, the present findings suggest that PAR1 is required for APC-mediated postischemic neovascularization and neurogenesis. Deletion of the PAR1 gene resulted in a complete loss of the effects of APC on brain repair. These findings are consistent with previous observations suggesting that PAR1 mediates the cellular effects of APC on endothelial cells, macrophages, and monocytes, and neurons both in vitro and in vivo in different animal models (Mosnier et al., 2007).
In sum, our data remarkably suggest that a delayed administration of multiple doses of APC at 6–72 h or a single-dose at 24 h after a transient ischemic insult to the mouse brain is neuroprotective and healing because this therapy (1) reduces infarct size and improves neurological functional outcome within 7 d, (2) promotes the survival and differentiation of neuronal progenitor cells and migration of neuroblasts from the SVZ to the periinfarct zone, and (3) improves postischemic survival of blood vessels and promotes angiogenesis in the ischemic border, which is primarily responsible for the observed postischemic vascularization effect. Our studies with multiple doses of APC at 72–144 h indicated that the effects of APC on neovascularization and neurogenesis are independent of its effects on controlling the spread of infarcted lesion. Multiple neuroprotective effects of APC could be interrelated in mediating brain tissue survival, repair, and regeneration after an ischemic insult depending on when after ischemia onset the APC therapy is administered.
A mouse model of transient brain ischemia is clinically relevant to prolonged transient ischemic attacks in humans or shorter-lasting spontaneously resolving strokes and does not reflect human conditions of permanent stroke. To show that our findings are not limited to only transient ischemia but could be more generalized, our ongoing studies show that late intervention with an APC analog has comparable neuroprotective effects in a model of permanent cerebral arterial occlusion (Y. Wang, H. Guo, D. Liu, J. A. Fernandez, J. H. Griffin, and B. V. Zlokovic, unpublished observations), as reported with APC in this study. Thus, the present findings may have important translational implications by offering the first experimental evidence in support of extending the current therapeutic window of intervention with APC in postischemic brain.
Footnotes
This work was supported by National Institutes of Health Grants HL63290 and HL081528 (B.V.Z.) and HL31950 and HL52246 (J.H.G.).
References
- Arenillas JF, Sobrino T, Castillo J, Dávalos A. The role of angiogenesis in damage and recovery from ischemic stroke. Curr Treat Options Cardiovasc Med. 2007;9:205–212. doi: 10.1007/s11936-007-0014-5. [DOI] [PubMed] [Google Scholar]
- Aronowski J, Samways E, Strong R, Rhoades HM, Grotta JC. An alternative method for the quantitation of neuronal damage after experimental middle cerebral artery occlusion in rats: analysis of behavioral deficit. J Cereb Blood Flow Metab. 1996;16:705–713. doi: 10.1097/00004647-199607000-00022. [DOI] [PubMed] [Google Scholar]
- Beck H, Acker T, Wiessner C, Allegrini PR, Plate KH. Expression of angiopoietin-1, angiopoietin-2, and tie receptors after middle cerebral artery occlusion in the rat. Am J Pathol. 2000;157:1473–1483. doi: 10.1016/S0002-9440(10)64786-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ., Jr Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- Breunig JJ, Arellano JI, Macklis JD, Rakic P. Everything that glitters isn't gold: a critical review of postnatal neural precursor analyses. Cell Stem Cell. 2007;1:612–627. doi: 10.1016/j.stem.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Alvarez D, González-Falcón A, García-Cabrera M, Martínez-Sánchez G, Merino N, Giuliani A, León OS. Neuroprotective efficacy of nimesulide against hippocampal neuronal damage following transient forebrain ischemia. Eur J Pharmacol. 2002;453:189–195. doi: 10.1016/s0014-2999(02)02422-6. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang ZG, Li Y, Wang L, Xu YX, Gautam SC, Lu M, Zhu Z, Chopp M. Intravenous administration of human bone marrow stromal cells induces angiogenesis in the ischemic boundary zone after stroke in rats. Circ Res. 2003a;92:692–699. doi: 10.1161/01.RES.0000063425.51108.8D. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang ZG, Li Y, Wang Y, Wang L, Jiang H, Zhang C, Lu M, Katakowski M, Feldkamp CS, Chopp M. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003b;53:743–751. doi: 10.1002/ana.10555. [DOI] [PubMed] [Google Scholar]
- Cheng T, Liu D, Griffin JH, Fernández JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernández JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- Connolly AJ, Ishihara H, Kahn ML, Farese RV, Jr, Coughlin SR. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996;381:516–519. doi: 10.1038/381516a0. [DOI] [PubMed] [Google Scholar]
- Dömötör E, Benzakour O, Griffin JH, Yule D, Fukudome K, Zlokovic BV. Activated protein C alters cytosolic calcium flux in human brain endothelium via binding to endothelial protein C receptor and activation of protease activated receptor-1. Blood. 2003;101:4797–4801. doi: 10.1182/blood-2002-12-3680. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- Fernández JA, Xu X, Liu D, Zlokovic BV, Griffin JH. Recombinant murine-activated protein C is neuroprotective in a murine ischemic stroke model. Blood Cells Mol Dis. 2003;30:271–276. doi: 10.1016/s1079-9796(03)00034-2. [DOI] [PubMed] [Google Scholar]
- Fernández JA, Lentz SR, Dwyre DM, Griffin JH. A novel ELISA for mouse activated protein C in plasma. J Immunol Methods. 2006;314:174–181. doi: 10.1016/j.jim.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280:17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- Griffin JH, Zlokovic B, Fernández JA. Activated protein C: potential therapy for severe sepsis, thrombosis, and stroke. Semin Hematol. 2002;39:197–205. doi: 10.1053/shem.2002.34093. [DOI] [PubMed] [Google Scholar]
- Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernández JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Noshita N, Sugawara T, Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab. 2003;23:166–180. doi: 10.1097/01.WCB.0000041283.53351.CB. [DOI] [PubMed] [Google Scholar]
- Heath DL, Vink R. Improved motor outcome in response to magnesium therapy received up to 24 hours after traumatic diffuse axonal brain injury in rats. J Neurosurg. 1999;90:504–509. doi: 10.3171/jns.1999.90.3.0504. [DOI] [PubMed] [Google Scholar]
- Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, Corat MA, Zeier M, Blessing E, Oh J, Gerlitz B, Berg DT, Grinnell BW, Chavakis T, Esmon CT, Weiler H, Bierhaus A, Nawroth PP. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Zhang RL, Zhang ZG, Ewing JR, Jiang P, Divine GW, Knight RA, Chopp M. Magnetic resonance imaging indexes of therapeutic efficacy of recombinant tissue plasminogen activator treatment of rat at 1 and 4 hours after embolic stroke. J Cereb Blood Flow Metab. 2000;20:21–27. doi: 10.1097/00004647-200001000-00005. [DOI] [PubMed] [Google Scholar]
- Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- Lin TN, Sun SW, Cheung WM, Li F, Chang C. Dynamic changes in cerebral blood flow and angiogenesis after transient focal cerebral ischemia in rats. Evaluation with serial magnetic resonance imaging. Stroke. 2002;33:2985–2991. doi: 10.1161/01.str.0000037675.97888.9d. [DOI] [PubMed] [Google Scholar]
- Liu D, Cheng T, Guo H, Fernández JA, Griffin JH, Song X, Zlokovic BV. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med. 2004;10:1379–1383. doi: 10.1038/nm1122. [DOI] [PubMed] [Google Scholar]
- Liu XS, Zhang ZG, Zhang RL, Gregg S, Morris DC, Wang Y, Chopp M. Stroke induces gene profile changes associated with neurogenesis and angiogenesis in adult subventricular zone progenitor cells. J Cereb Blood Flow Metab. 2007;27:564–574. doi: 10.1038/sj.jcbfm.9600371. [DOI] [PubMed] [Google Scholar]
- Manoonkitiwongsa PS, Jackson-Friedman C, McMillan PJ, Schultz RL, Lyden PD. Angiogenesis after stroke is correlated with increased numbers of macrophages: the clean-up hypothesis. J Cereb Blood Flow Metab. 2001;21:1223–1231. doi: 10.1097/00004647-200110000-00011. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. 2003;373:65–70. doi: 10.1042/BJ20030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- Nagai N, De Mol M, Lijnen HR, Carmeliet P, Collen D. Role of plasminogen system components in focal cerebral ischemic infarction: a gene targeting and gene transfer study in mice. Circulation. 1999;99:2440–2444. doi: 10.1161/01.cir.99.18.2440. [DOI] [PubMed] [Google Scholar]
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- Overgaard K, Sereghy T, Boysen G, Pedersen H, Diemer NH. Reduction of infarct volume and mortality by thrombolysis in a rat embolic stroke model. Stroke. 1992;23:1167–1173. doi: 10.1161/01.str.23.8.1167. discussion 1174. [DOI] [PubMed] [Google Scholar]
- Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita Y, Kubis N, Calando Y, Tran Dinh A, Méric P, Seylaz J, Pinard E. Long-term in vivo investigation of mouse cerebral microcirculation by fluorescence confocal microscopy in the area of focal ischemia. J Cereb Blood Flow Metab. 2005;25:858–867. doi: 10.1038/sj.jcbfm.9600077. [DOI] [PubMed] [Google Scholar]
- Uchiba M, Okajima K, Oike Y, Ito Y, Fukudome K, Isobe H, Suda T. Activated protein C induces endothelial cell proliferation by mitogen-activated protein kinase activation in vitro and angiogenesis in vivo. Circ Res. 2004;95:34–41. doi: 10.1161/01.RES.0000133680.87668.FA. [DOI] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. 1999a;2:266–270. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci U S A. 1999b;96:13427–13431. doi: 10.1073/pnas.96.23.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kittaka M, Sun N, Schreiber SS, Zlokovic BV. Chronic nicotine treatment enhances focal ischemic brain injury and depletes free pool of brain microvascular tissue plasminogen activator in rats. J Cereb Blood Flow Metab. 1997;17:136–146. doi: 10.1097/00004647-199702000-00002. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang Z, Wang Y, Zhang R, Chopp M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke. 2004;35:1732–1737. doi: 10.1161/01.STR.0000132196.49028.a4. [DOI] [PubMed] [Google Scholar]
- Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ninomiya M, Hernández Acosta P, García-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Friedman B, Cheng Q, Lyden PD. Stroke-evoked angiogenesis results in a transient population of microvessels. J Cereb Blood Flow Metab. 2007;27:755–763. doi: 10.1038/sj.jcbfm.9600378. [DOI] [PubMed] [Google Scholar]
- Zhang R, Wang Y, Zhang L, Zhang Z, Tsang W, Lu M, Zhang L, Chopp M. Sildenafil (Viagra) induces neurogenesis and promotes functional recovery after stroke in rats. Stroke. 2002;33:2675–2680. doi: 10.1161/01.str.0000034399.95249.59. [DOI] [PubMed] [Google Scholar]
- Zhang RL, LeTourneau Y, Gregg SR, Wang Y, Toh Y, Robin AM, Zhang ZG, Chopp M. Neuroblast division during migration toward the ischemic striatum: a study of dynamic migratory and proliferative characteristics of neuroblasts from the subventricular zone. J Neurosci. 2007;27:3157–3162. doi: 10.1523/JNEUROSCI.4969-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Tsang W, Soltanian-Zadeh H, Morris D, Zhang R, Goussev A, Powers C, Yeich T, Chopp M. Correlation of VEGF and angiopoietin expression with disruption of blood-brain barrier and angiogenesis after focal cerebral ischemia. J Cereb Blood Flow Metab. 2002;22:379–392. doi: 10.1097/00004647-200204000-00002. [DOI] [PubMed] [Google Scholar]
- Zivin JA, Fisher M, DeGirolami U, Hemenway CC, Stashak JA. Tissue plasminogen activator reduces neurological damage after cerebral embolism. Science. 1985;230:1289–1292. doi: 10.1126/science.3934754. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Zhang C, Liu D, Fernandez J, Griffin JH, Chopp M. Functional recovery after embolic stroke in rodents by activated protein C. Ann Neurol. 2005;58:474–477. doi: 10.1002/ana.20602. [DOI] [PubMed] [Google Scholar]