

Abstract
Endogenous levels of angiotensin II (Ang II) are increased in the cortex and hypothalamus following stroke, and Ang II type 1 receptor blockers (ARBs) have been shown to attenuate the deleterious effects in animal stroke models using middle cerebral artery (MCA) intraluminal occlusion procedures. However, the endothelin-1 (ET-1)-induced middle cerebral artery occlusion (MCAO) model of cerebral ischaemia is thought to more closely mimic the temporal events of an embolic stroke. This method provides rapid occlusion of the MCA and a gradual reperfusion that lasts for 16–22 h. The aim of the present study was to evaluate whether systemic administration of an ARB prior to ET-1-induced MCAO would provide cerebroprotection during this model of ischaemic stroke. Injection of 3 μl of 80 μm ET-1 adjacent to the MCA resulted in complete occlusion of the vessel that resolved over a period of 30–40 min. Following ET-1-induced MCAO, rats had significant neurological impairment, as well as an infarct that consisted of 30% of the ipsilateral grey matter. Systemic pretreatment with 0.2 mg kg−1 day−1 candesartan for 7 days attenuated both the infarct size and the neurological deficits caused by ET-1-induced MCAO without altering blood pressure. This study confirms the cerebroprotective properties of ARBs during ischaemic stroke and validates the ET-1-induced MCAO model for examination of the role of the brain renin–angiotensin system in ischaemic stroke.
While multiple factors influence stroke development and progression, the peptide angiotensin II (Ang II) appears to be a key player. For example, there are reports that Ang II levels are increased bilaterally in the cortex following stroke, and systemic treatment of spontaneously hypertensive rats (SHR) with Ang II type 1 receptor (AT1R) blockers (ARBs) reduces the occurrence of stroke (Stier et al. 1993; Inada et al. 1997; Kagiyama et al. 2004). Additionally, ARBs provide a 40–50% reduction of infarct volume and reduce the neurological deficits in normotensive rats and SHRs (Dai et al. 1999; Nishimura et al. 2000; Ito et al. 2002; Groth et al. 2003; Li et al. 2005; Lu et al. 2005). These actions of ARBs are, at least in part, independent of their blood pressure lowering actions (Dai et al. 1999; Li et al. 2005).
These animal studies have been substantiated clinically by the Losartan Intervention For Endpoint (LIFE) clinical trial, which demonstrated that ARBs are more effective as anti-stroke agents than the traditionally used β-adrenergic receptor (β-AR) blockers in patients with hypertension and left ventricular hypertrophy (Dahlöf et al. 2002). Other clinical trials, such as SCOPE (Study on Cognition and Prognosis in the Elderly), have substantiated the importance of AT1R blockade for stroke prevention (Papademetriou et al. 2004). Results from the Captopril Prevention Project (CAPPP) study demonstrated that angiotensin-converting enzyme (ACE) inhibitors, which reduce the levels of circulating Ang II, are inferior to conventional therapy for stroke prevention (Hansson et al. 1999). This observation led to the suggestion that increased levels of Ang II in response to ARB treatment may have a role in cerebroprotection (Hansson et al. 1999; Li et al. 2005).
Despite the focus on AT1R involvement in stroke, evidence suggests that Ang II type 2 receptors (AT2R) have a role in this disease. The AT2Rs often mediate effects of Ang II that are exactly opposite to those mediated by AT1R (Carey, 2005), and in fact the tissue levels of AT2R are dramatically increased following injury, such as in the heart following myocardial infarction, in atherosclerotic blood vessels, in wounded skin and in the peri-infarct region in the brain following ischaemia (Viswanathan & Saavedra, 1992; Makino et al. 1996; Horiuchi et al. 1999; Kagiyama et al. 2003; Li et al. 2005). Considering this, and the evidence that Ang II acts via AT2R in neurons to elicit differentiation, regeneration and neurotrophic actions (Lucius et al. 1998; Cote et al. 1999; Reinecke et al. 2003), Unger and colleagues hypothesized that the increased expression of AT2R within the peri-infarct region can (in the presence of ARBs to block AT1R) be activated by the raised endogenous levels of Ang II and serve a neuroprotective role (Li et al. 2005). These investigators have supported this theory by demonstrating that the beneficial action of ARBs after middle cerebral artery occlusion (MCAO)-induced cerebral ischaemia is prevented by specific AT2 receptor blockers (Li et al. 2005). Additional support is provided by the following experimental findings: (i) ARBs are more cerebroprotective than ACE inhibitors in a rat model of ischaemia and reperfusion (Krikov et al. 2008); (ii) MCAO produces greater ischaemic brain damage in AT2R knockout mice compared with wild-type control animals (Iwai et al. 2004); and (iii) CNS delivery of an AT2R agonist provides cerebroprotection during ischaemic stroke (McCarthy et al. 2009).
Considering that endogenous levels of Ang II are increased in the cortex and hypothalamus following stroke and that ARBs have been shown to attenuate the deleterious effects in animal stroke models using intraluminal occlusion, we have developed the general hypothesis that systemic administration of an ARB prior to ET-1-induced MCAO will provide cerebroprotection during ischaemic stroke. The ET-1-induced MCAO model of cerebral ischaemia is thought to more closely mimic the temporal events of an embolic stroke. This model provides rapid occlusion of the middle cerebral artery and a gradual reperfusion that lasts for 16–22 h (Biernaskie et al. 2001). Specifically, we determined whether systemic pretreatment of rats with the CNS-permeant ARB candesartan for 7 days would elicit a decrease in infarct size and in neurological deficits following ET-1-induced MCAO. This study aims to verify the cerebroprotective properties of ARBs during ischaemic stroke for the first time in the ET-1-induced MCAO model of ischaemia–reperfusion, as well as validate the ET-1-induced MCAO model for examination of the role of the brain renin–angiotensin system in ischaemic stroke.
Methods
Ethical approval
For the experiments described here, we used a total of 29 adult male Sprague–Dawley rats (250–275 g) purchased from Charles River Farms (Wilmington, MA, USA). All experimental procedures were approved by the University of Florida Institutional Animal Care and Use Committee. In addition, the principles governing the care and treatment of animals, as stated in the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (publication no. 85–323, revised 1996) and adopted by the American Physiological Society, were followed at all times during this study. Rats had ad libitum access to water and standard rat chow and were housed in a well-ventilated, specific pathogen-free, temperature-controlled environment (24 ± 1°C; 12 h–12 h light–dark cycle).
Surgical protocols
Rats were randomly allocated into treatment groups. Middle cerebral artery (MCA) visualization experiments (Fig. 1) were performed using three rats that underwent an MCAO induced by injection of 80 μM ET-1 adjacent to the MCA, two rats that underwent ET-1-induced MCAO plus subcutaneous pretreatment with candesartan (via osmotic mini-pump) for 7 days, and two rats that served as control animals (0.9% saline injected instead of ET-1). For the remainder of the experiments, rats underwent subcutaneous infusion with either candesartan (n = 8 rats) or 0.9% saline (n = 14 rats) via osmotic mini-pumps for 7 days, prior to undergoing ET-1-induced MCAO. Five rats from each of these two treatment groups received telemetry transducers (DSI; St Paul, MN, USA) implanted 10 days prior to the candesartan or 0.9% saline infusions. Two rats from the candesartan pretreatment group and two rats from the 0.9% saline pretreatment group died following the ET-1-induced MCAO. This is consistent with the approximately 20% mortality rate we have observed with this procedure. There was no significant difference in mortality rate between groups.
Figure 1. Visualization of MCA branches during ET-1-induced vasoconstriction.
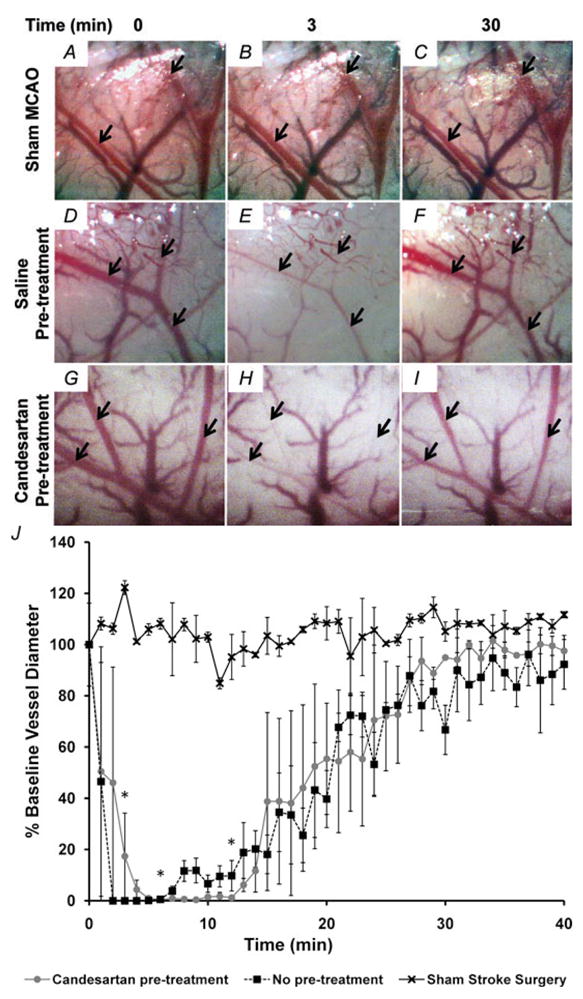
A–I, visualization of MCA branches (arrows) during ET-1-induced vasoconstriction. Primary and secondary branches of the MCA were visualized with a surgical microscope after temporal craniotomy to create a cranial window. Images were captured at a rate of 1 min−1, starting immediately prior to ET-1 injection (0 min), throughout the ET-1 injection (3 min) and for at least 40 min after completion of ET-1 injection. Shown here are representative images captured from rats that underwent a sham injection (0.9% saline, n = 2, A–C), an MCAO induced by 80 μM ET-1 injection (n = 3, C–E) and an ET-1-induced MCAO plus 7 days pretreatment with candesartan (0.2 mg kg−1 day−1, S.C., n = 2, G–I).
J, vasoconstriction of primary and secondary MCA branches was quantified as the percentage of baseline vessel diameter. Data are means ± S.E.M. Baseline vessel diameter was determined prior to ET-1 or 0.9% saline injection (time = 0 min). *P < 0.01 for both candesartan-pretreated and non-pretreated groups compared with 0.9% saline injection (one-way ANOVA followed by Tukey–Kramer multiple comparisons test).
Endothelin-1-induced middle cerebral artery occlusion
The ET-1-induced MCAO procedure used here has been modified slightly from that published previously (Biernaskie et al. 2001; Windle et al. 2006). Eight-week-old male Sprague–Dawley rats were anaesthetized with a mixture of O2 (1 l min −1) and 4% isoflurane, placed in a Kopf stereotaxic frame, and anesthesia was maintained for the duration of the surgery using an O2–isoflurane (2%) mixture delivered through a nose cone attached to the frame. The skull was exposed, and a small hole was drilled in the cranium dorsal to the right hemisphere using the following stereotaxic co-ordinates: 1.6 mm anterior and 5.2 mm lateral to bregma. A 26 gauge needle attached to a 5 μl Hamilton microsyringe was lowered 8.7 mm ventral to bregma, after which 3 μl of 80 μM ET-1 was infused adjacent to the MCA at a rate of 1 μl min−1. The needle was withdrawn 3 min after the injection was complete. Following this, the wound was closed and the rat was administered an analgesic agent (buprenorphine; 0.05 mg kg−1 S.C.) before waking.
Visualization of MCA branches via a cranial window
Rats were anaesthetized as described above, after which a temporal craniectomy was performed to visualize the primary and secondary branches of the MCA. An approximately 3–4 mm square piece of bone was removed from the left squamous portion of the temporal bone just caudal to the orbit. The dura was left in place, and debris was cleared away using sterile 0.9% saline. Next, ET-1-induced MCAO was performed as described in the previous subsection except that the needle was left in place until all images were captured so that the focal plane would not be disturbed. The cerebral cortex and associated vessels visible through this cranial window were imaged using a Moticam 1000 digital camera (Motic; Richmond, BC, Canada) coupled to a Revelation surgical microscope (Seiler Instrument and Manufacturing; St Louis, MO, USA). Vessel diameter was determined by averaging one primary and two secondary MCA branches per rat window using ImageJ software (NIH) by an individual who had been blinded as to the nature of the treatment. A baseline image was captured prior to ET-1 injection, and subsequent images were captured each 1 min interval for 40 min. Vessel diameter at each time point was normalized to the baseline vessel diameter so that comparisons could be made using multiple MCA branches of several rats.
Implantation of telemetry transducers
Rats were anaesthetized with a mixture of O2 (1 l min−1) and 4% isoflurane as described above (in the subsection ‘Endothelin-1-induced middle cerebral artery occlusion’), after which telemetry transducers (DSI) were implanted into the abdominal aorta, as detailed by us previously (Li et al. 2006). Rats were administered the analgesic (buprenorphine; 0.05 mg kg−1 S.C.) following surgery, and were left to recover for 10 days before the implantation of subcutaneous mini-osmotic pumps.
Implantation of osmotic mini-pumps
Rats were implanted subcutaneously between the shoulder blades with an osmotic mini-pump (model 2001, ALZET, Cupertino, CA, USA) as described previously (Grobe et al. 2007), which infused candesartan (0.2 mg kg−1 day−1) or 0.9% saline subcutaneously for 7 days prior to ET-1-induced MCAO.
Neurological deficits and infarct size
Neurological evaluations were performed using two separate scoring scales reported by Benderson et al. (1986) and Garcia et al. (1995), which cumulatively evaluate spontaneous activity, symmetry in limb movement, forepaw outstretching, climbing, body proprioception, response to vibrissae touch, resistance to lateral push and circling behaviour. Additionally, animals were evaluated for neurological deficits using a sunflower seed eating test (Gonzalez & Kolb, 2003), which provides an index of fine motor function. In this test, rats are timed while opening five sunflower seeds, and the number of shell pieces is recorded. Rats with less neurological impairment open the five seeds faster and efficiently by breaking the shell into fewer pieces to accomplish the task.
Infarct volume was assessed by staining brain sections with 0.05% 2,3,5-triphenyltetrazolium chloride (TTC) for 30 min at 37°C. Tissue ipsilateral to the occlusion, which was not stained, was assumed to be infarcted. After fixation with 10% formalin, brain sections were scanned on a flatbed scanner (Canon) and analysed using ImageJ software (NIH). To compensate for the effect of brain oedema, the corrected infarct volume was calculated using an indirect method (Kagiyama et al. 2004).
Cardiovascular measurements
Measurements of mean arterial pressure (MAP), systolic blood pressure (SBP), diastolic blood pressure (DBP), pulse pressure (PP) and heart rate (HR) were made via DSI telemetry transducers (Li et al. 2006). Measurements were made prior to candesartan or 0.9% saline pretreatment while awake, after 7 days of candesartan or 0.9% saline pretreatment while anaesthetized, during the ET-1-induced MCAO procedure while anesthetized, and 8 h after ET-1-induced MCAO when awake.
Data analysis
Data are expressed as means ± S.E.M. Statistical significance was evaluated, as specified in the figure legends, with the use of a one-way ANOVA, Mann–Whitney U test, Wilcoxon signed-rank test, or a Student’s unpaired t test, as well as Tukey–Kramer multiple comparisons test for post hoc analysis when appropriate. A Spearman non-parametric correlation was used to compare the relationship between infarct size and neurological testing data. Differences were considered significant at P < 0.05. Individual P values are noted in the Results and figure legends.
Chemicals
Candesartan was a gift from AstraZeneca (Alderley Park, UK). Endothelin-1 was purchased from American Peptide Company, Inc. (Sunnyvale, CA, USA). All other chemicals were purchased from Fisher Scientific (Pittsburgh, PA, USA).
Results
Intracranial injection of 3 μl of ET-1 (80 μM) into the brain parenchyma adjacent to the MCA resulted in abrupt constriction of the proximal MCA branches to 0% baseline vessel diameter within minutes, followed by recanalizaton of the vessel (Fig. 1D–F and J). Rats undergoing a sham MCAO were injected with 3 μl of 0.9% saline instead of ET-1. Vessel diameter remained relatively stable at baseline values following this saline injection (Fig. 1A–C and J). Additionally, rats were pretreated for 7 days with candesartan (0.2 mg kg−1 day−1, S.C.) prior to ET-1 injection. As with the group receiving no pretreatment, vessel diameter decreased to 0% baseline diameter in the candesartan-pretreated group (Fig. 1G–J). There was no significant difference in vessel diameter at any time point between candesartan-pretreated and non-pretreated rats that received an injection of ET-1 adjacent to the MCA. Both groups receiving an ET-1 brain injection displayed a decrease in vessel diameter 3 min after the start of injection that was significantly greater than the group undergoing sham MCAO (P < 0.01). These results indicate that ET-1 injection adjacent to the MCA causes constriction of the vessel followed by recanalization. In addition, pretreatment with candesartan does not alter the amplitude or time course of the ET-1-induced MCAO.
The effect of candesartan pretreatment on ET-1-induced cerebral damage was assessed by TTC staining, whereby non-infarcted grey matter is stained red after incubation in TTC, delineating the infarct in white (Fig. 2A). Pretreatment of rats for 7 days with candesartan (0.2 mg kg−1 day−1, S.C.) significantly reduced the infarct size to 14.35 ± 5.38% of the grey matter, compared with 28.98 ± 4.38% in the 0.9% saline-pretreated control group (P < 0.05; Fig. 2B). Neurological testing performed 48 h subsequent to the ET-1-induced MCAO indicated improved performance in rats pretreated with candesartan compared with those with 0.9% saline pretreatment (Fig. 3 and Table 1). Prior to ET-1-induced MCAO, all rats scored the maximum of 18 on the Garcia neurological examination, indicating that no deficit existed. After the ET-1-induced MCAO, the Garcia neurological examination score decreased significantly from pre-stroke values to 12.1 ± 1.1 in the 0.9% saline-pretreated group (P < 0.001) and 16.7 ± 0.7 in the candesartan-pretreated group (P < 0.05). Two days following ET-1-induced MCAO, the Garcia examination score was significantly higher in the candesartan-pretreated group compared with the 0.9% saline-pretreated group (P < 0.05). A similar pattern was observed with the Benderson neurological examination, in which all rats received the minimal score of 0 prior to ET-1-induced MCAO, indicating an absence of neurological deficit. After the ET-1-induced MCAO, the Benderson neurological examination score increased from pre-stroke values to 2.1 ± 0.2 in the 0.9% saline-pretreated group (P < 0.001) and 0.7 ± 0.4 in the candesartan-pretreated group. The post-stroke Benderson examination score was significantly higher in the candesartan-pretreated group compared with 0.9% saline-pretreated group (P < 0.01). Rats also participated in a sunflower seed-eating test, in which they were timed while opening five sunflower seeds. Prior to ET-1-induced MCAO, rats performed this task in 103.7 ± 14.4 s and broke the shell into 13.6 ± 0.7 pieces. Two days following ET-1-induced MCAO, the time to eat five sunflower seeds increased from pre-stroke values to 215.6 ± 28.3 s in the 0.9% saline-pretreated group (P < 0.01) and 114.7 ± 16.5 s in the candesartan-pretreated group (not significant, P = 0.06). In addition, the number of shell pieces increased from pre-stroke values to 23.8 ± 2.2 pieces in the 0.9% saline-pretreated group (P < 0.01) and 14.8 ± 0.8 pieces in the candesartan-pretreated group (not significant, P = 0.18). The post-stroke number of shell pieces was significantly lower for the candesartan-pretreated group compared with the 0.9% saline-pretreated group (P < 0.05), and a similar although non-significant pattern was observed for the time to eat five sunflower seeds (P = 0.08). Taken together, these results indicate that candesartan pretreatment prior to ET-1-induced MCAO can reduce the size of the cerebral infarct and the extent of neurological deficits produced by this model of focal cerebral ischaemia.
Figure 2. Candesartan pretreatment decreases cerebral infarct size after ET-1-induced MCAO.
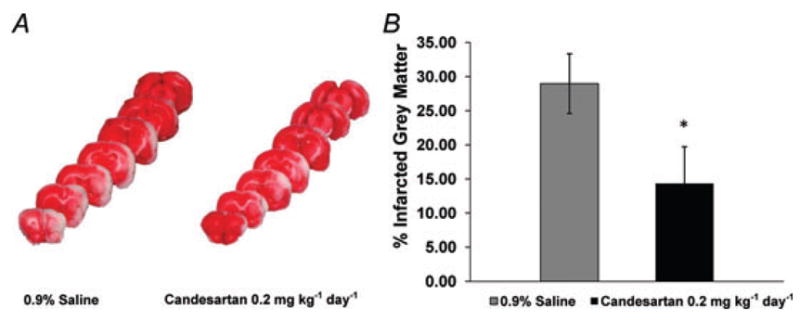
Rats were pretreated with candesartan or 0.9% saline for 7 days prior to ET-1-induced MCAO. Brains were removed and sectioned 48 h later. A, representative brain sections from both a control (0.9% saline-pretreated) and a candesartan-pretreated rat, showing infarcted (white) and non-infarcted (red) grey matter. B, bar graphs show the percentage infarcted grey matter in each treatment group. Data are means ± S.E.M. from 6 (candesartan) and 11 (0.9% saline) pretreated rats. *P < 0.05 compared with 0.9% saline-pretreated group (Mann–Whitney U test).
Figure 3. Pretreatment with candesartan leads to improvement of functional outcomes after ET-1-induced MCAO.
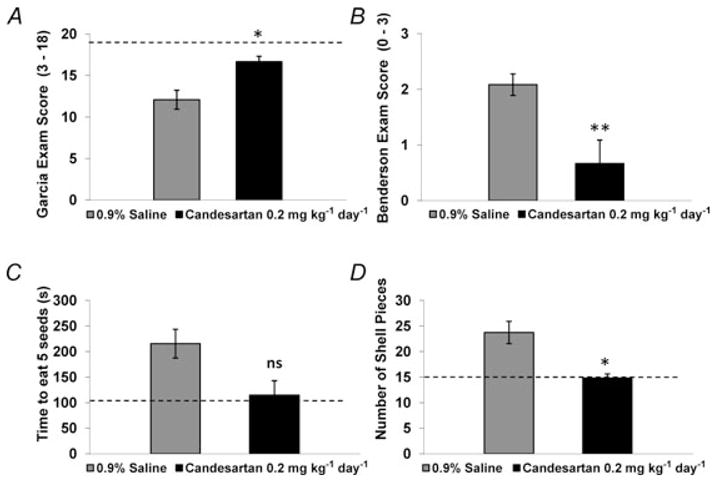
Rats were pretreated with candesartan or 0.9% saline for 7 days prior to ET-1-induced MCAO. Forty-eight hours later, neurological deficits were assessed via the Garcia neurological examination (A), the Benderson neurological examination (B) and the sunflower seed eating test (C and D). Bar graphs are means ± S.E.M. from 6 (candesartan) and 12 (0.9% saline) pretreated rats. The dashed horizontal lines in each panel indicate the scores obtained in each test prior to ET-1-induced MCAO. *P < 0.05, **P < 0.01 (Mann-Whitney U test) compared with 0.9% saline pretreatment.
Table 1.
Neurological examination results before and after ET-1-induced MCAO
| Benderson examination score |
Garcia examination score |
Sunflower seed test; time to eat five seeds |
Sunflower seed test; number of shell pieces |
|||||
|---|---|---|---|---|---|---|---|---|
| Treatment | 24 h Pre- stroke | 48 h Post- stroke | 24 h Pre- stroke | 48 h Post- stroke | 24 h Pre- stroke | 48 h Post- stroke | 24 h Pre- stroke | 48 h Post- stroke |
| 0.9% Saline (n = 12) | 0 ± 0 | 2.1 ± 0.2*** | 18 ± 18 | 12.1 ± 1.1*** | 120.8 ± 19.5 | 215.6 ± 28.3** | 14.1 ± .9 | 23.8 ± 2.2** |
| Candesartan (n = 6) | 0 ± 0 | 0.7 ± 0.4‡ | 18 ± 18 | 16.7 ± 0.7*† | 69.5 ± 10.5 | 114.7 ± 16.5 | 12.7 ± 1.1 | 14.8 ± 0.8† |
| Combined groups (n = 18) | 0 ± 0 | 1.6 ± 0.2*** | 18 ± 18 | 13.6 ± 0.9*** | 103.7 ± 14.4 | 181.9 ± 22.5** | 13.6 ± 0.7 | 20.8 ± 1.8** |
All values are reported as means ± SEM.
P < 0.05 or
P < 0.05 versus candesartan pretreatment (Mann–Whitney U test);
P < 0.05,
P < 0.01 or
P < 0.001 versus pre-stroke value (Wilcoxon signed-rank test).
In order to evaluate how closely our neurological examination scores predicted the extent of brain tissue damage, we grouped rats receiving both candesartan and 0.9% saline pretreatment together and compared pre-stroke and post-stroke neurological test scores, as well as post-stroke scores with the percentage infarcted grey matter. Prior to ET-1-induced MCAO, both candesartan-pretreated and 0.9% saline-pretreated rats showed no neurological deficits as indicated by a minimal score of 0 in the Benderson examination, and a maximal score of 18 in the Garcia examination. As expected, the rats displayed neurological deficits 48 h subsequent to ET-1-induced MCAO, with a Benderson examination score that increased to 1.6 ± 0.2 (P < 0.0001) and a Garcia examination score that decreased to 13.6 ± 0.9 (P < 0.0001) for both groups combined. Prior to ET-1-induced MCAO, rats from both treatment groups combined required an average of 103.7 ± 14.4 s to eat five seeds and broke the shells into an average of 13.6 ± 0.7 pieces. Rats displayed significant deficit in performing this task 48 h subsequent to ET-1-induced MCAO. The time to eat five seeds increased to 181.9 ± 22.5 s (P < 0.01) and the number of shell pieces increased to 20.8 ± 1.8 (P < 0.01) for both groups combined compared with pre-stroke outcomes. It is clear from these data that our model of ET-1-induced MCAO causes significant neurological deficits that can be measured 48 h after stroke induction (Table 1 and Fig. 3). For correlation between neurological examination results and infarct size, data showing the Garcia examination score, Benderson examination score, time to eat five sunflower seeds and number of shell pieces were plotted against the associated percentage infarcted grey matter for each rat (Fig. 4A–D). A strong and significant negative correlation was seen for percentage infarcted grey matter versus Garcia examination score (Spearman r = −0.9151, P < 0.0001). Additionally, strong and significant positive correlations were seen for percentage infarcted grey matter versus Benderson examination score (Spearman r = 0.8632, P < 0.0001), percentage infarcted grey matter versus time to eat five sunflower seeds (Spearman r = 0.6729, P < 0.005) and percentage infarcted grey matter versus number of shell pieces (Spearman r = 0.7241, P < 0.005).
Figure 4. Neurological examination scores correlate with percentage infarcted grey matter.
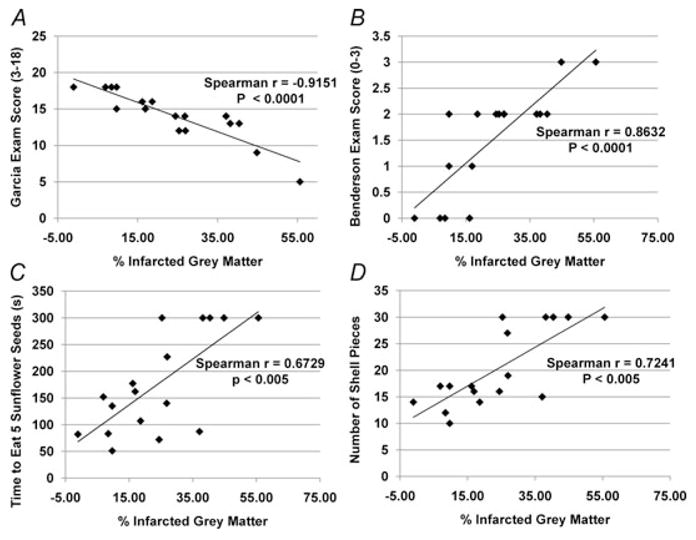
Neurological examinations were performed 48 h after ET-1-induced MCAO. Brains were removed, sectioned and TTC stained. Correlation of Garcia examination scores (A), Benderson examination scores (B), and the sunflower seed eating test scores (C and D) with percentage infarcted grey matter revealed strong correlations.
Finally, a subgroup of five rats was implanted with telemetry blood pressure transducers in the abdominal aorta in order to record arterial blood pressure and heart rate (Table 2). Cardiovascular variables consisting of MAP, SBP, DBP, PP and HR were measured. No differences in cardiovascular parameters were seen between treatment groups prior to candesartan or 0.9% saline pretreatment or after pretreatment for 7 days (Student’s unpaired t test). A slight increase in all cardiovascular variables was observed within 5 min of ET-1-induced MCAO, but no difference was observed between groups. Similarly, there were no differences in cardiovascular variables between groups recorded at 8 h subsequent to ET-1-induced MCAO.
Table 2.
Cardiovascular variables before, during and after ET-1-induced MCAO
| A. Prior to candesartan pretreatment for 7 days; awake | |||||
|---|---|---|---|---|---|
| Treatment | MAP | SBP | DBP | PP | HR |
| Candesartan | 87.19 ± 3.74 | 103.00 ± 4.91 | 74.82 ± 3.55 | 28.13 ± 3.54 | 333.66 ± 10.01 |
| 0.9% Saline | 92.36 ± 2.36 | 107.54 ± 1.82 | 80.35 ± 4.08 | 27.17 ± 4.22 | 328.08 ± 14.28 |
|
| |||||
| B. After candesartan pretreatment for 7 days; anaesthetized prior to ET-1-induced MCAO | |||||
| Treatment | MAP | SBP | DBP | PP | HR |
|
| |||||
| Candesartan | 80.19 ± 3.25 | 94.08 ± 2.70 | 69.35 ± 3.89 | 24.65 ± 1.80 | 336.23 ± 9.18 |
| 0.9% Saline | 83.98 ± 10.01 | 101.08 ± 8.78 | 66.51 ± 15.67 | 35.15 ± 13.65 | 311.43 ± 10.75 |
|
| |||||
| C. Change from baseline during 5 min period after ET-1-induced MCAO; anaesthetized | |||||
| Treatment | ΔMAP | ΔSBP | ΔDBP | ΔPP | ΔHR |
|
| |||||
| Candesartan | 7.89 ± 3.69 | 8.27 ± 3.69 | 8.15 ± 3.79 | 0.47 ± 0.27 | 19.56 ± 6.99 |
| 0.9% Saline | 10.57 ± 3.28 | 9.32 ± 2.42 | 19.51 ± 11.83 | 13.17 ± 10.75 | 27.09 ± 15.66 |
|
| |||||
| D. 8 h post ET-1-induced MCAO; awake | |||||
| Treatment | MAP | SBP | DBP | PP | HR |
|
| |||||
| Candesartan | 90.36 ± 6.45 | 107.06 ± 6.69 | 77.76 ± 6.60 | 29.28 ± 2.35 | 407.21 ± 19.20 |
| 0.9% Saline | 105.80 ± 21.41 | 117.77 ± 21.77 | 96.94 ± 21.20 | 20.82 ± 3.23 | 358.96 ± 16.17 |
All values are reported as means ± S.E.M. (n = 5 rats per treatmentgroup). No significant differences between groups (Student’s unpaired t test). Abbreviations: MAP, mean arterial pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure; PP, pulse pressure; and HR, heart rate.
Discussion
The significant findings of this study are that pretreatment with the CNS-permeant ARB, candesartan, attenuates the neurological deficits and CNS tissue damage produced in an ET-1-induced MCAO model of ischaemic stroke without altering blood pressure. This is the first example of cerebroprotection offered by ARBs in this model of ischaemia–reperfusion, which closely mimics the rapid occlusion and gradual reperfusion of human ischaemic stroke. In addition, we show that this model of stroke provides a rapid constriction, sustained occlusion and then gradual reperfusion of the proximal MCA. This constriction is not altered by pretreatment with candesartan, which is a concern when using the ET-1-induced MCAO as a disease model for investigating manipulations of the brain renin–angiotensin system during focal cerebral ischaemia. Lastly, we have shown that results of the neurological testing methods used here are strongly correlated with the amount of tissue damage produced by ET-1-induced MCAO. Taken together, our results confirm the deleterious action of Ang II in the presence of uninhibited AT1Rs in the pathophysiology of ischaemic stroke and validate the ET-1-induced MCAO model for examination of the role of the brain renin–angiotensin system in ischaemic stroke.
The ET-1-induced MCAO model has been used and characterized extensively in rodent models as a method for producing transient focal cerebral ischaemia (Sharkey & Butcher, 1995; Biernaskie et al. 2001; Windle et al. 2006). It has often been presented as a model that closely mimics the initial vessel occlusion in other stroke models, such as the intraluminal suture method, in that rapid and complete occlusion is initially achieved. A unique aspect of the ET-1-induced MCAO model is the reversibility of the chemically induced occlusion over time. Perfusion MRI studies in the ET-1-induced MCAO model have shown that cerebral blood flow (CBF) in the ipsilaterial hemisphere decreases to 30–50% of normal at 1 h and gradually returns to normal between 16 and 22 h. This is followed by a period of hyperperfusion that seems to peak between 24 and 48 h, at which point the study was ended (Biernaskie et al. 2001). This time course for CBF has been verified by Doppler flow studies of the cerebral cortex in the MCA territory (Glendenning et al. 2008). However, neither vasoconstriction nor regional blood flow of the proximal MCA have been examined in this stroke model. Our data indicate that ET-1 injection adjacent to the MCA does indeed provide an initial rapid occlusion of the proximal MCA. However, vessel diameter seems to return to baseline after approximately 30 min. This profile does not match that of CBF measured previously even though infarct sizes and neurological deficits are comparable. It is possible that a cerebrovascular event distal to the proximal MCA mediates ischaemia after the initial occlusion, but this fact is yet to be determined.
We have used the ET-1-induced MCAO model to verify the cerebroprotective actions of candesartan during ischaemic stroke. A few key facts have been revealed by previous studies using candesartan and other ARBs, given both centrally and peripherally for their cerebroprotective properties (Stier et al. 1993; Inada et al. 1997; Dai et al. 1999; Nishimura et al. 2000; Ito et al. 2002; Walther et al. 2002; Groth et al. 2003; Li et al. 2005; Lu et al. 2005; Krikov et al. 2008; Li et al. 2008). First, it is clear that ARB administration is cerebroprotective in intraluminal occlusion models of MCAO. In addition, it should be noted that certain hydrophobic ARBs, such as candesartan and valsartan, cross the blood–brain barrier readily and that systemic administration of these drugs for cerebroprotection has been used in non-blood-pressure-lowering doses in several cases. Therefore, the cerebroprotective effect of ARBs is likely to be independent of blood pressure changes associated with administration. In fact, several theories exist for the mechanism of ARB cerebroprotection. The site of action of ARBs can be grossly identified as either cerebrovascular or parenchymal. For example, there is some evidence that ARB pretreatment in a rat model of ischaemic stroke increases capillary density (Li et al. 2008), improves cerebrovascular reserve (Takada et al. 2006) and improves cerebral endothelial function (Liu et al. 2008). These cerebrovascular effects may be mediated by unopposed agonism of the AT2 and Ang II type 4 receptors (Li et al. 2005; Faure et al. 2008) or AT1R alone (Stenman & Edvinsson, 2004). In addition, AT2R stimulation has been shown to stimulate post-stroke neurite outgrowth and improve neuron survival in hypoxic conditions (Li et al. 2005). Similarly, inhibition of AT1R may protect neurons during hypoxia by decreasing oxidative stress (Yamamoto et al. 2008). Therefore, ARB administration can either increase the cerebrovascular reserve, allowing improved perfusion of tissues within the affected vascular territory, or act directly on neurons to improve their ability to survive an ischaemic insult. In reality, the plethora of evidence indicates a role for both vascular and parenchymal targets in the cerebroprotection offered by ARBs. Our observations provide support for a role of angiotensin peptides in the pathophysiology of ischaemic stroke and verify the need for further study of the renin–angiotensin system to identify potential targets for stroke therapy.
Acknowledgments
This work was supported by grants from the University of Florida McKnight Brain Institute, Clinical and Translational Science Institute, and the Medical Guild. Adam Mecca is a NIH/NINDS, NRSA predoctoral fellow. Timothy O’Connor was supported by a University of Florida HHMI Science for Life undergraduate research fellowship. Many thanks to Jeffrey Kleim, PhD for his assistance with development of the ET-1-induced MCAO stroke model and neurological testing methods, as well as to AstraZeneca (Alderley Park, UK) for their generous gift of candesartan for these studies.
References
- Benderson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Biernaskie J, Corbett D, Peeling J, Wells J, Lei H. A serial MR study of cerebral blood flow changes and lesion development following endothelin-1-induced ischemia in rats. Magn Reson Med. 2001;46:827–830. doi: 10.1002/mrm.1263. [DOI] [PubMed] [Google Scholar]
- Carey RM. Cardiovascular and renal regulation by the angiotensin type 2 receptor: the AT2 receptor comes of age. Hypertension. 2005;45:840–844. doi: 10.1161/01.HYP.0000159192.93968.8f. [DOI] [PubMed] [Google Scholar]
- Cote F, Do TH, Laflamme L, Gallo JM, Gallo-Payet N. Activation of the AT2 receptor of angiotensin II induces neurite outgrowth and cell migration in microexplant cultures of the cerebellum. J Biol Chem. 1999;274:31686–31692. doi: 10.1074/jbc.274.44.31686. [DOI] [PubMed] [Google Scholar]
- Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- Dai WJ, Funk A, Herdegen T, Unger T, Culman J. Blockade of central angiotensin AT1 receptors improves neurological outcome and reduces expression of AP-1 transcription factors after focal brain ischemia in rats. Stroke. 1999;30:2391–2398. doi: 10.1161/01.str.30.11.2391. discussion 2398–2399. [DOI] [PubMed] [Google Scholar]
- Faure S, Bureau A, Oudart N, Javellaud J, Fournier A, Achard JM. Protective effect of candesartan in experimental ischemic stroke in the rat mediated by AT2 and AT4 receptors. J Hypertens. 2008;26:2008–2015. doi: 10.1097/HJH.0b013e32830dd5ee. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- Glendenning ML, Lovekamp-Swan T, Schreihofer DA. Protective effect of estrogen in endothelin-induced middle cerebral artery occlusion in female rats. Neurosci Lett. 2008;445:188–192. doi: 10.1016/j.neulet.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez CL, Kolb B. A comparison of different models of stroke on behaviour and brain morphology. Eur J Neurosci. 2003;18:1950–1962. doi: 10.1046/j.1460-9568.2003.02928.x. [DOI] [PubMed] [Google Scholar]
- Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, Speth RC, Raizada MK, Katovich MJ. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1–7) Am J Physiol Heart Circ Physiol. 2007;292:H736–H742. doi: 10.1152/ajpheart.00937.2006. [DOI] [PubMed] [Google Scholar]
- Groth W, Blume A, Gohlke P, Unger T, Culman J. Chronic pretreatment with candesartan improves recovery from focal cerebral ischaemia in rats. J Hypertens. 2003;21:2175–2182. doi: 10.1097/00004872-200311000-00028. [DOI] [PubMed] [Google Scholar]
- Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, Luomanmäki K, Dahlöf B, de Faire U, Mörlin C, Karlberg BE, Wester PO, Björck JE. Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomised trial. Lancet. 1999;353:611–616. doi: 10.1016/s0140-6736(98)05012-0. [DOI] [PubMed] [Google Scholar]
- Horiuchi M, Akishita M, Dzau VJ. Recent progress in angiotensin II type 2 receptor research in the cardiovascular system. Hypertension. 1999;33:613–621. doi: 10.1161/01.hyp.33.2.613. [DOI] [PubMed] [Google Scholar]
- Inada Y, Wada T, Ojima M, Sanada T, Shibouta Y, Kanagawa R, Ishimura Y, Fujisawa Y, Nishikawa K. Protective effects of candesartan cilexetil (TCV-116) against stroke, kidney dysfunction and cardiac hypertrophy in stroke-prone spontaneously hypertensive rats. Clin Exp Hypertens. 1997;19:1079–1099. doi: 10.3109/10641969709083206. [DOI] [PubMed] [Google Scholar]
- Ito T, Yamakawa H, Bregonzio C, Terrón JA, Falcón-Neri A, Saavedra JM. Protection against ischemia and improvement of cerebral blood flow in genetically hypertensive rats by chronic pretreatment with an angiotensin II AT1 antagonist. Stroke. 2002;33:2297–2303. doi: 10.1161/01.str.0000027274.03779.f3. [DOI] [PubMed] [Google Scholar]
- Iwai M, Liu HW, Chen R, Ide A, Okamoto S, Hata R, Sakanaka M, Shiuchi T, Horiuchi M. Possible inhibition of focal cerebral ischemia by angiotensin II type 2 receptor stimulation. Circulation. 2004;110:843–848. doi: 10.1161/01.CIR.0000138848.58269.80. [DOI] [PubMed] [Google Scholar]
- Kagiyama T, Glushakov AV, Sumners C, Roose B, Dennis DM, Phillips MI, Ozcan MS, Seubert CN, Martynyuk AE. Neuroprotective action of halogenated derivatives of L-phenylalanine. Stroke. 2004;35:1192–1196. doi: 10.1161/01.STR.0000125722.10606.07. [DOI] [PubMed] [Google Scholar]
- Kagiyama T, Kagiyama S, Phillips MI. Expression of angiotensin type 1 and 2 receptors in brain after transient middle cerebral artery occlusion in rats. Regul Pept. 2003;110:241–247. doi: 10.1016/s0167-0115(02)00223-9. [DOI] [PubMed] [Google Scholar]
- Krikov M, Thone-Reineke C, Muller S, Villringer A, Unger T. Candesartan but not ramipril pretreatment improves outcome after stroke and stimulates neurotrophin BNDF/TrkB system in rats. J Hypertens. 2008;26:544–552. doi: 10.1097/HJH.0b013e3282f2dac9. [DOI] [PubMed] [Google Scholar]
- Li H, Gao Y, Freire CD, Raizada MK, Toney GM, Sumners C. Macrophage migration inhibitory factor in the PVN attenuates the central pressor and dipsogenic actions of angiotensin II. FASEB J. 2006;20:1748–1750. doi: 10.1096/fj.06-5836fje. [DOI] [PubMed] [Google Scholar]
- Li J, Culman J, Hörtnagl H, Zhao Y, Gerova N, Timm M, Blume A, Zimmermann M, Seidel K, Dirnagl U, Unger T. Angiotensin AT2 receptor protects against cerebral ischemia-induced neuronal injury. FASEB J. 2005;19:617–619. doi: 10.1096/fj.04-2960fje. [DOI] [PubMed] [Google Scholar]
- Li JM, Mogi M, Iwanami J, Min LJ, Tsukuda K, Sakata A, Fujita T, Iwai M, Horiuchi M. Temporary pretreatment with the angiotensin II type 1 receptor blocker, valsartan, prevents ischemic brain damage through an increase in capillary density. Stroke. 2008;39:2029–2036. doi: 10.1161/STROKEAHA.107.503458. [DOI] [PubMed] [Google Scholar]
- Liu H, Kitazato KT, Uno M, Yagi K, Kanematsu Y, Tamura T, Tada Y, Kinouchi T, Nagahiro S. Protective mechanisms of the angiotensin II type 1 receptor blocker candesartan against cerebral ischemia: in-vivo and in-vitro studies. J Hypertens. 2008;26:1435–1445. doi: 10.1097/HJH.0b013e3283013b6e. [DOI] [PubMed] [Google Scholar]
- Lu Q, Zhu YZ, Wong PT. Neuroprotective effects of candesartan against cerebral ischemia in spontaneously hypertensive rats. Neuroreport. 2005;16:1963–1967. doi: 10.1097/01.wnr.0000187636.13147.cd. [DOI] [PubMed] [Google Scholar]
- Lucius R, Gallinat S, Rosenstiel P, Herdegen T, Sievers J, Unger T. The angiotensin II type 2 (AT2) receptor promotes axonal regeneration in the optic nerve of adult rats. J Exp Med. 1998;188:661–670. doi: 10.1084/jem.188.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CA, Vinh A, Callaway JK, Widdop RE. Angiotensin AT2 receptor stimulation causes neuroprotection in a conscious rat model of stroke. Stroke. 2009;44:1482–1489. doi: 10.1161/STROKEAHA.108.531509. [DOI] [PubMed] [Google Scholar]
- Makino I, Shibata K, Ohgami Y, Fujiwara M, Furukawa T. Transient upregulation of the AT2 receptor mRNA level after global ischemia in the rat brain. Neuropeptides. 1996;30:596–601. doi: 10.1016/s0143-4179(96)90043-8. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Ito T, Saavedra JM. Angiotensin II AT1 blockade normalizes cerebrovascular autoregulation and reduces cerebral ischemia in spontaneously hypertensive rats. Stroke. 2000;31:2478–2486. doi: 10.1161/01.str.31.10.2478. [DOI] [PubMed] [Google Scholar]
- Papademetriou V, Farsang C, Elmfeldt D, Hofman A, Lithell H, Olofsson B, Skoog I, Trenkwalder P, Zanchetti A. Stroke prevention with the angiotensin II type 1-receptor blocker candesartan in elderly patients with isolated systolic hypertension: the Study on Cognition and Prognosis in the Elderly (SCOPE) J Am Coll Cardiol. 2004;44:1175–1180. doi: 10.1016/j.jacc.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Reinecke K, Lucius R, Reinecke A, Rickert U, Herdegen T, Unger T. Angiotensin II accelerates functional recovery in the rat sciatic nerve in vivo: role of the AT2 receptor and the transcription factor NF-κB. FASEB J. 2003;17:2094–2096. doi: 10.1096/fj.02-1193fje. [DOI] [PubMed] [Google Scholar]
- Sharkey J, Butcher SP. Characterisation of an experimental model of stroke produced by intracerebral microinjection of endothelin-1 adjacent to the rat middle cerebral artery. J Neurosci Methods. 1995;60:125–131. doi: 10.1016/0165-0270(95)00003-d. [DOI] [PubMed] [Google Scholar]
- Stenman E, Edvinsson L. Cerebral ischemia enhances vascular angiotensin AT1 receptor-mediated contraction in rats. Stroke. 2004;35:970–974. doi: 10.1161/01.STR.0000121642.53822.58. [DOI] [PubMed] [Google Scholar]
- Stier CT, Jr, Adler LA, Levine S, Chander PN. Stroke prevention by losartan in stroke-prone spontaneously hypertensive rats. J Hypertens Suppl. 1993;11:S37–S42. [PubMed] [Google Scholar]
- Takada J, Ibayashi S, Ooboshi H, Ago T, Ishikawa E, Kamouchi M, Kitazono T, Iida M. Valsartan improves the lower limit of cerebral autoregulation in rats. Hypertens Res. 2006;29:621–626. doi: 10.1291/hypres.29.621. [DOI] [PubMed] [Google Scholar]
- Viswanathan M, Saavedra JM. Expression of angiotensin II AT2 receptors in the rat skin during experimental wound healing. Peptides. 1992;13:783–786. doi: 10.1016/0196-9781(92)90187-8. [DOI] [PubMed] [Google Scholar]
- Walther T, Olah L, Harms C, Maul B, Bader M, Hörtnagl H, Schultheiss HP, Mies G. Ischemic injury in experimental stroke depends on angiotensin II. FASEB J. 2002;16:169–176. doi: 10.1096/fj.01-0601com. [DOI] [PubMed] [Google Scholar]
- Windle V, Szymanska A, Granter-Button S, White C, Buist R, Peeling J, Corbett D. An analysis of four different methods of producing focal cerebral ischemia with endothelin-1 in the rat. Exp Neurol. 2006;201:324–334. doi: 10.1016/j.expneurol.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Yamamoto E, Tamamaki N, Nakamura T, Kataoka K, Tokutomi Y, Dong YF, Fukuda M, Matsuba S, Ogawa H, Kim-Mitsuyama S. Excess salt causes cerebral neuronal apoptosis and inflammation in stroke-prone hypertensive rats through angiotensin II-induced NADPH oxidase activation. Stroke. 2008;39:3049–3056. doi: 10.1161/STROKEAHA.108.517284. [DOI] [PubMed] [Google Scholar]