

Abstract
Replacement of the constrained phenylalanine analogue 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (Tic) in the opioid Dmt-Tic-Gly-NH-Bn scaffold by the 4-amino-1,2,4,5-tetrahydro-indolo[2,3-c]azepin-3-one (Aia) and 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one (Aba) scaffolds has led to the discovery of novel potent μ-selective agonists (Structures 5 and 12) as well as potent and selective δ-opioid receptor antagonists (Structures 9 and 15). Both stereochemistry and N-terminal N,N-dimethylation proved to be crucial factors for opioid receptor selectivity and functional bioactivity in the investigated small peptidomimetic templates. In addition to the in vitro pharmacological evaluation, automated docking models of Dmt-Tic and Dmt-Aba analogues were constructed in order to rationalize the observed structure-activity data.
Conformationally constrained amino acids have found widespread application in search of novel peptidic opioid ligands with minored side-effects.1–8 Such residues, inducing enhanced receptor selectivity and affinity, can be subdivided in sterically (e.g., β-methylphenylalanine, β-methyltryptophan, β-methyl-2′,6′-dimethyltyrosine) and covalently constrained derivatives [e.g., 2-aminotetralin-2-carboxylic acid (Atc), 2-aminoindane-2-carboxylic acid (Aic), 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (Tic)].9 The Dmt-Tic scaffold 1, in particular, has been recognized as a firmly established template for opioid ligand design.3,4 Subtle changes in this scaffold have induced remarkable alterations in opioid receptor selectivity and/or activity, such as enhanced agonism, antagonism, or the acquisition of mixed activities at the opioid subtype receptors (i.e., μ, δ and κ receptors). Replacement of tyrosine by Dmt (2′,6′-dimethyl-L-tyrosine) markedly modified the pharmacological profile of numerous unrelated opioid ligands.3, 10, 11 Next to the crucial impact of Dmt on opioid potency, the introduction of Tic into naturally occurring opioid ligands,12–14 led to the conclusion that this conformationally restricted phenylalanine analogue was responsible for the receptor antagonism displayed by Tic-containing structures.
Earlier work in our laboratory involved the replacement of Tic2 in Dmt-Tic derivatives 1 by the dipeptidomimetic Aba-Gly (Aba: 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one). This substitution, yielding opioid ligands of type 2 (Figure 1), led to increased receptor selectivities as well as a δ to μ receptor affinity shift, relative to the reference Tic derivatives.15 The main structural difference between the constrained aromatic residues Tic and Aba, consists of the allowed low energy conformations for the amino acid χ1 space. The Tic residue allows g(−) and g(+) conformations (χ1 = −60° and +60°, respectively), whereas Aba limits the side chain orientation to g(+) and trans (χ1 = 60° and 180°, respectively).16 The same dihedral angles are favoured in the 4-amino-indolo[2,3-c]azepin-3-one (Aia) scaffold which is present in 3. The heterocyclic core of 3 was recently successfully used for sst4/5 selective somatostatin peptidomimetics.17
Figure 1.
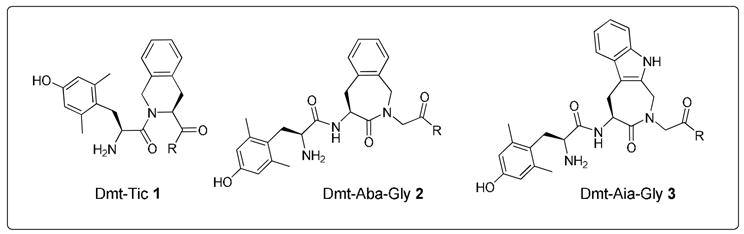
Structures of the Dmt-Tic 1, Dmt-Aba 2 and Dmt-Aia 3 scaffolds
In the present work, two approaches were followed to change potency, selectivity or functional properties of the previously reported Dmt-Aba-Gly analogues 2.15 In the first approach, the role of charge in the discrimination of opioid receptor selectivity and bioactivity was investigated by the preparation and evaluation of Aba-Asp (negative charge) and Aba-Lys (positive charge) derivatives. In the corresponding Dmt-Tic analogues, the charge of the C-terminal part had substantial effect on δ-selectivity and antagonism.18 In a second approach the effects of replacing the benzene ring in the Aba scaffold in 2 by an indole ring, to give 3, was investigated. This approach was motivated by the fact that, on the one hand, in the bioactive conformation of Tyr1-Tic2-Phe3-δ-antagonist peptides, the Tic2 aromatic ring would correspond to the indole ring in the non-peptide δ-antagonist naltrindole.19 On the other hand, the Tic2 residue in 1 can also correspond to the Phe3 residue in opioid peptides such as dermorphin (Tyr1-D-Ala2-Phe3-Gly4-Tyr5-Pro6-Ser7-NH2), deltorphin I or II (Tyr1-D-Ala2-Phe3-Asp4-Val5-Val6-Gly7-NH2 or Tyr1-D-Ala2-Phe3-Glu4-Val5-Val6-Gly7-NH2, resp.) or endomorphin-2 (EM-2: Tyr1-Pro2-Phe3-Phe4-NH2). The related analogue endomorphin-1 (EM-1: Tyr1-Pro2-Trp3-Phe4-NH2) contains a Trp residue at position 3 which may be mimicked by the Aia residue in scaffold 3 (Figure 1).
The reference structures for this work consisted of peptidomimetics 4 and 5 (Figure 2).15 Dmt-Tic-Gly-NH-Bn 4 displays a δ over μ selectivity (Table 1), whereas μ-opioid receptor binding is preferred for the Aba-containing analogue 5. Substitution of Tic2 by Aba in Dmt-Tic ligands reduced δ-opioid receptor affinity, but ligand 5 maintained high μ-opioid receptor affinity and functional bioactivity (Table 1). Peptide mimic 5 (Figure 2) possesses Kiμ and IC50(GPI) values comparable to those of the μ-selective endogenous tetrapeptides endomorphin-1 and endomorphin-2.
Figure 2.
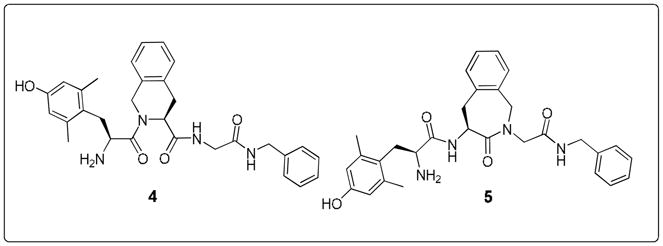
Reference compounds Dmt-Tic-NH-Bn 4 and Dmt-Aba-Gly-NH-Bn 5.15
Table 1.
Receptor affinities, selectivities and functional bioactivity of ligands 4 to 17
| Receptor affinity (nM)a | Selectivity | Functional Bioactivityb | ||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | Kiδ | Kiμ | Kiδ/Kiμ | Kiμ/Kiδ | MVD IC50 (nM) | MVD (pA2) | GPI IC50 (nM) | |
| 4 | H-Dmt-Tic-Gly-NH-Bnc | 0.031±0.002 | 0.16±0.018 | 5.2 | 9.3 | 2.69±0.07 | ||
| 5 | H-Dmt-Aba-Gly-NH-Bnc | 11.0±2.3 | 0.46±0.07 | 24 | 830±70 | 51±5 | ||
|
| ||||||||
| 6 | H-Dmt-Aba-Lys-NH-Bn | 290.1±16 (4) | 10.1±1.3 (5) | 29 | NT | 3272± 354 | ||
| 7 | H-Dmt-Aba-Asp-NH-Bn | 1478±189 (3) | 629.5±210 (3) | 2.3 | NT | NT | ||
| 8 | H-Dmt-Aba | 367.8±48 (5) | 12.4±1.2 (4) | 30 | NT | 631±54 | ||
| 9 | N,N(Me)2-Dmt-Aba | 20.5±0.67 (4) | 64.9±10 (5) | 3.2 | NA | 7.2 | NA | |
|
| ||||||||
| 10 | H-Dmt-Aia-Gly-NH-Bn | 357.9±43 (4) | 50.0±2.6 (3) | 7.2 | NT | 1795±215 | ||
| 11 | H-Dmt-Aia-Gly-O-Bn | 4065±410 (3) | 243.7±24 (3) | 17 | NT | NT | ||
| 1500±139 | ||||||||
| 12 | H-Dmt-D-Aia-Gly-NH-Bn | 160.7±23 (4) | 3.35±0.28 (3) | 48 | (Emax = 80%) | 14.9±1.6 | ||
| 13 | N,N(Me)2-Dmt-D-Aia-Gly NH-Bn | |||||||
| 681±36 (4) | 133±18 (5) | 5 | NT | NT | ||||
|
| ||||||||
| 14 | H-Dmt-Aia-Gly-OH | 805.9±79 (6) | 1450±130 (4) | 1.8 | NT | NA | ||
| 15 | N,N(Me)2-Dmt-Aia-Gly-OH | |||||||
| 6.64±0.89 (5) | 874.5±100 (5) | 132 | NA | 8.3 | NA | |||
| 16 | H-Dmt-D-Aia-Gly-OH | 720±31(4) | 2900±187 (5) | 4 | NT | NT | ||
| 17 | N,N(Me)2-Dmt-D-Aia-Gly-OH | 1150±112 (5) | 10100±982(6) | 8.8 | NT | NT | ||
The Ki values (nM) were determined according to Cheng and Prusoff,38 using published methods.39 The mean ± SEM values of three to six repetitions are based on independent binding assays conducted in duplicate using five to eight grade doses of peptides with several different synaptosomal preparations.
Agonism was expressed as IC50 obtained from dose response curves.40 These values represent the mean ± SEM of at least six fresh tissue samples. Deltorphin C and dermorphin were the internal standards for MVD (δ-opioid receptor bioactivity) and GPI (μ-opioid receptor bioactivity) tissue preparations, respectively.
Data taken from Ballet et al.15 NA: not active (>20.000nM); NT: not tested
Three-dimensional structural models of the δ-opioid (DOR) and μ-opioid receptor (MOR) were constructed and refined according to a GPCR modeling procedure previously described,20 using the beta 2 adrenergic receptor crystal structure21 as a homology modeling template and using the experimental constraints defined by Mosberg et al.22, 23 Surflex24 automated docking simulations25 yielded binding modes of the known δ-agonist JOM13 (Tyr1-c[D-Cys2-Phe3-D-Pen4]OH) in DOR (Fig. 3A) and of the known μ-agonist JOM6 (Tyr1-c(S-Et-S)[D-Cys2-Phe3-d-Pen4]NH2) in MOR (Fig. 4A) which are in line with experimental studies.22, 23, 26–34 In both receptors, the protonated amine group of the ligand forms a complementary H-bond interaction network with D3.3228, 31 (Ballesteros-Weinstein numbering) and Y7.43,30, 31 while the Tyr1 phenol ring binds in the hydrophobic pocket between Y3.3330, 33 and W6.4830 and forms an H-bond with H6.52.26 The Phe3 group of JOM6 adopts a trans χ1 orientation and forms aromatic interactions with W7.35 in MOR,22 while the Phe3 group of JOM13 adopts a gauche (+) orientation and binds in the hydrophobic pocket near H7.36 in DOR.23 The C-terminal carboxamide group of JOM6 is in close proximity of E5.35 in MOR,22 whereas the C-terminal carboxylate group of JOM13 forms a salt bridge with K5.39 in DOR.23 Docking poses25 of reference compounds 4 (in DOR, see Fig. 3B) and 5 (in MOR, see Fig. 4B) involved in the same receptor-ligand interactions as JOM1323 (in DOR, Fig. 3A) and JOM622 (in MOR, Fig. 4A), respectively, were selected using a receptor-ligand interaction fingerprint (IFP) scoring method35 as described earlier.20 The N-terminal Dmt groups of compounds 4 and 5 form the same H-bond interactions with D3.32 and H6.52 as the N-terminal tyramine groups of JOM6 and JOM13 and bind in the same hydrophobic pocket between Y3.33 and W6.48. The Tic group of DOR-specific compound 4 occupies the same binding pocket near H7.36 as the Phe3 group of JOM13, while the Bn group of compound 4 stacks with W6.58, which plays an important role in DOR-specific agonist binding34 (Fig. 3B). The Aba group of the MOR-specific compound 5, on the other hand, stacks with W7.35, a residue which plays an important role in MOR-specific agonist binding,32 and occupies the same binding pocket as the Phe3 ring of JOM622 (Fig. 4). The Bn aromatic ring of compound 5 stacks with the Aba group of the ligand and forms a cation-pi interaction with the positively charged nitrogen atom of K5.3927 of MOR (Fig. 4B).
Figure 3.

Docking poses of: (A) JOM13 (green carbon atoms) and (B) compound 4 (yellow carbon atoms) in the DOR receptor model. The backbone of transmembrane helices 5, 6, and 7 are represented by magenta ribbons (TM3 is not shown for clarity). Important binding residues are depicted as ball-and-sticks with grey carbon atoms. Oxygen, nitrogen, sulphur and hydrogen atoms are coloured red, blue, orange and cyan, respectively. H-bonds described in the text are depicted by white dots.
Figure 4.

Docking poses of: (A) JOM6 (green carbon atoms) and (B) compound 5 (yellow carbon atoms) in the MOR receptor model. Rendering and colour coding are the same as defined for Fig. 3.
To obtain positively and negatively charged scaffolds, respectively, Aba-Lys 6 and Aba-Asp 7 derivatives were prepared according to previously reported methods.15, 36 The presence of a positive charge in ligand 6 seemed to be detrimental for binding to the active sites of both μ-opioid and δ-opioid receptors, as witnessed by a 22 to 26-fold decrease in affinity (Table 1, Kiδ = 290.1 ± 16 nM and Kiμ = 10.1 ± 1.3 nM). Because of the nanomolar range of the μ-affinity, μ-receptor activity was nonetheless verified. The in vitro GPI assay of ligand 6, measuring the activation of the μ-opioid receptor, reflected this loss in binding by an equivalent observed decrease in potency IC50μ = 3272 ± 354 nM).
The negative charge in the aspartic acid side chain of the analogous Dmt-Aba-Asp-NH2 ligand 7, was even less tolerated by both subtype receptors Kiδ = 1478 ± 189 nM and Kiμ = 629 ± 210 nM). Next to a loss in binding, the negative charge also eliminates μ-receptor selectivity (5→7: Kiδ/Kiμ24 → 2.3) The binding orientation of compound 5 in the μ-receptor presented in Fig. 4B suggests that introduction of a positively (compound 6) or negatively (compound 7) charged side chain at the Gly position indeed does not enable beneficial interactions with the charged residues at positions 5.35 (Asp in DOR, Glu in MOR), 5.39 (Lys in DOR and MOR), nor 6.58 (Lys in MOR), but instead cause the ligand to clash with TM6 lower in the binding pocket.
The truncation of the Dmt-Aba-Gly-NH-Bn 5, presenting ligand 8, results in a moderate binding to and activation of MOR (Kiμ = 12.4 ± 1.2 nM, IC50μ = 631 ± 54 nM), but affinity for DOR is significantly decreased (Kiδ = 367.8 ± 48 nM). According to our docking model, this affinity loss in MOR of compound 8 compared to 5 can be ascribed to the elimination of the favourable cation-pi interaction between the C-terminal benzyl group in 5 and the positively charged amine group of K5.39, and the loss of stabilizing intramolecular pi-pi stacking between the Aba and Bn rings (Fig. 4B). The N,N-dimethylation of Dmt-Tic sequences was initially introduced to develop more stable structures, as Tyr-Tic and Dmt-Tic peptides are prone to form diketopiperazines.3 The N,N-dimethyl analogue of 8, ligand 9, shifts receptor selectivity from the μ- to the δ- opioid receptor and concomitantly converts the activity profile, creating a δ-opioid receptor antagonist, an effect that was also observed in Dmt-Tic analogues.37
The 4-amino-indolo[2,3-c]azepin-3-one scaffold (Aia) in 3 can be regarded as a constrained tryptophan residue that limits both χ1 and χ2 dihedral angles in the same way as the Aba scaffold does for a phenylalanine residue.17, 41 Structurally, the introduction of Aia into the opioid pharmacophore can, for example, mimic Trp3 as was shown in the endogenous endomorphin-1 (EM-1: H-Tyr1-Pro2-Trp3-Phe4-NH2). Although the peptidomimetic [Aia3]EM-1 was determined to be a full agonist in the GPI and MVD assays, a loss in functional bioactivity was observed (IC50μ = 223.8 ± 3.64 nM vs. 9.7 ± 2.21 nM for EM-1).41 To further evaluate the constrained Aia template in opioid ligands, we prepared compounds 10 to 17 according to literature methodologies.15, 41 The aminobenzazepinone moiety in the most active compound of the series, Dmt-Aba-Gly-NH-Bn 5, was replaced by the Trp-counterpart Aia and yielded ligand 10 (Table 1). Both δ- and μ-opioid receptor affinities of 10 are situated in the moderate to high nanomolar range, while maintaining MOR selectivity. Only micromolar range potency was displayed when this structure was submitted to the functional GPI assay. The C-terminal ester equivalent of 10, compound 11, showed even less affinity for both μ- and δ-opioid receptors, and suggests the amide in 10 to be involved in hydrogen bonding and/or favorable directing of the terminal benzyl group. The substitution of the benzene ring of the Aba residue in 5 by the indole ring in 10 positions the benzene ring of the Aia residue further away from the MOR specific binding pocket between W7.35 and H7.36 (Figure 4B), but also results in a slightly different orientation of the N-benzyl substituent, which may weaken the cation -π interaction with the K5.39 residue of MOR. This is shown in the superpositions of the binding conformation of 5 and 10 in Figure 5. The combination of both effects may explain the loss in binding affinity observed for 10 versus 5.
Figure 5.
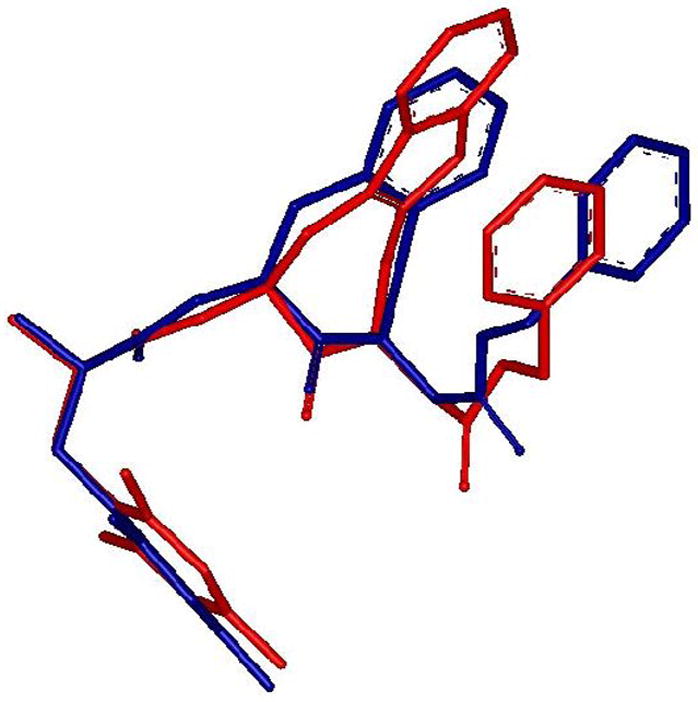
Superposition of the docking poses of compound 5 (blue) and compound 10 (red) in the MOR receptor model.25
The importance of the Aia α-stereochemistry was investigated by switching to the D-isomer of Aia. D-Aia analogue 12 not only yielded a highly selective derivative, its μ-bioactivity is comparable to that of the potent endogenous peptide EM-1 (IC50μ(12)= 14.9 ± 1.6 nM and IC50μ(EM–1)= 9.7 ± 2.21 nM41). This observation suggests that the D-configuration of the Aia residue might be able to restore a binding pose as observed for 5. N,N-dimethylation of 12, resulting in ligand 13, significantly decreased μ-receptor binding and was detrimental for receptor selectivity, an observation which was also made N,N(Me)2-Dmt-Tic-amides.37 Apparently the extensive hydrogen bonding network that is observed between the primary amine of Dmt in 5 (Figure 4 B) is disturbed by the N,N-dimethylation.
The presence of a C-terminal carboxylic acid in opioid ligands is known to induce δ-receptor selectivity due to an unfavorable interaction between the ligands’ carboxylate and residue E5.35 in the μ-opioid receptor.42 This electrostatic interaction is most probably responsible for the δ-opioid receptor preference of ligands 14 to 17. Only one of these ligands, compound 15, efficiently binds to DOR (Kiδ = 6.64 ± 0.89 nM). It showed potent antagonist properties(pA2δ = 8.3), confirming that N,N-dimethylation of Dmt is able to convert a δ-agonist into an antagonist.37 The comparison of ligands 15 and 17 clearly shows the crucial importance of stereochemistry in receptor binding efficiency.
In conclusion, of our two approaches to change the pharmacological profile of Dmt-Aba-Gly analogues, the one involving the introduction of both positive (Aba-Lys ligand 6) and negative (Aba-Asp ligand 7) charge proved to be detrimental for binding to and activation of the μ- and δ-opioid receptors, relative to the reference compound 5. This is in sharp contrast to previous observations in the Dmt-Tic series of analogues, where a positive charge in the side chain of the C-terminal amino acid considerably increased the μ-affinity while maintaining δ-affinity. A negative charge in these Dmt-Tic analogues was shown to be important for δ-affinity, but prevented these ligands from interacting with the μ-receptor.18 These findings could be rationalized by the different binding modes that were observed for the receptor-docked structures between the Tic-containing versus the Aba-containing analogues. The C-terminal benzyl group in 5, on the other hand, was shown to be important for bioactivity at the MOR as demonstrated by the reduced potency of truncated ligand 8, a feature that was also rationalized by the binding mode of these ligands to the MOR. In contrast, our second approach to investigate the importance of replacing the benzene ring in the Aba scaffold by an indole ring, which is equivalent to replacing a constrained Phe residue by a constrained Trp residue, resulted in some interesting new opioid ligands. The replacement of Aba in 5 by Aia in 10 resulted in a drop of affinity for both receptors, However, the change of chirality to D-Aia in 12 resulted in a μ-opioid receptor selective agonist with a potency comparable to the endogenous opioid endomorphin-1. The indole ring of Aia is therefore is more likely to mimic the Trp3 residue in the μ-agonist endomorphin-1 than the indole in the δ-antagonist naltrindole. Finally, two novel N,N-dimethylated scaffolds 9 and 15 proved to be δ-selective antagonists, which confirms that N,N-dimethylation of Dmt is able to induce antagonist properties.18
Acknowledgments
D. Feytens is a Research Assistant for the Fund for Scientific Research - Flanders (Belgium). This work was supported in part by the Intramural Research Program of NIH and NIEHS and by the Fund for Scientific Research - Flanders (Belgium, grants G.0036.04 and G.0008.08) and European grant “Normolife” (LSHC-CT-2006-037733).
Abbreviations
- Aba
4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one
- Aia
4-amino-1,2,4,5-tetrahydro-indolo[2,3-c]azepin-3-one
- Aic
2-aminoindane-2-carboxylic acid
- Atc
2-aminotetralin-2-carboxylic acid
- Dmt
2′,6′-dimethyl-L-tyrosine
- DOR
δ-opioid receptor
- EM-1
endomorphin-1
- GPI
guinea-pig ileum
- MOR
μ-opioid receptor
- MVD
mouse vas deferens
- Tic
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
- 7TM
α-helical transmembrane domain 7
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Hruby VJ, Li GG, HaskellLuevano C, Shenderovich M. Biopolymers. 1997;43:219. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 2.Birkas E, Kertesz I, Toth G, Bakota L, Gulya K, Szucs M. Neuropeptides. 2008;42:57. doi: 10.1016/j.npep.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Bryant SD, Jinsmaa Y, Salvadori S, Okada Y, Lazarus LH. Biopolymers. 2003;71:86. doi: 10.1002/bip.10399. [DOI] [PubMed] [Google Scholar]
- 4.Schiller PW, Weltrowska G, Berezowska I, Nguyen TMD, Wilkes BC, Lemieux C, Chung NN. Biopolymers. 1999;51:411. doi: 10.1002/(SICI)1097-0282(1999)51:6<411::AID-BIP4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 5.Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguyen TMD, Lemieux C, Chung NN, Coderre TJ. J Med Chem. 1999;42:3520. doi: 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- 6.Schiller PW, Nguyen TMD, Berezowska I, Dupuis S, Weltrowska G, Chung NN, Lemieux C. Eur J Med Chem. 2000;35:895. doi: 10.1016/s0223-5234(00)01171-5. [DOI] [PubMed] [Google Scholar]
- 7.Schiller PW. Aaps Journal. 2005;7:E560. doi: 10.1208/aapsj070356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Töth G, Ioja E, Tömböly C, Ballet S, Tourwé D, Péter A, Martinek T, Chung NN, Schiller PW, Benyhe S, Borsodi A. J Med Chem. 2007;50:328. doi: 10.1021/jm060721u. [DOI] [PubMed] [Google Scholar]
- 9.Gibson SE, Guillo N, Tozer MJ. Tetrahedron. 1999;55:585. [Google Scholar]
- 10.Guerrini R, Capasso A, Sorrentino L, Anacardio R, Bryant SD, Lazarus LH, Attila M, Salvadori S. Eur J Pharmacol. 1996;302:37. doi: 10.1016/0014-2999(96)00067-2. [DOI] [PubMed] [Google Scholar]
- 11.Okada Y, Tsuda Y, Fujita Y, Yokoi T, Sasaki Y, Ambo A, Konishi R, Nagata M, Salvadori S, Jinsmaa Y, Bryant SD, Lazarus LH. J Med Chem. 2003;46:3201. doi: 10.1021/jm020459z. [DOI] [PubMed] [Google Scholar]
- 12.Tancredi T, Salvadori S, Amodeo P, Picone D, Lazarus LH, Bryant SD, Guerrini R, Marzola G, Temussi PA. Eur J Biochem. 1994;224:241. doi: 10.1111/j.1432-1033.1994.tb20017.x. [DOI] [PubMed] [Google Scholar]
- 13.Lu YX, Nguyen TMD, Weltrowska G, Berezowska I, Lemieux C, Chung NN, Schiller PW. J Med Chem. 2001;44:3048. doi: 10.1021/jm0101186. [DOI] [PubMed] [Google Scholar]
- 14.Guerrini R, Capasso A, Marastoni M, Bryant SD, Cooper PS, Lazarus LH, Temussi PA, Salvadori S. Bioorg Med Chem. 1998;6:57. doi: 10.1016/s0968-0896(97)10008-6. [DOI] [PubMed] [Google Scholar]
- 15.Ballet S, Salvadori S, Trapella C, Bryant SD, Jinsmaa Y, Lazarus LH, Negri L, Giannini E, Lattanzi R, Tourwé D, Balboni G. J Med Chem. 2006;49:3990. doi: 10.1021/jm0603264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tourwé D, Verschueren K, Frycia A, Davis P, Porreca F, Hruby VJ, Töth G, Jaspers H, Verheyden P, Van Binst G. Biopolymers. 1996;38:1. doi: 10.1002/(sici)1097-0282(199601)38:1<1::aid-bip1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Feytens D, Cescato R, Reubi JC, Tourwé D. J Med Chem. 2007;50:3397. doi: 10.1021/jm070246f. [DOI] [PubMed] [Google Scholar]
- 18.Balboni G, Salvadori S, Guerrini R, Negri L, Giannini E, Bryant SD, Jinsmaa Y, Lazarus LH. J Med Chem. 2004;47:4066. doi: 10.1021/jm040033f. [DOI] [PubMed] [Google Scholar]
- 19.Wilkes BC, Schiller PW. Biopolymers. 1995;37:391. doi: 10.1002/bip.360370606. [DOI] [PubMed] [Google Scholar]
- 20.de Graaf C, Foata N, Engkvist O, Rognan D. Proteins. 2008;71:599. doi: 10.1002/prot.21724. [DOI] [PubMed] [Google Scholar]
- 21.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fowler CB, Pogozheva ID, Lomize AL, LeVine H, Mosberg HI. Biochemistry. 2004;43:15796. doi: 10.1021/bi048413q. [DOI] [PubMed] [Google Scholar]
- 23.Mosberg HI. Biopolymers. 1999;51:426. doi: 10.1002/(SICI)1097-0282(1999)51:6<426::AID-BIP5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 24.Jain AN. J Comput Aided Mol Des. 2007;21:281. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 25.Ligands were automatically docked into the DOR and MOR receptor models using standard parameters of Surflex v2.11. Twenty poses were generated for each ligand. The binding site (protomol file) was defined by providing a list of cavity residues in 5 Ångstrom distance from JOM6 (in MOR) or JOM13 (in DOR) in the refined receptor-ligand structure.
- 26.Spivak CE, Beglan CL, Seidleck BK, Hirshbein LD, Blaschak CJ, Uhl GR, Surratt CK. Mol Pharmacol. 1997;52:983. doi: 10.1124/mol.52.6.983. [DOI] [PubMed] [Google Scholar]
- 27.Chen CG, Yin YL, deRiel JK, DesJarlais RL, Raveglia LF, Zhu JM, LiuChen LY. J Biol Chem. 1996;271:21422. doi: 10.1074/jbc.271.35.21422. [DOI] [PubMed] [Google Scholar]
- 28.Li JG, Chen CG, Yin JL, Rice K, Zhang Y, Matecka D, de Riel JK, DesJarlais RL, Liu-Chen LY. Life Sci. 1999;65:175. doi: 10.1016/s0024-3205(99)00234-9. [DOI] [PubMed] [Google Scholar]
- 29.Surratt CK, Johnson PS, Moriwaki A, Seidleck BK, Blaschak CJ. JBiol Chem. 1994;269:20548. [PubMed] [Google Scholar]
- 30.Befort K, Tabbara L, Kling D, Maigret B, Kieffer BL. J Biol Chem. 1996;271:10161. doi: 10.1074/jbc.271.17.10161. [DOI] [PubMed] [Google Scholar]
- 31.Befort K, Zilliox C, Filliol D, Yue SY, Kieffer BL. J Biol Chem. 1999;274:18574. doi: 10.1074/jbc.274.26.18574. [DOI] [PubMed] [Google Scholar]
- 32.Bonner G, Meng F, Akil H. Eur J Pharmacol. 2000;403:37. doi: 10.1016/s0014-2999(00)00578-1. [DOI] [PubMed] [Google Scholar]
- 33.Xu H, Lu YF, Partilla JS, Zheng QX, Wang JB, Brine GA, Carroll FI, Rice KC, Chen KX, Chi ZQ, Rothman RB. Synapse. 1999;32:23. doi: 10.1002/(SICI)1098-2396(199904)32:1<23::AID-SYN3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 34.Valiquette M, Vu HK, Yue SY, Wahlestedt C, Walker P. JBiol Chem. 1996;271:18789. doi: 10.1074/jbc.271.31.18789. [DOI] [PubMed] [Google Scholar]
- 35.Marcou G, Rognan D. J Chem Inf Model. 2007;47:195. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- 36.Van Rompaey K, Van den Eynde I, De Kimpe N, Tourwé D. Tetrahedron. 2003;59:4421. [Google Scholar]
- 37.Salvadori S, Guerrini R, Balboni G, Bianchi C, Bryant SD, Cooper PS, Lazarus LH. JMed Chem. 1999;42:5010. doi: 10.1021/jm990165m. [DOI] [PubMed] [Google Scholar]
- 38.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 39.Salvadori S, Balboni G, Guerrini R, Tomatis R, Bianchi C, Bryant SD, Cooper PS, Lazarus LH. J Med Chem. 1997;40:3100. doi: 10.1021/jm9607663. [DOI] [PubMed] [Google Scholar]
- 40.Kosterlitz HW, Watt AJ. Br J Pharmacol. 1968;33:266. doi: 10.1111/j.1476-5381.1968.tb00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pulka K, Feytens D, Van den Eynde I, De Wachter R, Kosson P, Misicka A, Lipkowski A, Chung NN, Schiller PW, Tourwé D. Tetrahedron. 2007;63:1459. [Google Scholar]
- 42.Mosberg HI, Fowler CB. J Pept Res. 2002;60:329. doi: 10.1034/j.1399-3011.2002.21061.x. [DOI] [PubMed] [Google Scholar]