

Abstract
A current popular model to explain phosphorylation of smooth muscle myosin (SMM) by smooth muscle myosin light chain kinase (MLCK) proposes that MLCK is bound tightly to actin but weakly to SMM. We found that MLCK and calmodulin (CaM) co-purify with unphosphorylated SMM (up-SMM) from chicken gizzard, suggesting that they are tightly bound. Although the MLCK:SMM molar ratio in SMM preparations was well below stoichiometric (1:73 ± 9), the ratio was ~ 23–37% of that in gizzard tissue. Fifteen to 30% of MLCK was associated with CaM at ~1 nM free [Ca2+]. There were two MLCK pools that bound unphosphorylated SMM with Kd ~10 μM and 0.2 μM and phosphorylated SMM with a Kd ~ 20 μM and 0.2 μM. Using an in vitro motility assay to measure actin sliding velocities, we showed that the co-purifying MLCK-CaM was activated by Ca2+ and phosphorylation of SMM occurred at a pCa50 of 6.1 and Hill coefficient of 0.9. Similar properties were observed from reconstituted MLCK-CaM-SMM. Using motility assays, co-sedimentation assays, and on-coverslip ELISA assays to quantify proteins on the motility assay coverslip, we provide strong evidence that most of the MLCK is bound directly to SMM through the telokin domain and some may also be bound to both SMM and to co-purifying actin through the N-terminal actin binding domain. These results suggest that this MLCK may play a role in the initiation of contraction.
Keywords: smooth muscle, myosin light chain kinase, smooth muscle myosin, actin motility, phosphorylation
Introduction
In smooth muscles, contraction is initiated by phosphorylation of the smooth muscle myosin (SMM) regulatory light chain (RLC) at Ser19 by Ca2+-CaM-dependent myosin light chain kinase (MLCK). 1; 2; 3 This phosphorylation increases the actin-activated ATPase activity of SMM leading to cyclic interaction with actin and force development. MLCK is ubiquitously expressed in smooth muscles 4 and its only known substrate in vivo is myosin. 5 Smooth muscle MLCK (the short ~130 kDa isoform) remains bound tightly to detergent-washed myofilaments 6 and detergent/glycerol-skinned smooth muscle tissue strips. 7 This MLCK isoform has an actin-binding domain, composed of 3 DFRXXL motifs, at the N-terminus. 8; 9; 10; 11 This domain binds to purified F-actin,12 but has a much higher affinity for myofilaments,8; 9; 10; 11; 13; 14 which contain many other proteins including SMM. Mutating the actin-binding domain significantly decreases the association of MLCK with myofilaments in vitro and with actin-containing filaments in smooth muscle cells.9
MLCK also contains myosin- and CaM-binding sites. MLCK binds to SMM filaments, 12; 15; 16 the predominant arrangement of SMM in vivo, at the junction between the two SMM heads and the tail, 15 most likely through the C-terminal telokin domain of the kinase.17; 18 The CaM-binding site on MLCK, responsible for activation of the enzyme, lies between the catalytic domain and the C-terminal telokin domain.19 Ca2+/CaM induces a conformational change in MLCK, which involves displacement of the auto-inhibitory sequence on MLCK from the surface of the catalytic core, thereby allowing substrate (RLC) access.20 Purified MLCK can only bind to CaM in the presence of Ca2+, 21 whereas CaM can bind to myofilaments in a Ca2+-independent manner. 22
Elucidating the mechanisms of smooth muscle contraction and relaxation is central for understanding smooth muscle hypercontractility associated with diseases such as asthma and hypertension. Since MLCK is required to initiate contraction, an understanding of its location and binding partners in the muscle is critical to understand the rate limiting steps and other kinetic parameters of muscle activation. A current working model emphasizes that MLCK is an elongated and potentially flexible molecule. 15; 20 Even if the N-terminus is firmly anchored to actin, 13; 14 the molecular dimensions may allow the catalytic core to reach the myosin in the thick filaments. If the affinity of the C-terminal myosin-binding domain of MLCK is weak, 12; 17; 18 it may serve only to steer the kinase towards the myosin filaments, increasing the probability of phosphorylation of the RLC. Since MLCK remains attached to thin filaments in the presence of Ca2+/CaM, 14 it may localize close to thin and thick filaments during contractile activity. This model raises interesting questions about how an enzyme that is present in very low amounts relative to its substrate myosin could actually cause the initiation of contraction. By some estimates there are only 1–2 MLCK per 1 μm long thin filaments, and assuming a random distribution, there may be many thin filaments that have no bound MLCK. 8 It is known that the level of SMM phosphorylation at the peak of a contraction is 30–80%. 23 Therefore to obtain this level of phosphorylation, the model implies that more and more SMM will be phosphorylated as the thin filament-bound MLCK slides relative to the thick filament. However, SMM is a very low (0.05) duty ratio motor, 24 meaning that it requires the presence of many activated (phosphorylated) heads to actually move actin in a directed manner. Therefore the above model may have an internal inconsistency in that phosphorylation is needed to make the muscle contract, but phosphorylation cannot occur without contraction.
Clues to these questions may be found by noting that the behavior of the MLCK in muscle may be quite different from the purified proteins. For example, it appears that MLCK binds much more tightly to thin filaments than to purified actin, but it is not known which other thin filament proteins are needed for the high affinity. Similarly, there are conflicting reports of the binding affinity of MLCK and CaM to SMM filaments depending upon the purity of the proteins, suggesting that other proteins could be important to the interaction in muscle. These studies and the relationship to the present work will be further elaborated in the discussion.
Our objective is to study the regulation of smooth muscle contraction using a reconstituted system in which both the kinetics and mechanics of activation and relaxation can be simultaneously measured. Our preliminary studies showed unexpectedly that highly purified up-SMM 25 could be induced to form filaments upon addition of Ca2+ and ATP. This suggested the formation of phosphorylated filaments, which are known to be more stable in the presence of ATP. 26; 27; 28 We also observed actin motility in vitro upon addition of Ca2+ to up-SMM, without addition of MLCK or CaM, and confirmed that phosphorylation did occur (see below). These initial observations suggested that MLCK and CaM may bind tightly to upSMM, either directly or through interaction with other protein(s). Clearly if these complexes are present in the muscle in appreciable quantities, they could be relevant to the mechanism of activation and relaxation of smooth muscle and are therefore the focus of this study.
Our main findings are that SMM co-purified with surprisingly large amounts of both MLCK and CaM in the absence of added Ca2+. The co-purified MLCK in a solution of SMM or attached to immobilized SMM was catalytically inactive at low [Ca2+], but could be induced to phosphorylate the SMM in response to physiological [Ca2+], as evidenced by MLCK enzyme assays and by actin filament motion in an in vitro motility assay. We enhanced the power of this approach by developing a novel assay, referred to as “the on-coverslip ELISA”, whereby the relative amounts of various proteins, on the same coverslips on which the motility assays were performed, were quantified using antibodies. This allowed us to show that the amount of actin motility was correlated with the level of SMM phosphorylation in a manner expected from other studies, showing that the MLCK-SMM complexes are both enzymatically and mechanically functional. Equilibrium binding studies reveal the presence of a pool of MLCK that binds with high affinity to both up- and pSMM. These tightly-associated complexes could play a role in the initiation of contraction in smooth muscles.
Results
Quantification of MLCK, CaM and actin content of up-SMM
Western blotting was used to determine the concentrations of MLCK (Fig. 1(a)), CaM (Fig. 1(b)) and actin (Fig. 1(c)) that co-purified with SMM. The following molar ratios (± SD) were calculated from standard curves: MLCK:SMM = 1:73 ± 9, CaM:SMM = 1:371 ± 153, actin:SMM = 1:19 ± 11. The large deviation in CaM ratio was due to a low signal-to-noise ratio. The surprisingly high levels of MLCK and CaM suggested that the proteins may be tightly bound to SMM even in the absence of added Ca2+.
FIGURE 1. . MLCK, CaM and actin content in upSMM preparation.
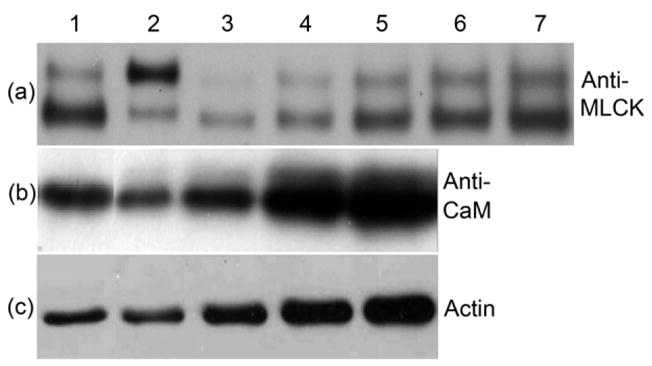
(a), Anti-MLCK western blot. Comparison of an aged SMM sample (Lane 1, 10 μg) and a sample analyzed immediately after preparation (Lane 2, 10 μg), showing the upper full length band at 130 kDa and the lower band resulting from proteolytic cleavage; lanes 3–7, aged chicken gizzard MLCK standards (5, 10, 15, 20, 25 ng), showing the same proteolytic cleavage product as found in the SMM sample. Proteolysis is slow, occurring on the timescale of weeks at 4 °C. There is no evidence that the two forms of MLCK bind differently to SMM. Quantitative data are representative of 4 upSMM preparations and 2–3 independent measurements. (b), Anti-CaM western blot. Lane 1, upSMM, 25 μg; lanes 2–5, chicken gizzard CaM standards (2.1, 4.2, 8.4, 12.6 ng). Data are representative of 2 upSMM preparations and 4 independent measurements. (c), Anti-actin western blot. Lane 1, upSMM, 10 μg; lanes 2–5, smooth muscle actin standards (25, 50, 80, 100 ng). Data are representative of 4 upSMM preparations and 4 independent measurements.
MLCK enzyme activities
The specific activity of MLCK in SMM preparations was 0.7 ± 0.2 μmol min−1 mg−1 (at 2.1 μM SMM) at saturating added CaM (0.1 μM and 0.5mM Ca2+). After adjusting for the fact that the experiment was done below the Km of MLCK for SMM (~14–17 μM),29 our estimate for the Vmax is ~ 5.1 +/− 1.4 μmol min−1 mg−1, which is in accord with the previously measured Vmax of 4 μmol min−1 mg−1. 29 In the absence of added CaM (pCa4), the activity was ~ 8–10 fold less. These values are consistent with the protein ratios estimated in Figure 1 and suggest that CaM present in the SMM preparations is binding to the MLCK and not to other proteins. These data suggest that the SMM contains CaM-bound MLCK and free MLCK that is capable of binding CaM and that the CaM binding enhances the activity of the MLCK as expected. Therefore the MLCK that co-purifies with SMM is enzymatically similar to purified MLCK and appears to be fully active.
Functional assessment of MLCK and CaM by in vitro motility assay for actin sliding
We used an in vitro motility assay30 to establish whether or not MLCK and CaM formed a functional complex that could phosphorylate SMM to generate actin motility. The in vitro motility assay for measurement of actin sliding velocities is a well-established protocol to study the behavior of molecular motors. 31; 32; 33 The actin sliding velocity is thought to be a measure of the unloaded shortening velocity of intact muscle. SMM is attached to a nitrocellulose-coated coverslip and the ATP-activated motion of fluorescently-labeled actin filaments is observed microscopically and quantified by image analysis. Whereas up-SMM does not move actin filaments, phosphorylation triggers motion. 34
UpSMM was applied to the coverslip in Ca2+-free buffer and unbound SMM and any weakly bound protein was removed during the subsequent washing steps in the protocol totaling 320 μl of solutions (40 μl × 8), not including the motility buffer, over a period of approximately 5 min. The velocity of actin motion was Ca2+-dependent (Fig. 2(a)) with a pCa50 of 6.1 and a Hill coefficient of 0.9, whereas the velocity for pre-thiophosphorylated SMM was Ca2+-independent (Fig. 2(b)). Note that there was a time-dependent increase (data not shown) in motility with a parallet increase in phosphorylation (see Figure 3 for Method) for upSMM, but not for pSMM. We are currently investigating the mechanism for the time-dependent increase in phosphorylation using single molecule approaches.
FIGURE 2. Actin sliding velocities of upSMM and pre-thiophosphorylated SMM at different Ca2+ concentrations.

(a), UpSMM (0.1–0.2 mg/ml) was applied to the coverslip and the motility assay was performed at varying [Ca2+]free. Velocities (mean ± SD; n = 5 from 3 upSMM preparations) were normalized to the velocity at pCa 4 (0.3 μm/s). The line is a fit to the Hill equation; pCa50 = 6.1 and Hill coefficient = 0.9. (b), Pre-thiophosphorylated SMM (0.1 mg/ml, n = 3 from 2 preparations). Data were collected at ~ 5 min after loading motility buffer. [Ca2+]free at no added Ca2+ was estimated to be 1 nM. The absolute velocity at all Ca2+ concentrations averaged to ~0.6 μm/s. Note that there was a time-dependent increase (data not shown) in motility with a parallet increase in phosphorylation (see Figure 3 for Method) for upSMM, but not for pSMM. We are currently investigating the mechanism for the time-dependent increase in phosphorylation using single molecule approaches.
FIGURE 3. Quantification of p-RLC on the coverslip after the motility assay by on-coverslip ELISA.
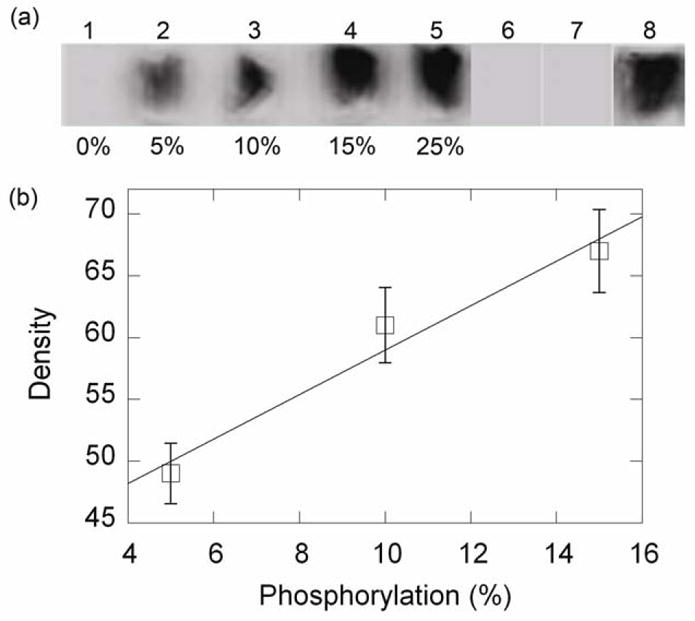
(a), Validation of ELISA method. Images 1–5, mixtures of pSMM and upSMM (values indicate % p-SMM) were applied to the coverslips. Unattached SMM was washed away before the ELISA (see Experimental Procedures) was performed. Image 6, upSMM; Image 7, upSMM subjected to motility conditions for 7–8 min at room temperature at pCa ~ 9; Image 8, same as image 7, except that motility buffer contained 100 μM free Ca2+. (b), standard curve from the data in (a).
No motion was observed if wortmannin (a MLCK inhibitor) was incubated with the up-SMM prior to application to the coverslip, followed by the motility assay at pCa 4, confirming that the SMM was phosphorylated by the MLCK present in the SMM sample (data not shown). The maximum actin velocity with up-SMM at pCa 4 (0.3 μm/s) was half of the velocity with pre-thiophosphorylated SMM (~0.6 μm/s) under the conditions. Since it is known that MLCK is activated by Ca2+-CaM, these results suggest that the kinase activity of the MLCK in the sample was facilitated by pre-existing tightly-bound CaM. Therefore the observed actin motility due to phosphorylated SMM appears to be due to the result of enzymatically functional MCLK/CaM complexes. This result is consistent with the observed MLCK activities in solution.
On-coverslip ELISA to measure p-RLC after the motility assay
From Fig. 2(a) it can be seen that maximal [Ca2+] (pCa 4) generated ~ half of the maximal velocity, suggesting that not all of the SMM was phosphorylated under these conditions. To measure the level of phosphorylation that occurred on the coverslip during the motility assay, we developed a new semi-quantitative ELISA that measures the amount of any protein of interest by applying primary antibody and secondary antibody to the coverslip followed by detection of a chemiluminescence signal using X-ray film. The method was validated with an anti-p-RLC (phospho-specific) antibody by using mixtures of pre-phosphorylated- and up-SMM at varying ratios (Fig. 3(a), images 1–5), which generated a linear standard curve (Fig. 3(b)) up to ~15% p-SMM. Fig. 3(a), image 6, shows that up-SMM gave no detectable phosphorylation signal with this method. Similarly, up-SMM exposed to the exact conditions of the motility assay without adding Ca2+ in the motility buffer, also gave no detectable signal (Fig. 3(a), image 7) but at pCa 4 (Fig. 3(a), image 8) phosphorylation was 13 ± 1% (n = 3). These data, together with those from Figures 1 and 2, show that with a ratio of MLCK:SMM of 1:73, 50% maximal motility can be generated after 10 min by phosphorylation of ~ 13% of the SMM RLCs on the coverslips under the conditions specified. Further studies on the kinetics of MLCK phosphorylation are in progress.
Reconstitution of up-SMM with MLCK and CaM
If the activity of the MLCK/CaM that co-purified with SMM is not an artifact of the in vitro system or of the protein preparation itself, we should be able to reconstitute a similar activity using purified components. To reconstitute the proteins, we applied highly purified up-SMM (molar ratio of MLCK to SMM was ~ 1:490, and molar ratio of actin to SMM was ~ 1:130–260) to the coverslip, washed and blocked with BSA, and performed a motility assay. No motion was observed at pCa 4. This showed that this low level of kinase was not sufficient to generate motility and also that there is no detectable kinase activity in the TRITC-actin preparation used in the motility assay. However, if purified MLCK and CaM (both at 0.1 μM) were added in the absence of added Ca2+ (with EGTA) followed by extensive washing. Ca2+-dependent motility was observed and the velocity was 0.36 μm/s at pCa 4, which is similar to that observed in Fig. 2(a). Therefore, the MLCK/CaM complexes that co-purify with SMM in the absence of Ca2+ are similar to those that can be reconstituted from mixtures of purified proteins in the absence of Ca2+. Further reconstitution experiments will be required to firmly establish the relationship between bound MLCK/CaM and the level of phosphorylation and motility. Actin in the MLCK was below our detection limit using quantitative western blotting analysis (data not shown; contained < 1 actin per 25 MLCK). Even with the actin level very low, we cannot rule out the possibility that only actin-bound MLCK was responsible for the reconstituted motility. However, it is very unlikely due to the extremely low actin concentration in the added MLCK (< 25 times less than the MLCK concentration of 100 nM or ≤ 4 nM actin), which promotes actin depolymerization to the G form, which does not bind to MLCK.
Estimate of the Kd for MLCK binding to SMM by co-sedimentation
The data above suggest that at least some of the MLCK binds to SMM with high affinity. To estimate the binding affinity of the MLCK to the SMM preparation, we centrifuged the SMM under conditions where essentially all (>95%) the SMM forms filaments and is found in the pellet and determined the [MLCK] in the supernatant by quantitative western blot. Figure 4 shows that for upSMM, 46% of the MLCK remains bound to SMM (2.1 μM) (spin 1). This data suggests that MLCK binds to SMM with an apparent Kd of approximately 2.5 μM. To determine whether there was evidence for an additional more tightly bound pool of MLCK, the protocol was repeated by resuspending the pellet from the first spin to the same volume and centrifuging again (spin 2), and continuing to spin 4. Figure 4 shows that 75% of the total MLCK in the tube for spin 2 (not the initial MLCK) remains bound to SMM. The fact that Fb for spins 2–4 is higher than 46% shows that there must be a more tightly bound pool than the original estimate of 2.5 μM (derived from spin 1), in addition to a more weakly bound pool. If all the MLCK was binding with a Kd of 2.5 μM, 46% of the MLCK in each spin should have pelleted with the SMM for spin 1–4. To estimate the amounts and affinities of these two pools, we fit the data to Equation 4, which predicts the amount of MLCK bound to SMM for each spin, relative to the total MLCK present in that spin, if there are two Kds with Kd1≫Kd2. The results of the fits to the data for both upSMM and pSMM are shown in Table 1. In both upSMM and pSMM, about 60–75% of the MLCK appears to be more weakly bound, with Kds in the 10–20 μM range, whereas the remaining MLCK is more tightly bound with Kds in the 0.2 μM range. It is likely that the motility observed in Figure 2 is generated mostly from the tightly bound species. Interestingly, the MLCK that co-purifies with SMM binds with similar affinity to upSMM and pSMM.
FIGURE 4. Estimate of binding affinity of MLCK for upSMM and pSMM.

Data for upSMM (squares) are from 4 independent measurements on 3 SMM preparations (error bars show the standard error). The fit to equation 4 is shown as a solid line. Data for pSMM (diamonds) are from 2 independent measurements from one SMM preparation (error bars show the range). The endogenous kinase was used to phosphorylate SMM. The fit to Equation 4 is shown as a dotted line. For comparison, the plot shows the expected data (circles) for one binding constant with Kd = 2 μM, showing that Fb remains constant for every spin number.
Table 1.
Equilibrium binding of MLCK pools to upSMM and pSMM
| SMM | Equilibrium Dissociation Constants (μM) | Fraction of Total MLCK (%) |
|---|---|---|
| upSMM | 9.5 ± 2.7 | 61 ± 4 |
| 0.20 ± 0.01 | 39 ± 2 | |
| pSMM | 23.8 ± 1.4 | 76 ± 1 |
| 0.24 ± 0.01 | 24 ± 1 |
Data are the results of the fits to Equation 4 of the data in Figure 4. See Figure 4 for definitions of errors.
Configuration of protein complexes responsible for SMM phosphorylation
To understand the nature of the MLCK complexes that are responsible for phosphorylation of SMM and subsequent ability of the SMM to generate actin motility, we have performed a series of experiments with four different reagents, telokin, GST-N1-75MLCK, ATP, and Mg2+. Experiments were performed in solution using co-sedimentation to directly quantify equilibrium binding, or on the coverslip using the ELISA protocol to quantify protein binding and to measure the effects on actin motility. For clarity, the results will be presented in the context of Scheme 1, which describes the configuration of possible protein complexes containing actin, SMM and MLCK. The Scheme depicts the complexes attached through SMM to the coverslip, but the data for the co-sedimentation experiments can be similarly interpreted because the SMM is induced to form filaments and sediments to a pellet. Scheme 1 summarizes the predicted effects of the four reagents upon both actin dissociation and MLCK dissociation from the coverslip or from SMM in a co-sedimentation experiment. On the right side of the Scheme is what we observed, as described in the following Figures.
Scheme 1. Potential configurations of proteins bound to coverslip.

Possible configurations (A–H) of SMM, MLCK and actin bound to the coverslip (horizontal line) indicating binding interactions. X’s on SMM heads indicate dead heads that cannot dissociate from actin. Aligned below A–H are predicted abilities of N1-75, telokin, and Mg2+ to dissociate actin (upper) or MLCK (lower) from the coverslip or from sedimented SMM. A “+” sign means that the reagent is predicted to cause dissociation of that protein from that configuration, and a “−”sign means it is not predicted. The observed effects are shown on the right side. Dotted boxes indicate regions of agreement between observation and prediction. The data suggest that most of the MLCK is in Form G or H, whereas most of the actin is in configuration C or bound directly alone to the coverslip.
Effects of telokin
It is known that telokin, like MLCK, binds to up-SMM at the head-tail junction. 18 This is consistent with the fact that the sequence of telokin is identical to the COOH-terminal domain of MLCK. If the MLCK that co-purified with SMM is binding to SMM (as in Scheme 1, G and H), telokin should be effective at reducing the velocity of actin motion by competing with MLCK for SMM binding, resulting in displacing the MLCK from the coverslip. All other potential configurations that contain MLCK in Scheme 1, with respect to both actin and MLCK dissociation, should be insensitive to telokin. Up-SMM was applied to the coverslip, washed, blocked and incubated with varying concentrations of telokin for 20 min at room temperature. After washout, a motility assay was performed at pCa 4. Fig. 5(a) shows that telokin inhibited the phosphorylation of SMM, as evidenced by a decrease in observed actin sliding velocity. We were unable to show directly using the ELISA that telokin displaced MLCK from the coverslip, because our MLCK antibodies had a slight affinity for telokin (data not shown). Therefore, instead we incubated telokin with filamentous SMM, pelleted the SMM, and analyzed the supernatant for displaced MLCK and actin by western blot. Figure 5(b) shows that telokin displaced the MLCK from the SMM in a concentration dependent manner. Telokin (50 μM) displaced approximately 5% of the actin in a co-sedimentation experiment and ~ 9% as measured by ELISA (Figure 5(b)). These data suggest that much of the MLCK that co-purified with SMM responds to telokin as predicted in the SMM-bound configurations G and H depicted in Scheme 1. The fact that only a small amount of actin could be displaced with telokin suggests that most of the actin that co-purified with SMM is not in form H. This might be expected because the ratio of MLCK to actin is typically 1/5 to 1/8, so all the MLCK would not be expected to be bound to actin (as in H).
FIGURE 5. Effects of telokin.

(a), UpSMM (0.2 mg/ml) was applied to the coverslip, washed, blocked with BSA, and incubated with telokin for 20 min at room temperature, followed by washout and motility assay at pCa 4. The control was pSMM. Data are from 3–5 replicates. (b), left axis, SMM (1 mg/ml) was incubated in binding buffer with varying concentrations of telokin at room temperature for 20 min. After removing an aliquot representing the total MLCK or actin, SMM filaments were pelleted at 200,000 × g for 20 min at 4 °C. Less than 5% of the SMM remained in the supernatant. The amount of MLCK (circles) or actin (squares) in the pellet was calculated and plotted relative to the amount in the pellet at zero [telokin]. Data are from 3 titrations on 2 independent SMM preparations. (b) right axis (diamonds) ELISA signal for actin after treatment with telokin or buffer (10mM MOPS, pH7.0, 50mM NaCl, 1mM MgCl2, 0.1mM EGTA, 3mM DTT; 3 × 40ul) on SMM loaded coverslips, followed by an incubation for 20min at room temperature before washing out and continuing with the normal ELISA assay. Data are from 3 independent measurements on 2 preparations, ± standard error.
Effects of ATP pre-wash
To determine whether actin was present as configurations A, B or C, SMM was washed on the coverslip with MgATP (without Ca2+), prior to a motility assay. If for example, configuration A was largely responsible for SMM phosphorylation, ATP should dissociate the actin from the SMM taking the MLCK with it, therefore decreasing the motility due to a lower level of SMM phosphorylation. However, MgATP (without Ca2+) did not remove a significant amount of actin as assayed by the ELISA (103 ± 6 %). These data suggest that much of the actin that co-purified with SMM may be irreversibly bound to dead SMM heads (Scheme 1, B and C), or directly bound to the coverslip (Scheme 1, D).
Effects of an expressed N-terminal actin-binding domain of MLCK
Scheme 1 summarizes the predicted effects of adding the actin-binding domain of MLCK (N1-75) to the putative protein configurations. This domain should compete with MLCK binding to actin, but have no effect on the binding of MLCK to SMM or the coverslip. We expressed the actin-binding domain of MLCK domain (denoted GST-N1-75MLCK), which consists of the first 75 residues of MLCK that includes the 3 actin-binding motifs, as a GST fusion protein13 in bacteria (Fig. 6(a)) and confirmed that it binds to chicken gizzard myofilaments (Fig. 6(b), lane 1). Treatment of the myofilaments with 50 mM MgCl2, which is known to displace MLCK, prior to adding GST-N1-75MLCK, allowed for further binding of GST-N1-75MLCK (Fig. 6(b), lane 3). Fig. 6(c) shows that ~1/3 of MLCK is dissociated by GST-N1-75MLCK (lanes 3 and 4), whereas the control without GST-N1-75MLCK resulted in dissociation of very little of the MLCK (lanes 1 and 2). Interestingly, further washing with GST-N1-75MLCK maximally dissociated only ½ of the MLCK (data not shown), suggesting that not all MLCK in myofilaments is bound to actin.
FIGURE 6. Characterization of expressed actin-binding domain of MLCK (GST-N1-75MLCK).

(a), SDS-PAGE with Coomassie Blue staining. Lane 1, markers (numbers at left indicate kDa); lane 2, purified GST-N1-75MLCK (abbrev. N1-75); lane 3, thrombin digest of GST-N1-75MLCK producing GST and N1-75MLCK. (b), Western blot analysis (anti-GST) demonstrating binding of GST-N1-75MLCK to myofilaments. Lane 1, myofilaments were incubated with 100 μM GST-N1-75MLCK in 25 mM Tris-HCl, pH 7.5, 80 mM KCl, 1 mM DTT and centrifuged to pellet the myofilaments and bound proteins; lane 2, myofilaments were treated with 50 mM MgCl2 to remove endogenous MLCK; lane 3, myofilaments treated with 50 mM MgCl2 then GST-N1-75 MLCK (note increased binding of GST-N1-75MLCK); lane 4, purified GST-N1-75MLCK. (c), Myofilaments were stirred for 10 min with 0.5 ml of 25 mM Tris-HCl, pH 7.5, 80 mM KCl, 1 mM DTT without or with 100 μM GST-N1-75MLCK and centrifuged at 10,000 × g for 30 min. Pellets were resuspended in 0.5 ml buffer. Lanes 1 (pellet) and 2 (supernatant): incubated without GST-N1-75MLCK; lanes 3 (pellet) and 4 (supernatant): myofilaments were extracted with GST-N1-75MLCK. Results represent n = 3–5. GST-N1-75MLCK treatment dissociates ~ ½ of the MLCK under these conditions.
Having shown that GST-N1-75MLCK functions as expected in myofilaments, we incubated N1-75MLCK (GST removed; 4 μM; Kd ~ 0.8 μM; 13) with SMM in solution before applying the mixture to the coverslip followed by a motility assay Fig. 7(a). Fig. 7(b) shows an independent experiment in which SMM was applied to the coverslip and the coverslip was blocked with BSA before the N1-75MLCK peptide was applied. For either method, the values for the treated samples were not significantly different from the controls. There was also no significant change in the MLCK signal on the coverslip by ELISA (Fig. 7(b)). To confirm these results, Fig. 7(c) shows the results of yet another independent experiment using 20 μM N1-75MLCK. The results of all experiments in Fig. 7 were consistent, indicating that N1-75MLCK had no statistically significant effect on motility or on MLCK bound to the coverslip. These data are inconsistent with significant quantities of configurations A and B in Scheme 1. The fact that a small but significant amount of actin was displaced by N1-75 MLCK (Fig. 7(b)) is consistent with some of the actin being in the E or H form (Scheme 1).
FIGURE 7. Effect of N1-75MLCK on actin sliding velocity and amount of MLCK on coverslip.

A standard motility assay was conducted at pCa 4 after the following treatments. Velocities were collected ~ 5 min after loading motility buffer. (a), N1-75MLCK (abbrev. N1-75) (4 μM) and up-SMM (0.1 mg/ml) in myosin buffer were incubated for 30 min at room temperature before applying the mixture to the coverslip. Values indicate mean ± SD. Data are from 2 upSMM preparations and 4 independent measurements. There was no significant difference between the control and experimental groups (p < 0.05). (b), velocity, left axis, (open bars); after applying 0.2 mg/ml upSMM to the coverslip, followed by BSA blocking, N1-75MLCK was added to the coverslip and incubated for 10 min at room temperature, prior to washing and the standard motility assay protocol. Data are from 1 upSMM preparation and 4 independent measurements. There was no significant difference between the control and experimental groups (p < 0.05); right axis, ELISA signal for MLCK (solid bars), and actin (hatched bars) (averages from two independent experiments). (c), Independent experiment similar to (b), using a different SMM preparation (3 measurements).
Effects of Mg2+
Fifty mM MgCl2 is known to displace MLCK from actin. 35 Figure 8 shows that 50 mM MgCl2 did not reduce the amount of MLCK attached to the coverslip as measured by the on-coverslip ELISA, nor did it decrease the actin sliding velocity under two different conditions. These data are inconsistent with the presence of a significant amount of the MLCK in configurations A, B, or D in Scheme 1. These data are consistent with the presence of configurations G and H. It is of interest that the SMM used in this study was prepared by a method that uses a MgCl2 (150 mM)-treatment step. The fact he fact that we observe a relatively large amount of MLCK bound to our SMM using this very protocol is consistent with Figure 8 and Scheme 1 in that that MgCl2 does not appear to affect the MLCK that is bound to myosin.
FIGURE 8. Effect of MgCl2 on amount of MLCK on coverslip and actin sliding velocity.
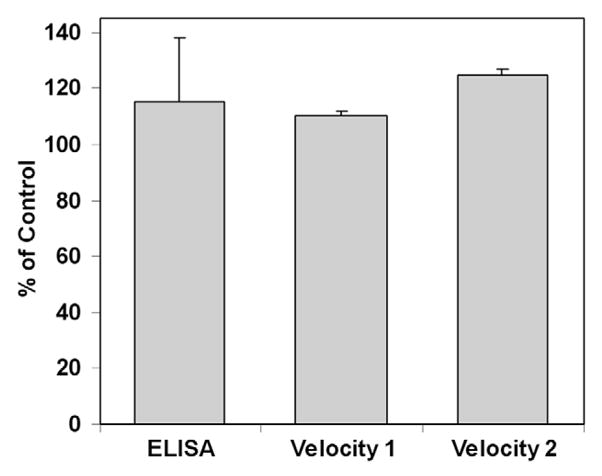
For the ELISA, SMM (0.1 mg/ml) and MgCl2 (50 mM), or for the control, KCl (50 mM), were mixed and incubated on ice for 30 min prior to applying to the slides in 3 × 40 μl additions. For Velocity 1, SMM (0.3 mg/ml) was attached to the coverslip, blocked, washed and treated with MgCl2 (50 mM), or for the control KCl (50 mM), for 10 min at room temperature. For Velocity 2, SMM (0.3 mg/ml) and MgCl2 (50 mM) were incubated for 30 min on ice prior to applying to the slides in 3 × 40 μl additions. Values are plotted as % of the KCl control. Error bars represent the range of two independent measurements.
Discussion
We have shown that a surprisingly large amount of MLCK reproducibly co-purifies with up-SMM prepared from chicken gizzards (MLCK:SMM = 1:73 ± 9, Fig. 1). This becomes evident considering the relative amounts of the two proteins in smooth muscle tissue. [MLCK] is ~ 2–4 μM 36; 37; 38 and [SMM] ~ 50–80 μM in gizzard tissue. 37; 39 Assuming [MLCK] is 3 μM, this gives a molar ratio of MLCK:SMM of 1:17 to 1:27. Therefore, the MLCK:SMM ratio in our SMM preparation is ~ 23–37% of the MLCK:SMM ratio in tissue. The actin:SMM, unlike the MLCK:SMM, molar ratio in our up-SMM preparation was highly variable (1:19 ± 11; Fig. 1(c)) and represents only 0.3% of the molar ratio of actin (830 μM) to SMM (50 μM) in tissue (17:1). There are approximately 4 actin monomers per MLCK. Since the actin-binding site of MLCK is thought to span 3 actin monomers of F-actin, it is unlikely that the MLCK is in a functional complex with the actin. The CaM:SMM ratio in our preparation (1:371±153, Fig. 1(b)) was more difficult to assess due to low-intensity signals on western blots. Our best estimate is that there are 3–7 MLCK per CaM. MLCK enzyme activity was observed with added Ca2+ suggesting strongly that the CaM was bound or could bind to the MLCK, because MLCK requires CaM for enzyme activity. The MLCK that was not bound to CaM could be enzymatically activated by adding exogenous CaM. The observed enzyme activities were reasonably consistent with the known kinetics of MLCK with SMM as the substrate.
These data were consistent with our assessment of the MLCK and CaM that co-purified with SMM using the in vitro motility assay. The dependence of actin motility on [Ca2+] gave a pCa50 of 6.1 and a Hill coefficient of 0.9 (Fig. 2). These values are similar to those found in vitro 40 and in vivo. 41 Calcium-activated motion was also observed by reconstituting MLCK-free SMM with purified MLCK and CaM. Together, these data argue against the notion the co-purified complex is a damaged or otherwise non-physiologically-modified complex arising from aspects of the purification protocol.
To determine whether all the SMM that was phosphorylated by the co-purified MLCK was capable of generating motion in the in vitro motility assay, we measured the amount of RLC phosphorylation on the coverslip after the motility assay was performed using a new ELISA protocol (Fig. 3(a)). We showed that a ratio of MLCK:SMM of ~1:75 after 10 min incubation on the coverslip under phosphorylation conditions (ATP and Ca2+) generated 50% maximal motility by phosphorylation of ~ 13% of the SMM RLCs. These values of motility and phosphorylation are in perfect agreement with the nonlinear relationship between motility and extent of phosphorylation found by Harris et al. 42 using mixtures of up-SMM and p-SMM (without Ca2+). We conclude that all the SMM that was phosphorylated by the co-purified MLCK/CaM had normal mechanical interactions with actin. Not only could it bind to actin, but it could generate the actin velocity expected under these conditions.
We estimated that the co-purified MLCK appears to represent two pools with different affinities for SMM filaments (Table 1). One pool constitutes about 60–75% of the total and has a Kd between 10–20 μM, and the other pool binds more tightly with a Kd ~ 200 nM in the absence of Ca2+. Our protein quantification results (Figure 1) suggest the possibility that the more tightly bound pool is MLCK bound to CaM, but we have no direct evidence for this at this time. It is possible that the more tightly bound pool is the predominant form on the coverslip during motility assays, but we have no method to quantify the pools on the coverslip at this time. We are not the first to suggest that MLCK (and CaM) can bind to SMM with nM affinity in EGTA-buffered solutions. For example, Ngai and Walsh 43 have shown that SMM, purified by a different method than used here, also has tightly bound MLCK and CaM that were removed only after hydrophobic interaction and affinity chromatography, although the relative quantities of the proteins were not determined. Sobieszek16 using hydroxyapatite-purified SMM from which the tightly-bound MLCK had been removed, concluded that SMM filaments bind to MLCK with two affinities, 30 nM and 1.3 μM, although others 12 have found weaker affinity binding of MLCK to SMM filaments (0.8 μM without Ca2+ or CaM). Cross and Sobieszek 44 showed that both MLCK and CaM remain bound to SMM both in the presence and absence of Ca2+. This is different from the binding of CaM to purified MLCK, which is absolutely dependent upon Ca2+.21 Interestingly, there is evidence that MLCK, CaM and SMM phosphatase (fragments of) form a tightly-bound complex in smooth muscle 45 although the relative ratios of these proteins to others, for example actin, has not been firmly established.46
Interestingly, the MLCK that co-purified with SMM bound with similar affinity to upSMM (10 μM) and pSMM (Table 1). We did not observe the large difference in Kd found previously for MLCK to pSMM (0.77 μM for upSMM and >100 μM for pSMM). 12 Instead, both forms of SMM appeared to bind to MLCK with similar affinity within a factor of 2.
We have performed a series of experiments (Figures 5–8; See Scheme 1 for summary) designed to determine the binding configurations of SMM, MLCK and actin in our preparations that are responsible for SMM phosphorylation and subsequent ability to generate actin motility. The data taken together strongly suggest that most of the MLCK that co-purifies with SMM and that is responsible for phosphorylation and motility is bound to SMM directly (G and H, Scheme 1), although some of this SMM-bound MLCK may also be binding to actin (as in H). This is evidenced by the combined agreement between the predicted and observed effects of telokin, N1-75MLCK, ATP and Mg2+ on both MLCK and actin binding to SMM (dotted boxed area, Scheme 1).
The strongest evidence that most of the MLCK is in forms G and H (Scheme 1) is that telokin reduces the velocity of motion (Fig. 4(a)) and the amount of MLCK bound to myosin filaments (Fig. 4(b)). Since telokin and MLCK have common myosin binding domains, these data strongly suggest that MLCK is binding to SMM. Configurations E and F are not likely because telokin would not displace them from the coverslip. At the same time, telokin only displaces a small % of the total actin from SMM (Figure 5(b)), which is consistent with at least some of the MLCK existing in Form H. Further evidence that MLCK exists in Forms G and H is that N1-75MLCK (Figure 7(b)) and Mg2+ (Figure 8) did not dissociate MLCK. This peptide should only have dissociated MLCK from Forms A, B or D, suggesting they are not present is significant amounts.
Our data also allow us to make some conclusions about the distribution of actin amongst the configurations in Scheme 1. Since ATP, which should weaken the SMM-actin bonds, did not measurably dissociate actin from the coverslip, little of the actin exists in configuration A. And since only a small amount of actin is likely to be in configuration H (see above), it is likely to be attached irreversibly to “dead” SMM heads in configurations B and C or bound to the coverslip directly, as in D, or alone. The fact that N1-75MLCK did dissociate a small but measurable amount of actin (Figure 7(b)) is consistent with the presence of only configurations E and H. Since E is largely ruled out by the telokin results (see above), the actin that was dissociated was most likely in configuration H. This suggests that remainder of the actin that was not dissociable with N1-75MLCK, ATP or telokin, is in configuration C, or bound alone directly to the coverslip (not shown in Scheme).
Physiological significance
The physiological significance of our findings will require further studies. It is possible that MLCK that is bound tightly to SMM and remains bound to pSMM plays a role in the initiation of contraction. This MLCK could rapidly access and phosphorylate several local myosin heads, which could then interact with actin thus promoting relative motion between the thin and thick filaments, which could then expose more heads to actin-bound MLCK. Although MLCK binds to SMM filaments in vitro, 12; 15; 16 it has not been established that it binds to SMM in vivo. The prevailing evidence suggests that most of the MLCK in smooth muscle cells is binding to actin through 3 actin-binding motifs in the N-terminal actin-binding domain. 8; 13 Since the binding to myofilaments is extremely tight compared to purified actin filaments,14 other proteins are thought to be required for tight binding. Strong evidence against MLCK binding to SMM is that the N-terminal actin-binding domain of MLCK is necessary and sufficient for binding to myofilaments. 35 However, a potential caveat is that these studies were done with myofilaments that were extracted with 50 mM MgCl2 to remove the endogenous kinase. It is possible that proteins critical for tight binding of MLCK to SMM filaments were also extracted from the myofilaments under these conditions. For example, myosin phosphatase can also be extracted under these conditions. 47 Consistent with this idea is that the actin-binding domain of MLCK (GST-N1-75MLCK), even at concentrations much higher than the measured Kd, displaces only about 50% of the endogenous MLCK from gizzard myofilaments that have not been treated with 50 mM MgCl2 (Fig. 6(c); and see 35). However, if the endogenous kinase is extracted with MgCl2 first, followed by incubation of the myofilaments with excess GST-N1-75MLCK, binding of exogenously added MLCK is completely prevented.35 Other important observations of localization of fluorescently-labeled MLCK in transfected A7r5 cells strongly suggest that MLCK binds to cellular stress fibers only through the actin-binding domain.14 However, in these experiments GFP was fused to the C-terminus of MLCK, which could potentially affect SMM filament binding. In addition, some experiments were performed under conditions that strongly promote depolymerization of SMM to the 10S conformation, which can diffuse out of permeabilized cells 48 and which binds weakly to MLCK. 44; 49 Other experiments with Cy3-labeled MLCK in Swiss 3T3 cells or in primary bovine tracheal smooth muscle cells in culture 50 do not exclude the possibility that some of the MLCK co-localizes with SMM. It is known, for example, that the expression of SMM is down-regulated in such cells. 51 Other studies have shown that the 130 kDa isoform of MLCK co-localized with myosin IIA but not with myosin IIB or F-actin in bovine pulmonary artery endothelial cells.51 Similarly, antibodies to smooth MLCK co-localize with the periodic distribution of myosin along the stress fibers of gerbil fibroma nonmuscle cells. 52 In relation to purification of SMM from smooth muscles, a key step is to depolymerize the SMM in the presence of ATP at low ionic strength to the 10S conformation. This might explain why so little of the SMM studied here has tightly-bound MLCK. Further studies will be required to clarify these issues. In summary, we believe that prior studies are not sufficient to exclude the possibility that MLCK binds tightly to SMM, in addition to binding to actin.
In this paper, we have established that MLCK/CaM binds tightly and functionally to SMM isolated from chicken gizzards. This observation has significant implications for our understanding of the critical complexes involved in smooth muscle regulation. Moreover, our characterization of these complexes and the techniques we’ve established here to study the chemistry and mechanics of these complexes represent an important first step in addressing key issues such as i) the mechanism by which relatively few MLCK can phosphorylate many SMM in our in vitro system (as is the case in muscle) and ii) the effects of other proteins on both MLCK/CaM affinity (to both actin and myosin) and on the calcium sensitivity of motility. For example, to determine the mechanism by which relatively few MLCK can phosphorylate many SMM on a coverslip (13%; Fig. 3), consider the area around a single actin filament of 10 μm in length and 6 nm in width. If all SMM heads within a 10 nm distance from the filament can interact to generate force, then the actin-accessible zone is~ 260,000 nm2.53 If the density of SMM is ~1100/μm2 54 there are ~ 300 SMM molecules within this zone. Based on the data in Fig. 1, this zone could contain as few as 1 MLCK/CaM in complex with SMM. It will be of interest to determine whether the distribution of phosphorylated heads about the coverslip requires the presence of the moving actin filaments or whether a simple non-assisted diffusion mechanism is sufficient to explain the measured levels and kinetics of phosphorylation of SMM.
Summary
In summary, we have quantified and characterized MLCK and CaM that co-purify with SMM prepared from chicken gizzards. Enzymatic measurements suggest that some of the MLCK has bound CaM and some does not, but both forms of MLCK are fully active with regard to SMM phosphorylation in the presence of added CaM and Ca2+. Equilibrium binding measurements in the absence of added Ca2+ (~ 1 nM) clearly reveal the presence of a major pool of MLCK that binds with Kds equal to ~10 μM (upSMM) and ~20 μM (pSMM), and a minor pool (30–40%) that binds with Kds equal to ~0.2 μM for both upSMM and pSMM,. The MLCK/CaM is activated by Ca2+ in a concentration-dependent manner and, at physiological [Ca2+], phosphorylates SMM to generate actin movement in an in vitro system. Similar behavior could be reconstituted from purified proteins. We provide strong evidence that most of the MLCK is bound directly to SMM through the telokin domain and some may also be bound to both SMM and to co-purifying actin though the N-terminal actin binding domain. Further experiments are in progress to study the kinetics and mechanism of phosphorylation of SMM using the in vitro system described here and to address whether these high affinity MLCK-CaM-SMM complexes exist in the muscle and have an effect upon muscle activation.
Materials and Methods
Materials
4–20% Tris-glycine gels (1.0 mm thick, 10 or 12 wells) are from Invitrogen (Carlsbad, CA). D(+)-glucose, glucose oxidase, Type VII BSA, and ATP are from Sigma-Aldrich (St. Louis, MO). Adenosine-5′-O-(3-thiotriphosphate) (ATP-γ-S) is from Roche Diagnostics (Indianapolis, IN).
Antibodies and western blotting
Goat anti-mouse IgG (H+L)-HRP conjugate (1:2,000, 170-6516; Bio-Rad Laboratories, Hercules, CA) was the secondary antibody for all blots. Anti-phospho-smooth muscle RLC 2 monoclonal (1:1000, Ser19-specific, 3675L; Cell Signaling, Danvers, MA), anti-smooth muscle MLCK monoclonal (1:10,000, M-7905, clone k36; Sigma), and anti-actin monoclonal (1:500, A4700, clone AC-40; Sigma) were used with SuperSignal West Pico chemiluminescence substrate (Thermo Scientific, Rockford, IL). Anti-CaM monoclonal (1:5000, 05-173; Upstate, Lake Placid, NY) was used with the secondary antibody at 1:20,000 dilution and with SuperSignal West Femto chemiluminescence substrate (Thermo Scientific). All blots were performed at 50V (Powerpac Power Supply, Bio-Rad) using nitrocellulose with the transfer buffer (33 mM Tris, 192 mM glycine, 20% methanol) at 4 °C for 1 h (CaM) or 2–2.5 h (MLCK and actin). Labworks software (UVP Inc, Upland, CA) was used to determine band intensities and the concentrations of samples were computed by use of linear standard curves.
Proteins
UpSMM was isolated from frozen chicken gizzards as described 55 except that the last polymerization-depolymerization step was excluded. Telokin in the SMM preparation was below our detection limit using quantitative western blotting analysis (data not shown; contained less than 1 telokin per 10 SMM). The SMM with a low MLCK/SMM ratio was prepared as above, except that the preparation was done with only a few grams of gizzard tissue, which we find results in the retention of less MLCK. UpSMM was thiophosphorylated by incubation in HMM buffer (see below) containing 10 mM MOPS, 50 mM NaCl, 0.2 mM EGTA, 2.0 mM MgCl2, 1mM DTT, 3.0 mM CaCl2, 1 mM ATP-γ-S, 10 μg/ml CaM, and 40 μg/ml MLCK at 25 °C for 0.5 h, followed by overnight incubation on ice. 56 The extent of thiophosphorylation was quantified by urea/glycerol-PAGE 57. MLCK was isolated from frozen chicken gizzards by the method of Adelstein and Klee 6 except that Superdex 200 (GE Healthcare) was used for gel filtration and Super Q (Tosohass) for ion exchange chromatography. Actin in the MLCK preparation was below our detection limit using quantitative western blotting analysis (data not shown; contained less than 1 actin per 25 MLCK). Actin for in vitro motility was purified from chicken pectoralis muscle 58 and incubated with TRITC-labeled phalloidin (Alexis Corp., San Diego, CA) overnight 34 and stored on ice at 4 °C. Cow brain CaM and smooth muscle actin standards were purchased from Sigma. Expressed chicken gizzard CaM was a gift from Dr. Albert Wang (Boston Biomedical Research Institute). Myofilaments were prepared from chicken gizzard. 59 The construct for human telokin was a gift from Vladimir Shirinsky. Telokin was expressed in E. coli essentially as described.18 It bound to SMM with an apparent Kd of ~5 μM and promoted 50% SMM filament formation at ~7 μM (data not shown), similar to that described by Silver et al. 18 Protein concentrations were determined using the following extinction coefficients (1 mg/ml): SMM (280 nm) = 0.56; RLC (277 nm) = 0.337; MLCK (280 nm) = 1.14; CaM (276 nm) = 0.18; actin (280 nm) = 1.1; telokin (280 nm) = 0.78.
GST-N1-75MLCK (the N-terminal actin-binding domain of MLCK, residues 1-75, with glutathione S-transferase (GST) fused to the N-terminus) in pGEX-2TK was expressed in BL21 cells. Cultures (5 l) were grown in LB medium to an OD of 0.4 prior to IPTG (0.1 mM) induction for 3 h. Bacterial pellets were suspended in 500 ml PBS containing leupeptin (2.5 μg/ml), pepstatin A (2.5 μg/ml) and PMSF (0.1 mM). Lysozyme was added to 5 mg/l and the culture was stirred for 30 min at room temperature. DTT (5 mM) was added and the mixture was centrifuged at 30,000 × g for 45 min, the supernatant was diluted 3-fold with PBS and 5 ml of a 1:1 slurry of glutathione-Sepharose 4FF beads in PBS added. After gentle shaking for 30 min and centrifugation at 150 × g for 5 min, the supernatant was removed and the beads were washed with 100 ml PBS. The binding to beads was repeated twice more. The 3 sets of beads were combined, poured into a column (1 × 20 cm), washed with 10 column volumes of PBS and eluted with 10 mM glutathione, 50 mM Tris-HCl, pH 8, 5 mM DTT. GST-N1-75MLCK-containing fractions were identified by SDS-PAGE (see Fig. 6), pooled and dialyzed against 30 mM Tris-HCl, pH 7.5, 20 mM NaCl, 1 mM EGTA, 0.1 mM DTT. Thrombin (Novagen, restriction grade) digestion (8 units/μl) was carried out for 2 h at room temperature and stopped by addition of 0.1 mM PMSF. The solution was clarified by centrifugation at 39,000 × g for 30 min. The supernatant was applied to an SP-Sepharose column (1 × 10 cm), equilibrated with 30 mM Tris-HCl, pH 7.5, 20 mM NaCl, 1 mM EGTA, 0.1 mM DTT, at a flow rate of 25 ml/h. After unbound proteins were eluted, 0.5 M NaCl in the same buffer, followed by a step to 1 M NaCl eluted the N1-75MLCK. Fractions were identified by NEXT GEL™ (Amresco) electrophoresis (15% acrylamide), pooled and dialysed against 30 mM Tris-HCl, pH 7.5, 20 mM NaCl, 1 mM EGTA, 0.1 mM DTT using 3.5 kDa cut-off dialysis tubing, concentrated in Aquacide II and dialysed again. Protein concentration was determined by the bicinchoninic acid assay. N1-75MLCK has the following sequence (corresponding to the N-terminal domain of rabbit smooth muscle MLCK):LVPR/GSRRASVGSGGGMDFRANLQRQVKPKTVSEEERKVHSPQQVDFRSVL-AKKGTPKTPVPEKAPPPKPATPDFRSVLGSKKKLPAENGS. Mr = 9,348 Da after thrombin cleavage. The/indicates a thrombin cleavage site, an engineered PKA phosphorylation site is underlined and a linker to the beginning of the MLCK sequence is in italics.
In vitro motility assay
Myosin buffer: 300 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, 10 mM DTT, pH 7.0. Actin buffer: 50 mM KCl, 50 mM imidazole, 2 mM EGTA, 8 mM MgCl2, 10 mM DTT, pH 7.0. Motility buffer: 50 mM KCl, 50 mM imidazole, pH 7.0, 2 mM EGTA, 8 mM MgCl2, 10 mM DTT, 2 mM ATP, 0.5% methylcellulose, 0–100 μM free Ca2+, as indicated in the Figures. Free Ca2+ concentrations were calculated using an algorithm based on 60. Motility assays were performed as previously described 30 at 30 °C. Briefly, 0.1–0.2 mg/ml SMM in myosin buffer was applied to a nitrocellulose-coated coverslip. After 1 min, 40 μl myosin buffer was used to wash the coverslip two times. The nitrocellulose surface was blocked with 2 × 40 μl 0.5% BSA in actin buffer for 1 min, followed by 2 × 40 μl 10 nM TRITC-actin in actin buffer for 1 min, and two washes with 40 μl actin buffer. Forty μl motility buffer (at [Ca2+] indicated in the figures) was loaded twice and movies were recorded at 1–5 fields for 30 s. Data obtained from these 1–5 fields constitute one (n = 1) experiment. Velocities for each actin filament were collected and averaged.
Quantification of protein by on-coverslip ELISA
The on-coverslip ELISA procedure was performed at room temperature. All solutions were added to the motility assay chambers (made from standard microscope slides and nitrocellulose-coverslips separated by spacers) with the slides oriented at 45 degrees, whereas the slides were horizontal for all incubation steps. The spacers were as long as or longer than the coverslips. The assay was used to quantify proteins on the coverslip after the completion of a standard motility assay or under similar conditions to a motility assay. The coverslips were blocked with 1% BSA in PBST (degassed) for 30 min before primary antibody (see above; diluted in PBST containing 1% BSA) was applied to the coverslip and incubated for 1 h. Further antibody was added as needed during the incubation to prevent evaporation or the slides can be incubated in a container to retain moisture. The coverslip was washed with 200 μl 1% BSA in PBST at least 3 times, each time for 5 min. The secondary antibody (goat anti-mouse 1:2000 in PBST containing 1% BSA) was applied to the coverslip and incubated for 1 h followed by washing with PBS (3 times with 200 μl). SuperSignal West Pico Chemiluminescence Substrate was applied according to the manufacturer and the signal was detected with film (High Contrast X-Ray film, Konica Minolta Medical Imaging USA Inc, Wayne, NJ) without use of a cassette. Care was taken to avoid evaporation of the substrate prior to film exposure. The film was scanned, inverted, and the relative amounts of signal were compared using a box of equal dimensions that was placed over the most uniform region (using Image J, NIH).
Effect of ATP pre-wash on actin ELISA signals
UpSMM (0.1 mg/ml) in myosin buffer was applied to the coverslip. After 1 min, the coverslip was washed twice with 40 μl myosin buffer, followed by blocking with 2 × 40 μl 0.5% BSA in actin buffer for 1 min. The buffer pre-wash and ATP pre-wash coverslips were treated twice with 40 μl of actin buffer or 2 mM ATP (in actin buffer), respectively, and then twice with 40 μl actin buffer. This buffer or ATP application and subsequent removal sequence was repeated 3 times, prior to performing the ELISA procedure as described above.
Estimate of MLCK and actin binding to SMM by co-sedimentation
Up-SMM filaments (1 mg/ml in 10 mM MOPS, 50 mM NaCl, 0.2 mM EGTA, 1 mM DTT, 5 mM MgCl2, pH 7.0) were pelleted at 16,100 g for 10 min at 4°C. The amounts of SMM, MLCK and actin before and after sedimentation were determined by western blot analysis using purified proteins as standards. Under these conditions essentially all the SMM sediments to the pellet. Samples were appropriately diluted to ensure that the values were within the linear range of the standards. CaM could not be determined due to a low signal from the supernatant.
If there are two species or pools of MLCK in the SMM, K1 and K2, and one binds more tightly than the other Kd1≫Kd2, the relative amounts and limits for the two Kd values can be determined from an analysis of the data produced in a sequential multi-step spin-down experiment. The amount of MLCK before (total) and after spin (supernatant) is quantified by quantitative western blot and the fraction of the total MLCK that is bound to SMM (M = 2.1 μM) is determined by:
| Equation 1 |
The pellet is resuspended in an equal volume of buffer and the procedure is repeated 3 times. The MLCK that is measured will be a combination of the two putative MLCK pools, K1 and K2, with respective equilibrium dissociation constants, Kd1 and Kd2, and
| Equation 2 |
| Equation 3 |
where, n= the number of spins and spin zero is the initial sample. The fraction of bound MLCK, relative to the amount of MLCK present in the resuspended pellet prior to the next spin is
| Equation 4 |
Figure 4 is a plot of Fb versus spin number and the data were fitted to Equation 4. As the spin number increases, the Fb will increase if more than one species is present with Kd1≫Kd2. If only one species is present, then Fb will remain constant.
The abbreviations used are
- BSA
bovine serum albumin
- CaM
calmodulin
- DTT
dithiothreitol
- EGTA
ethylene glycol-bis(2-aminoethylether)-N, N, N′, N′-tetraacetic acid
- ELISA
enzyme-linked immunosorbent assay
- GST-N1-75MLCK
GST fusion protein of the N-terminal 75 residues of MLCK
- N1-75MLCK
cleaved fragment of fusion protein lacking GST
- HMM
heavy meromyosin
- MLCK
smooth muscle myosin light chain kinase
- PBST
phosphate-buffered saline containing 0.5% Tween-20
- p-RLC
phosphorylated regulatory light chain
- SMM
smooth muscle myosin
- pSMM and upSMM
phosphorylated and unphosphorylated smooth muscle myosin, respectively
- TRITC
tetramethylrhodamine isothiocyanate
- TRITC-actin
actin labeled at Cys-374 with tetramethylrhodamine
Footnotes
This work was supported in part by grants from the National Institute of Arthritis and Musculoskeletal Disease (5R01AR040917-19 to C.R.C and 1R21AR055749-01 to J.E.B) and the Canadian Institutes of Health Research (MT13101 to M.P.W.). M.P.W. is an Alberta Heritage Foundation for Medical Research Scientist and holder of a Canada Research Chair (Tier 1) in Vascular Smooth Muscle Research. The authors are grateful to Susan Li, Cindy Sutherland, Mike Carter, Amy Schneck, and Bill Wiehler for expert technical assistance. We thank Dr. Albert Wang (Boston Biomedical Research Institute) for expressed chicken gizzard CaM and Vladimir Shirinsky for the human telokin construct.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kamm KE, Stull JT. Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 2.Kamm KE, Stull JT. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- 3.Stull JT, Lin P, Krueger JK, Trewhella J, Zhi G. Myosin light chain kinase: functional domains and structural motifs. Acta Physiol Scand. 1998;164:471–482. doi: 10.1111/j.1365-201x.1998.tb10699.x. [DOI] [PubMed] [Google Scholar]
- 4.Herring BP, Dixon S, Gallagher PJ. Smooth muscle myosin light chain kinase expression in cardiac and skeletal muscle. Am J Physiol Cell Physiol. 2000;279:C1656–1664. doi: 10.1152/ajpcell.2000.279.5.C1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallagher PJ, Herring BP, Stull JT. Myosin light chain kinases. J Muscle Res Cell Motil. 1997;18:1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- 6.Adelstein RS, Klee CB. Purification of smooth muscle myosin light-chain kinase. Methods Enzymol. 1982;85(Pt B):298–308. doi: 10.1016/0076-6879(82)85029-5. [DOI] [PubMed] [Google Scholar]
- 7.Wirth A, Schroeter M, Kock-Hauser C, Manser E, Chalovich JM, de Lanerolle P, Pfitzer G. Inhibition of contraction and myosin light chain phosphorylation in guinea-pig smooth muscle by p21-activated kinase 1. J Physiol (Paris) 2003;549:489–500. doi: 10.1113/jphysiol.2002.033167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanoh S, Ito M, Niwa E, Kawano Y, Hartshorne DJ. Actin-binding peptide from smooth muscle myosin light chain kinase. Biochemistry. 1993;32:8902–8907. doi: 10.1021/bi00085a023. [DOI] [PubMed] [Google Scholar]
- 9.Smith L, Su X, Lin P, Zhi G, Stull JT. Identification of a novel actin binding motif in smooth muscle myosin light chain kinase. J Biol Chem. 1999;274:29433–29438. doi: 10.1074/jbc.274.41.29433. [DOI] [PubMed] [Google Scholar]
- 10.Gallagher PJ, Stull JT. Localization of an actin binding domain in smooth muscle myosin light chain kinase. Mol Cell Biochem. 1997;173:51–57. doi: 10.1023/a:1006876318155. [DOI] [PubMed] [Google Scholar]
- 11.Ye LH, Hayakawa K, Kishi H, Imamura M, Nakamura A, Okagaki T, Takagi T, Iwata A, Tanaka T, Kohama K. The structure and function of the actin-binding domain of myosin light chain kinase of smooth muscle. J Biol Chem. 1997;272:32182–32189. doi: 10.1074/jbc.272.51.32182. [DOI] [PubMed] [Google Scholar]
- 12.Sellers JR, Pato MD. The binding of smooth muscle myosin light chain kinase and phosphatases to actin and myosin. J Biol Chem. 1984;259:7740–7746. [PubMed] [Google Scholar]
- 13.Smith L, Stull JT. Myosin light chain kinase binding to actin filaments. FEBS Lett. 2000;480:298–300. doi: 10.1016/s0014-5793(00)01931-1. [DOI] [PubMed] [Google Scholar]
- 14.Lin P-j, Luby-Phelps K, Stull JT. Properties of Filament-bound Myosin Light Chain Kinase. J Biol Chem. 1999;274:5987–5994. doi: 10.1074/jbc.274.9.5987. [DOI] [PubMed] [Google Scholar]
- 15.Numata T, Katoh T, Yazawa M. Functional role of the C-terminal domain of smooth muscle myosin light chain kinase on the phosphorylation of smooth muscle myosin. J Biochem (Tokyo) 2001;129:437–444. doi: 10.1093/oxfordjournals.jbchem.a002875. [DOI] [PubMed] [Google Scholar]
- 16.Sobieszek A. Phosphorylation reaction of vertebrate smooth muscle myosin: an enzyme kinetic analysis. Biochemistry. 1985;24:1266–1274. doi: 10.1021/bi00326a032. [DOI] [PubMed] [Google Scholar]
- 17.Shirinsky VP, Vorotnikov AV, Birukov KG, Nanaev AK, Collinge M, Lukas TJ, Sellers JR, Watterson DM. A kinase-related protein stabilizes unphosphorylated smooth muscle myosin minifilaments in the presence of ATP. J Biol Chem. 1993;268:16578–16583. [PubMed] [Google Scholar]
- 18.Silver DL, Vorotnikov AV, Watterson DM, Shirinsky VP, Sellers JR. Sites of interaction between kinase-related protein and smooth muscle myosin. J Biol Chem. 1997;272:25353–25359. doi: 10.1074/jbc.272.40.25353. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi H, Inoue A, Mikawa T, Kuwayama H, Hotta Y, Masaki T, Ebashi S. Isolation of cDNA for bovine stomach 155 kDa protein exhibiting myosin light chain kinase activity. J Biochem. 1992;112:786–791. doi: 10.1093/oxfordjournals.jbchem.a123976. [DOI] [PubMed] [Google Scholar]
- 20.Krueger JK, Olah GA, Rokop SE, Zhi G, Stull JT, Trewhella J. Structures of calmodulin and a functional myosin light chain kinase in the activated complex: a neutron scattering study. Biochemistry. 1997;36:6017–6023. doi: 10.1021/bi9702703. [DOI] [PubMed] [Google Scholar]
- 21.Adelstein RS, Klee CB. Purification and characterization of smooth muscle myosin light chain kinase. J Biol Chem. 1981;256:7501–7509. [PubMed] [Google Scholar]
- 22.Wilson DP, Sutherland C, Walsh MP. Ca2+ activation of smooth muscle contraction: evidence for the involvement of calmodulin that is bound to the triton insoluble fraction even in the absence of Ca2+ J Biol Chem. 2002;277:2186–2192. doi: 10.1074/jbc.M110056200. [DOI] [PubMed] [Google Scholar]
- 23.Barany M, Barany K. In: Biochemistry of Smooth Muscle Contraction. Barany M, editor. Vol. 25. Academic Press, Inc; San Diego: 1996. pp. 321–339. [Google Scholar]
- 24.Tyska MJ, Warshaw DM. The myosin power stroke. Cell Motil Cytoskeleton. 2002;51:1–15. doi: 10.1002/cm.10014. [DOI] [PubMed] [Google Scholar]
- 25.Olney JJ, Sellers JR, Cremo CR. Structure and Function of the 10 S Conformation of Smooth Muscle Myosin. J Biol Chem. 1996;271:20375–20384. doi: 10.1074/jbc.271.34.20375. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki H, Onishi H, Takahashi K, Watanabe S. Structure and function of chicken gizzard myosin. J Biochem (Tokyo) 1978;84:1529–1542. doi: 10.1093/oxfordjournals.jbchem.a132278. [DOI] [PubMed] [Google Scholar]
- 27.Trybus KM, Lowey S. Conformational states of smooth muscle myosin. Effects of light chain phosphorylation and ionic strength. J Biol Chem. 1984;259:8564–8571. [PubMed] [Google Scholar]
- 28.Ikebe M, Hinkins S, Hartshorne DJ. Correlation of enzymatic properties and conformation of smooth muscle myosin. Biochemistry. 1983;22:4580–4587. doi: 10.1021/bi00288a036. [DOI] [PubMed] [Google Scholar]
- 29.Persechini A, Hartshorne DJ. Ordered phosphorylation of the two 20 000 molecular weight light chains of smooth muscle myosin. Biochemistry. 1983;22:470–476. doi: 10.1021/bi00271a033. [DOI] [PubMed] [Google Scholar]
- 30.Hooft AM, Maki EJ, Cox KK, Baker JE. An accelerated state of myosin-based actin motility. Biochemistry. 2007;46:3513–3520. doi: 10.1021/bi0614840. [DOI] [PubMed] [Google Scholar]
- 31.Lippincott-Schwartz J. Current Protocols in Cell Biology. Chapter 13. John Wiley & Sons, Inc; New York: 2005. [Google Scholar]
- 32.Sellers JR. Current Protocols in Cell Biology. Chapter 13. John Wiley & Sons, Inc; 1998. [Google Scholar]
- 33.Cremo CR, Sellers JR, Facemyer KC. Two Heads are Required for Phosphorylation-Dependent Regulation of Smooth Muscle Myosin. J Biol Chem. 1995;270:2171–2175. doi: 10.1074/jbc.270.5.2171. [DOI] [PubMed] [Google Scholar]
- 34.Warshaw DM, Desrosiers JM, Work SS, Trybus KM. Smooth muscle myosin cross-bridge interactions modulate actin filament sliding velocity in vitro. J Cell Biol. 1990;111:453–463. doi: 10.1083/jcb.111.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith L, Su X, Lin P, Zhi G, Stull JT. Identification of a novel actin binding motif in smooth muscle myosin light chain kinase. J Biol Chem. 1999;274:29433–29438. doi: 10.1074/jbc.274.41.29433. [DOI] [PubMed] [Google Scholar]
- 36.Walsh MP, Hinkins S, Dabrowska R, Hartshorne DJ. Smooth muscle myosin light chain kinase. Methods Enzymol. 1983;99:279–288. doi: 10.1016/0076-6879(83)99063-8. [DOI] [PubMed] [Google Scholar]
- 37.Dabrowska R, Hinkins S, Walsh MP, Hartshorne DJ. The binding of smooth muscle myosin light chain kinase to actin. Biochem Biophys Res Commun. 1982;107:1524–1531. doi: 10.1016/s0006-291x(82)80172-1. [DOI] [PubMed] [Google Scholar]
- 38.Tansey MG, Luby-Phelps K, Kamm KE, Stull JT. Ca(2+)-dependent phosphorylation of myosin light chain kinase decreases the Ca2+ sensitivity of light chain phosphorylation within smooth muscle cells. J Biol Chem. 1994;269:9912–9920. [PubMed] [Google Scholar]
- 39.Ruegg JC. Calcium in Muscle Activation: A Comparative Approch. Springer-Verlag; Berlin: 1986. pp. 201–235. [Google Scholar]
- 40.Malencik DA, Anderson SR. Calmodulin-linked equilibria in smooth muscle myosin light chain kinase. Biochemistry. 1986;25:709–721. doi: 10.1021/bi00351a031. [DOI] [PubMed] [Google Scholar]
- 41.Geguchadze R, Zhi G, Lau KS, Isotani E, Persechini A, Kamm KE, Stull JT. Quantitative measurements of Ca(2+)/calmodulin binding and activation of myosin light chain kinase in cells. FEBS Lett. 2004;557:121–124. doi: 10.1016/s0014-5793(03)01456-x. [DOI] [PubMed] [Google Scholar]
- 42.Harris DE, Work SS, Wright RK, Alpert NR, Warshaw DM. Smooth, cardiac and skeletal muscle myosin force and motion generation assessed by cross-bridge mechanical interactions in vitro. J Muscle Res Cell Motil. 1994;15:11–19. doi: 10.1007/BF00123828. [DOI] [PubMed] [Google Scholar]
- 43.Ngai PK, Walsh MP. Purification of smooth-muscle myosin free of calmodulin and myosin light-chain kinase. Susceptibility to oxidation. Biochem J. 1987;246:205–211. doi: 10.1042/bj2460205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cross RA, Sobieszek A. Influence of smooth muscle myosin conformation on myosin light chain kinase binding and on phosphorylation. FEBS Lett. 1985;188:367–374. doi: 10.1016/0014-5793(85)80404-x. [DOI] [PubMed] [Google Scholar]
- 45.Sobieszek A, Borkowski J, Babiychuk VS. Purification and characterization of a smooth muscle myosin light chain kinase-phosphatase complex. J Biol Chem. 1997;272:7034–7041. doi: 10.1074/jbc.272.11.7034. [DOI] [PubMed] [Google Scholar]
- 46.Sobieszek A, Andruchov OY, Grabarek Z, Kulikova N, Liebetrau C, Matusovsky OS. Modulation of myosin filament activation by telokin in smooth muscle liberation of myosin kinase and phosphatase from supramolecular complexes. Biophys Chem. 2005;113:25–40. doi: 10.1016/j.bpc.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 47.Shimizu H, Ito M, Miyahara M, Ichikawa K, Okubo S, Konishi T, Naka M, Tanaka T, Hirano K, Hartshorne DJ, et al. Characterization of the myosin-binding subunit of smooth muscle myosin phosphatase. J Biol Chem. 1994;269:30407–30411. [PubMed] [Google Scholar]
- 48.Kolega J. Fluorescent analogues of myosin II for tracking the behavior of different myosin isoforms in living cells. J Cell Biochem. 1998;68:389–401. doi: 10.1002/(sici)1097-4644(19980301)68:3<389::aid-jcb9>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 49.Ikebe M, Inagaki M, Naka M, Hidaka H. Correlation of conformation and phosphorylation and dephosphorylation of smooth muscle myosin. J Biol Chem. 1988;263:10698–10704. [PubMed] [Google Scholar]
- 50.Lin P-j, Luby-Phelps K, Stull JT. Binding fo Myosin Light Chain Kinase to Cellular Actin-Myosin Filaments. J Biol Chem. 1997;272:7412–7420. doi: 10.1074/jbc.272.11.7412. [DOI] [PubMed] [Google Scholar]
- 51.Blue EK, Goeckeler ZM, Jin Y, Hou L, Dixon SA, Herring BP, Wysolmerski RB, Gallagher PJ. 220- and 130-kDa MLCKs have distinct tissue distributions and intracellular localization patterns. Am J Physiol Cell Physiol. 2002;282:C451–460. doi: 10.1152/ajpcell.00333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Lanerolle P, Adelstein RS, Feramisco JR, Burridge K. Characterization of antibodies to smooth muscle myosin kinase and their use in localizing myosin kinase in nonmuscle cells. Proc Natl Acad Sci U S A. 1981;78:4738–4742. doi: 10.1073/pnas.78.8.4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uyeda TQ, Kron SJ, Spudich JA. Myosin step size. Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J Mol Biol. 1990;214:699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 54.Harris DE, Warshaw DM. Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J Biol Chem. 1993;268:14764–14768. [PubMed] [Google Scholar]
- 55.Ikebe M, Hartshorne DJ. Effects of Ca2+ on the conformation and enzymatic activity of smooth muscle myosin. J Biol Chem. 1985;260:13146–13153. [PubMed] [Google Scholar]
- 56.Ellison PA, Sellers JR, Cremo CR. Kinetics of smooth muscle heavy meromyosin with one thiophosphorylated head. J Biol Chem. 2000;275:15142–15151. doi: 10.1074/jbc.275.20.15142. [DOI] [PubMed] [Google Scholar]
- 57.Wahlstrom JL, Randall MA, Jr, Lawson JD, Lyons DE, Siems WF, Crouch GJ, Barr R, Facemyer KC, Cremo CR. Structural model of the regulatory domain of smooth muscle heavy meromyosin. J Biol Chem. 2003;278:5123–5131. doi: 10.1074/jbc.M206963200. [DOI] [PubMed] [Google Scholar]
- 58.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85(Pt B):164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 59.Weber LP, Van Lierop JE, Walsh MP. Ca2+-independent phosphorylation of myosin in rat caudal artery and chicken gizzard myofilaments. J Physiol. 1999;516 (Pt 3):805–824. doi: 10.1111/j.1469-7793.1999.0805u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J Physiol (Paris) 1979;75:463–505. [PubMed] [Google Scholar]