

Abstract
HMG CoA reductase produces mevalonate, an important intermediate in the synthesis of cholesterol and essential nonsterol isoprenoids. The reductase is subject to an exorbitant amount of feedback control through multiple mechanisms that are mediated by sterol and nonsterol end-products of mevalonate metabolism. Here, I will discuss recent advances that shed light on one mechanism for control of reductase, which involves rapid degradation of the enzyme. Accumulation of certain sterols triggers binding of reductase to endoplasmic reticulum (ER) membrane proteins called Insig-1 and Insig-2. Reductase-Insig binding results in recruitment of a membrane-associated ubiquitin ligase called gp78, which initiates ubiquitination of reductase. This ubiquitination is an obligatory reaction for recognition and degradation of reductase from ER membranes by cytosolic 26S proteasomes. Thus, sterol accelerated degradation of reductase represents an example of how a general cellular process (ER-associated degradation) is used to control an important metabolic pathway (cholesterol synthesis).
Introduction
3-Hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase catalyzes conversion of HMG CoA to mevalonate (Figure 1), the precursor of isoprenoid groups that are incorporated into many end-products including cholesterol, ubiquinone, heme, dolichol, and the farnesyl and geranylgeranyl groups that can become attached to many cellular proteins 1. HMG CoA reductase has been long-recognized as the rate-limiting enzyme in cholesterol synthesis and as such, is a primary focus of regulation. This is underscored by a multivalent system mediated by sterol and nonsterol isoprenoids that exerts stringent feedback control on reductase through multiple mechanisms 2. The complexity of this regulatory system was first revealed in the late 1970s through the use of compactin, a member of the statin family of drugs that are potent competitive inhibitors of reductase 3. Treatment of cultured cells with compactin blocks production of mevalonate, thereby reducing levels of sterol and nonsterol isoprenoids that normally govern feedback regulation of reductase. Cells respond to the inhibition of reductase by developing a drastic increase in reductase protein (∼200-fold), owing to the combined effects of enhanced transcription of the reductase gene, efficient translation of reductase mRNA, and extended half-life of reductase protein. Complete reversal of this compensatory increase in reductase requires regulatory actions of both sterol and nonsterol end-products of mevalonate metabolism 2, 4. Sterols inhibit the activity of sterol regulatory element-binding proteins (SREBPs), a family of membrane-anchored transcription factors that enhance cholesterol synthesis and uptake by modulating genes encoding cholesterol biosynthetic enzymes (including reductase) and the low density lipoprotein (LDL)-receptor 5. An unknown nonsterol mevalonate-derived product(s) control the translational effects through a poorly understood mechanism that may be mediated by the complex 5′-untranslated region of the reductase mRNA 4. Both sterol and nonsterol end-products of mevalonate metabolism combine to accelerate degradation of reductase protein through a mechanism mediated by the ubiquitin-proteasome pathway 6-8. Through these mechanisms, the multivalent regulation of reductase coordinates mevalonate metabolism such that essential nonsterol isoprenoids can be constantly supplied without risking the potentially toxic overproduction of cholesterol or one of its sterol precursors.
Figure 1.
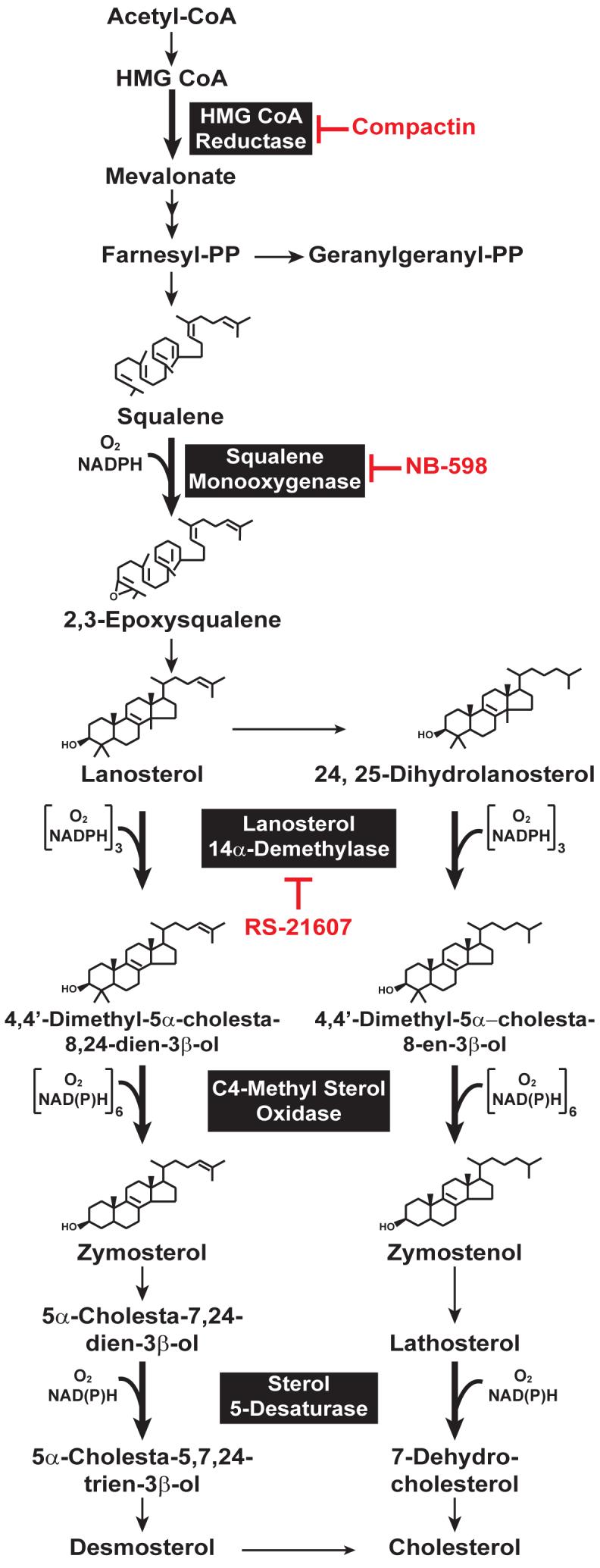
Schematic Representation of the Cholesterol Synthetic Pathway in Animal Cells. Reactions that require molecular oxygen are indicated, and specific inhibitors of various enzymes in the pathway are highlighted in red.
In all mammalian species studied to date (i.e., human, hamster, rat, and mouse), reductase localizes to membranes of the endoplasmic reticulum (ER) and consists of 887 or 888 amino acids that can be separated into two contiguous domains (Figure 2A) 9-12. The N-terminal domain of reductase is comprised of 339 amino acids and is integrated into ER membranes by virtue of eight membrane-spanning segments that are separated by short loops (Figure 2B) 13. The 548-amino acid C-terminal domain of reductase projects into the cytosol and exerts all of the enzymatic activity 12. The amino acid sequence of the membrane domain of reductase is strikingly conserved among mammalian species 14, 15, which suggested early on that the region may be important for more than just membrane anchorage. Indeed, two key observations have disclosed an important role for the membrane domain in sterol-accelerated degradation of reductase. 1) Expression of the truncated, cytosolic C-terminal domain of reductase produced a stable, catalytically active protein whose degradation was not influenced by sterols 16. 2) A chimeric protein consisting of a fusion between the membrane domain of reductase and soluble β-galactosidase exhibited sterol-accelerated degradation similar to the wild type full-length reductase 17. These observations led to the hypothesis that the membrane domain of reductase somehow senses levels of membrane-embedded sterols, which triggers reactions that render the enzyme susceptible to proteolytic degradation 16. This degradation occurs from ER membranes and can be blocked by inhibitors of the 26S proteasome, which leads to the accumulation of ubiquitinated forms of reductase 8, 18.
Figure 2.

Domain Structure of Hamster HMG CoA Reductase.
(A) HMG CoA reductase consists of two distinct domains: a hydrophobic N-terminal domain with eight membrane-spanning segments that anchor the protein to ER membranes; and a hydrophilic C-terminal domain that projects into the cytosol and exhibits all of the enzyme’s catalytic activity.
(B) Amino acid sequence and topology of the membrane domain of hamster HMG CoA reductase. The lysine residues implicated as sites of Insig-dependent, sterol-regulated ubiquitination are highlighted in red and denoted by arrows. The YIYF sequence in the second membrane-spanning helix that mediates Insig binding is highlighted in yellow.
Ubiquitin and proteasomes have been implicated in degradation of reductase in the yeast Saccharomyces cerevisiae 19. HMG2p, one of two reductase isozymes in yeast, is rapidly degraded when flux through the mevalonate pathway is high. Degradation of the other isozyme, HMG1p, is not regulated. Although the catalytic domains of yeast HMG2p and mammalian reductase show strong similarity (>50% identity over 540 amino acids), the membrane domains bear limited resemblances (<20% identity over 340 amino acids). Considering that the membrane domains of yeast and mammalian reductase are necessary and sufficient for accelerated degradation 17, 20, limited conservation between these regions provides an explanation for the observation that degradation is not triggered by sterols in yeast, but rather by nonsterol isoprenoids 21. Despite these differences, regulated ubiquitination and degradation of reductase is employed by yeast and mammals to modulate flux through the mevalonate pathway. For further insight into the pathway for degradation of HMG2p in yeast, readers are referred to several excellent reviews 19, 21, 22.
Insigs, polytopic proteins of the ER that mediate sterol-accelerated degradation of HMG CoA reductase
Crucial insights into the mechanism for sterol-accelerated degradation of reductase have emerged from comparisons made between reductase and Scap (the SREBP cleavage-activating protein). Similar to reductase, Scap contains two distinct domains: a hydrophobic N-terminal domain that spans the membrane eight times and a hydrophilic C-terminal domain that projects into the cytosol 23. The C-terminal domain of Scap mediates a constitutive association with SREBPs; this interaction is required for Scap-dependent translocation of SREBPs from the ER to Golgi in sterol-deprived cells (Figure 3). Upon arrival in the Golgi, SREBPs encounter a pair of proteases that act successively to release soluble fragments from the membrane into the cytosol 24-28. These processed forms of SREBPs then migrate from the cytosol into the nucleus and stimulate target gene expression, which results in increased synthesis and uptake of sterols 5. The subsequent accumulation of sterols in ER membranes prevents proteolytic activation of SREBPs by blocking exit of Scap-SREBP complexes from the ER; transcription of SREBP target genes decline and cholesterol synthesis and uptake is suppressed. Inhibition of ER to Golgi transport of SREBPs results from sterol-induced binding of Scap to ER retention proteins called Insig-1 and Insig-2 29, 30. Insig binding occludes a cytosolic binding site in Scap recognized by COPII proteins, which incorporate cargo molecules into vesicles that deliver ER-derived proteins to the Golgi 31. Scap-Insig binding is mediated by a segment of Scap’s membrane domain that includes transmembrane helices 2-6 24, 29. A similar stretch of transmembrane helices is found in at least four other polytopic membrane proteins (including the Niemann Pick C1 protein, Patched, Dispatched, and reductase) that have been postulated to interact with sterols. Thus, the region has become known as the sterol-sensing domain 32. The importance of the sterol-sensing domain in regulation of Scap is illustrated by findings that point mutations within the region disrupt Insig binding, which relieves sterol-mediated retention of mutant Scap-SREBP complexes in the ER 29, 30, 33-35.
Figure 3.

Model for Sterol-Regulated Scap-SREBP Pathway.
SCAP is a sensor of sterols and an escort of SREBPs. In sterol-depleted cells, Scap facilitates export of SREBPs from the ER to the Golgi apparatus, where two proteases, Site-1 protease (S1P) and Site-2 protease (S2P), act to release the transcriptionally active, N-terminal bHLH-Zip domain of SREBPs from the membrane. The released bHLH-Zip domain migrates into the nucleus and binds to a sterol response element (SRE) in the enhancer/promoter region of target genes, activating their transcription. Accumulation of sterols in ER membranes trigger binding of Scap to one of two retention proteins called Insigs, which blocks incorporation of Scap-SREBP complexes into ER transport vesicles. As a result, SREBPs no longer translocate to the Golgi apparatus, the bHLH-Zip domain cannot be released from the membrane, and transcription of all target genes declines.
The recognition of sequence resemblances between the sterol-sensing domains of Scap and reductase stimulated an appraisal of a role for Insigs in degradation of reductase. This effort led to the following observations, which considered together divulge the action of at least one of the Insig proteins in sterol-accelerated degradation of reductase. First, when overexpressed by transfection in Chinese hamster ovary (CHO) cells, reductase cannot be degraded when the cells are treated with sterols 36. Co-expression of Insig-1 restores sterol-accelerated degradation of reductase, suggesting the saturation of endogenous Insigs by the overexpressed reductase. Second, reduction of both Insig-1 and Insig-2 by RNA interference (RNAi) abolishes sterol-accelerated degradation of endogenous reductase 37. Third, mutant CHO cells lacking both Insigs are impervious to sterol-stimulated degradation of reductase as well as sterol-mediated inhibition of SREBP processing 38.
Degradation of reductase coincides with sterol-induced binding of its membrane domain to Insigs 36, an action that requires a tetrapeptide sequence, YIYF, located in the second transmembrane segment of reductase (see Figure 2B) 37. A mutant form of reductase in which the YIYF sequence is mutated to alanine residues no longer binds to Insigs and the enzyme is not subject to rapid degradation. The YIYF sequence is also present in the second transmembrane domain of Scap, where it mediates sterol-dependent formation of Scap-Insig complexes 29, 30. In fact, overexpressing the sterol-sensing domain of Scap in cells blocks Insig-mediated, sterol-accelerated degradation of reductase. Mutation of the YIYF sequence in the Scap sterol-sensing domain ablates this inhibition. This indicates that Scap and reductase bind to the same site on Insigs and the two proteins compete for limiting amount of Insigs when intracellular sterols levels rise.
Insig-Mediated Ubiquitination of HMG CoA Reductase
Evidence supporting a major role for the ubiquitin-proteasome pathway in sterol-accelerated degradation of reductase was first provided by the observation that proteasome inhibition blocks the process 18, leading to the accumulation of ubiquitinated forms of reductase on ER membranes 8. This ubiquitination is obligatory for degradation of reductase and exhibits an absolute requirement for the presence of Insigs 37, 38. Reduction of Insig-1 and Insig-2 mRNA by genetic mutation or RNAi-mediated knockdown abrogates sterol-dependent ubiquitination of endogenous reductase, rendering the enzyme refractory to accelerated degradation. Moreover, sterol-induced ubiquitination of reductase exhibits an absolute Insig requirement in transient transfection assays. Mutation of the YIYF sequence in reductase, which blocks Insig binding, prevents regulated ubiquitination and slows the enzyme’s degradation. In contrast, conservative substitutions of arginine for lysines 89 and 248 in the membrane domain of reductase (Figure 2B) do not block Insig binding, but the substitutions rather abolish ubiquitination and subsequent degradation of reductase. Thus, lysines 89 and 248 in reductase are implicated as sites for Insig-mediated, sterol-induced ubiquitination. It is important to note that mutation of lysines 89 and 248 blocks ubiquitination and degradation of reductase in the context of the full-length enzyme, suggesting that the catalytic domain does not contribute to ubiquitination. This is consistent with the observation that the soluble catalytic domain is dispensable for sterol-regulated degradation 16.
How might Insig binding impart recognition of reductase by the ubiquitinating machinery? This question was addressed by examining reductase ubiquitination in a permeabilized cell system 39. Sterol-depleted cells were permeabilized with low concentrations of the mild detergent digitonin such that nearly all of the cytosolic proteins were released into the supernatant upon centrifugation, whereas membrane proteins such as reductase remained associated with the pellet fraction. The pellet of permeabilized cells supports Insig-dependent ubiquitination of reductase that is stimulated by additions of ATP, sterols, and rat liver cytosol in vitro. Surprisingly, reductase ubiquitination is potently stimulated by oxygenated derivatives of cholesterol, including 24-, 25-, and 27-hydroxycholesterol, but not by cholesterol itself. The significance of this finding will be discussed in more detail below.
Ubiquitination of proteins is a multistep process, involving the action of at least three types of enzymes 40. In the first step, ubiquitin is activated by the ubiquitin-activating enzyme (E1), which forms a thiol ester between a reactive cysteine residue in E1 and the C-terminus of ubiquitin. Next, ubiquitin is transferred from E1 to a catalytic cysteine of the ubiquitin-conjugating enzyme (E2) thiol ester. The third type of enzyme, ubiquitin ligase (E3) facilitates transfer of activated ubiquitin from E2 to a lysine residue in the substrate (or a previously attached ubiquitin). Once a poly-ubiquitin chain of sufficient size is built, the substrate is recognized and subsequently degraded by proteasomes. Only two E1 enzymes exist, and both are cytosolic proteins. In contrast, a variety of E2s and E3s, both soluble and membrane-bound have been described 41. The exquisite sensitivity of substrate ubiquitination is ultimately determined by the E3, either alone or in combination with its cognate E2.
Fractionated S100 from Hela cells was utilized to determine which component of the reductase ubiquitinating machinery (E1, E2 and E3) is provided by rat liver cytosol in the permeabilized cell system 39. These fractions were first described by Hershko and co-workers 42, 43 and were generated by separating Hela cell S100 into fractions that bind (Fraction II) or do not bind (Fraction I) an anion exchange resin. It was subsequently determined that Fraction I contained ubiquitin, whereas Fraction II contains E1 44. Fraction II effectively replaces rat liver cytosol for regulated ubiquitination of reductase in permeabilized cells, but Fraction I does not. Immunodepletion of E1 eliminates the reductase ubiquitinating activity of rat liver cytosol. Consistent with this, purified E1 replaces rat liver cytosol for sterol-regulated ubiquitination of reductase in permeabilized cells. Thus, E1 is the only cytosolic protein required for reductase ubiquitination, which indicates the reductase E2 and E3 are membrane-associated proteins. This notion is consistent with the localization of apparent sites of reductase ubiquitination, lysines 89 and 248, which are cytosolically exposed and are predicted to lie immediately adjacent to transmembrane helices three and seven (Figure 2B).
Results from the analysis of reductase ubiquitination in permeabilized cells indicated that Insig binding results in recruitment of enzymes that ubiquitinate reductase. Coimmunoprecipitation experiments, coupled with tandem mass spectroscopy, were utilized to identify membrane proteins that associate with the sterol-dependent reductase-Insig complex. These studies revealed that Insig-1 binds to a known membrane-anchored ubiquitin ligase called gp78 45. The cDNA for gp78 predicts a 643-amino acid protein that can be divided into four domains. The N-terminal domain of 298 amino acids contains five to seven membrane-spanning helices that anchors the protein to ER membranes and mediates association with Insig-1. The membrane attachment region of gp78 is followed by a 43-amino acid region with a RING finger consensus sequence that confers ubiquitin ligase activity 46. Following the RING domain is a 42-amino acid region homologous to Cue1p, an ER membrane protein in yeast that serves as a membrane anchor for Ubc7p, a cytosolic ubiquitin-conjugating enzyme 47. Recently, this region of gp78 has been shown to directly bind to Ufd1, a cytosolic protein that modulates gp78 ubiquitin ligase activity, thereby enhancing ubiquitination and degradation of the enzyme’s substrates 48. Finally, the extreme C-terminus of gp78 (48 amino acids) mediates an interaction with VCP (Valosin-containing protein, also known as p97), an ATPase that has been implicated in the post-ubiquitination steps of ER-associated degradation (ERAD) 49.
At least three lines of evidence indicate that gp78, through its binding to Insig-1, initiates sterol-accelerated degradation of reductase. 1) Overexpression of the membrane domain of gp78 blocks Insig-mediated, sterol-accelerated degradation of reductase, suggesting that the membrane domain of gp78 competes with full-length gp78 for binding to Insig-1, thereby abolishing reductase ubiquitination. 2) Sterols trigger binding of gp78 to reductase in an Insig-dependent, sterol-regulated manner. The specificity of this binding is illustrated by the inability of gp78 to bind Scap, regardless of the presence or absence of sterols and/or Insigs. 3) RNAi-mediated knockdown of gp78 prevents sterol-regulated ubiquitination and degradation of endogenous reductase. Importantly, the effect of gp78 knockdown is specific inasmuch as knockdown of a related membrane-bound ubiquitin ligase, Hrd1, does not effect reductase ubiquitination. Another function of gp78, besides its role as a ubiquitin ligase, is to couple ubiquitination of reductase to degradation through its association with VCP. Indeed, coimmunoprecipitation experiments show that gp78 is an intermediary in association of VCP and Insig-1. Moreover, knockdown of VCP by RNAi prevents sterol-accelerated degradation of endogenous reductase and a dominant-negative ATPase-deficient mutant of VCP blocks sterol-regulated degradation of transfected reductase.
The identification of gp78 as an E3 ubiquitin ligase that mediates reductase ubiquitination has important implications for yet another mode of sterol regulation. The regulation of Insig-1 contrasts that of reductase in that Insig-1 becomes ubiquitinated and is rapidly degraded by proteasomes in sterol-depleted cells 50. Ubiquitination of Insig-1 is mediated by gp78 51. When sterols induce reductase to bind Insig-1, ubiquitination is diverted toward reductase and the enzyme becomes rapidly degraded. However, when sterols cause Scap to bind Insig-1, gp78 is displaced and no longer ubiquitinates Insig-1, thereby stabilizing the protein. This reaction helps to explain why reductase is degraded when it binds to Insig-1, whereas Scap binding to Insig-1 leads to retention in the ER. In addition, gp78-mediated ubiquitination and degradation of Insig-1 provides a mechanism for a recently appreciated process termed “convergent feedback inhibition” 50. In sterol-depleted cells, Scap-SREBP complexes no longer bind Insig-1, which in turn becomes ubiquitinated and degraded. Thus, Scap-SREBP complexes are free to exit the ER and translocate to the Golgi, where the SREBPs are processed to the nuclear form that stimulates transcription of target genes, including the Insig-1 gene. Increased transcription of the Insig-1 gene leads to increased synthesis of Insig-1 protein, but the protein is ubiquitinated and degraded until sterols build up to levels sufficient to trigger Scap binding. Thus, inhibition of SREBP processing requires convergence of newly synthesized Insig-1 and newly acquired sterols.
The HMG CoA Reductase Sterol-Sensing Reaction
Oxysterols are derivatives of cholesterol that contain hydroxyl groups at various positions in the iso-octyl side chain 52, 53. These compounds are synthesized in many tissues by specific enzymes called hydroxylases; oxysterols play key roles in cholesterol export and they are also intermediates in the synthesis of bile acids 54. Oxysterols are significantly more soluble than cholesterol in aqueous solution and thus, can readily pass across the plasma membrane and enter cells. This property renders oxysterols such as 24-, 25-, and 27-hydroxycholesterol extremely potent in inhibiting cholesterol synthesis by stimulating binding of both reductase and Scap to Insigs. Oxysterols are present at very low concentrations (104 - to 106-fold less than cholesterol) in tissues and blood, which raises questions as to whether they act through a similar mechanism as LDL-derived cholesterol to block cholesterol synthesis. In the case of Scap, the mode of action of these two classes of sterols is becoming clear. Cholesterol directly binds to the membrane domain of Scap in a specific and saturable fashion 55. The interaction causes a conformational change in Scap that promotes Insig binding 56. The addition of cholesterol in vitro to membranes isolated from sterol-depleted cells causes exposure of a cryptic trypsin cleavage site, thereby altering the tryptic digestion pattern of Scap that can be monitored by immunoblot analysis 57. Co-expression of Insigs lowers the amount of cholesterol required to induce the conformational change in Scap. Oxysterols neither alters Scap’s conformation in vitro nor binds to the protein’s membrane domain, leading to the postulation of the existence of a membrane-bound oxysterol binding protein. Remarkably, Insig-2 has been recently defined as a membrane-bound oxysterol binding protein with binding specificity that correlates with the ability of oxysterols to inhibit SREBP processing 31, 58. Thus, formation of the Scap-Insig complex can be initiated by either binding of cholesterol to the membrane domain of Scap or by binding of oxysterols to Insigs. Both events prevent incorporation of Scap-SREBP into vesicles that bud from the ER en route to the Golgi. By analogy, the likely mechanism by which oxysterols stimulate degradation of reductase is through their binding to Insigs.
In striking contrast to results obtained with Scap, the analysis of reductase ubiquitination in permeabilized cells revealed that the reaction was potently stimulated by oxysterols, but not by cholesterol 39. These results led to a search for endogenous sterol regulators of reductase ubiquitination and degradation. Previous indirect studies implicated that lanosterol, the first sterol produced in the cholesterol biosynthetic pathway (Figure 1), or one of its metabolites participates in feedback inhibition of reductase. For example, genetic mutation or pharmacologic inhibition of lanosterol 14α-demethylase, which catalyzes the first step in conversion of lanosterol to cholesterol (Figure 1), markedly reduces the amount of reductase activity in cells 59, 60. These observations led to the evaluation of lanosterol and its metabolite 24,25-dihydrolanosterol as endogenous regulators of reductase ubiquitination and degradation 61. When added to intact cells, lanosterol and 24,25-dihydrolanosterol potently stimulate ubiquitination and degradation of reductase through a reaction that requires the presence of Insigs. The activity of both sterols is specific inasmuch as they do not inhibit processing of SREBPs. This is consistent with the inability of lanosterol to directly bind to Scap and Insig or alter Scap’s conformation in vitro 57. The action of lanosterol and 24,25-dihydrolanosterol is direct and does not require their conversion into an active metabolite as indicated by the reconstitution of reductase ubiquitination by simply incubating isolated membranes with the sterols and purified E1. Using this in vitro assay, the action of lanosterol and 24,25-dihydrolanosterol in stimulating ubiquitination and degradation of reductase was traced to methyl groups present in the 4α, 4β, and 14α positions of the sterol ring.
Insig-mediated regulation of reductase is controlled by three classes of sterols: oxysterols, cholesterol, and methylated sterols such as lanosterol and 24,25-dihydrolanosterol. Oxysterols, which are derived from cholesterol, have dual actions in that they accelerate degradation of reductase and block ER to Golgi transport of Scap-SREBP through their direct binding to Insigs. Cholesterol does not regulate reductase stability directly, but binds to Scap and triggers Insig binding, thereby preventing escape of Scap-SREBP from the ER. On the other hand, lanosterol selectively accelerates degradation of reductase without an effect on ER to Golgi transport of Scap-SREBP. Notably, the demethylation of lanosterol has been implicated as a rate-limiting step in the post-squalene portion of cholesterol synthesis, situating the reaction as a potential focal point in sterol regulation 62, 63. Considering that lanosterol is the first sterol produced in cholesterol synthesis, it seems reasonable that it controls early steps in the pathway (i.e., the nonsterol branch) by stimulating reductase degradation. The accumulation of lanosterol is avoided, owing to its inability to block SREBP processing through Scap. This assures that mRNAs encoding enzymes catalyzing reactions subsequent to lanosterol remain elevated and lanosterol is metabolized to cholesterol. The importance of this conversion is highlighted by the observation that lanosterol cannot support cell growth in the absence of cholesterol and may be toxic 64. This toxicity is likely due to the inability to optimize certain physiologic properties of cell membranes with regard to biological functions.
Oxygen sensing in the cholesterol biosynthetic pathway
The physiologic relevance of lanosterol as an endogenous regulator of reductase ubiquitination and degradation was deduced by the recognition that cholesterol synthesis is a highly oxygen-consumptive process. The synthesis of one molecule of cholesterol from acetyl-CoA requires eleven molecules of dioxygen, nine of which are consumed during the removal of the 4α, 4β, and 14α methyl groups in lanosterol and its metabolite 24,25-dihydrolanosterol by the successive actions of lanosterol 14α-demethylase and C4-methyl sterol oxidase (Figure 1). This led to speculation that oxygen deprivation (hypoxia) might block demethylation of lanosterol and 24,25-dihydrolanosterol and thereby stimulate degradation of reductase. Indeed, a recent study shows that hypoxia blunts cholesterol synthesis by inhibiting lanosterol and 24,25-dihydrolanosterol demethylation, causing both sterols to accumulate in cells 65. Rapid degradation of reductase parallels hypoxia-induced accumulation of lanosterol and 24,25-dihydrolanosterol. This Insig-mediated degradation requires de novo sterol synthesis as indicated by its inhibition by compactin and the squalene monooxygenase inhibitor NB-598 but not the lanosterol 14α-demethylase inhibitor RS-21607 (see Figure 1). Although hypoxia accelerates degradation of reductase, processing of SREBPs remains unaffected. This finding is consistent with the observation described above that exogenous lanosterol stimulates degradation of reductase without inhibiting SREBP processing.
In addition to the accumulation of methylated sterols, the degradation of reductase in hypoxic cells also requires the action of the oxygen-sensitive transcription factor, HIF-1α. In oxygenated cells, HIF-1α is rapidly degraded owing to hydroxylation of specific proline residues in the protein 66. Prolyl hydroxylation enhances binding of HIF-1α to the von Hippel Lindau tumor suppressor protein (pVHL), which is the recognition component of a ubiquitin ligase that targets HIF-1α for proteasomal degradation. Prolyl hydroxylation of HIF-1α is catalyzed by a family of dioxygenases that use 2-oxoglutarate as a co-substrate and exhibit strict dependence for molecular oxygen 67-69. When cells are deprived of oxygen, prolyl hydroxylation is inhibited, allowing HIF-1α to escape degradation and accumulate to high levels. The stabilized HIF-1α subunits associate with the constitutive HIF-1β subunit, forming a heterodimeric transcription factor (HIF) that modulates expression more than 70 genes involved in both systemic and cellular responses to oxygen deprivation 70.
Evidence implicating a major role for HIF-1α in the hypoxia-induced degradation of reductase is provided by both pharmacologic and genetic data. Treatment of oxygenated cells with dimethyloxalylglycine (DMOG), a non-specific inhibitor of 2-oxoglutarate-dependent dioxygenases, not only stabilizes HIF-1α 71 but also triggers rapid degradation of reductase through an Insig-dependent mechanism that requires de novo sterol synthesis. Genetic evidence for a role of HIF-1α in reductase degradation is provided by the observation that the enzyme is refractory to hypoxia- and DMOG-induced degradation in mutant cells that are deficient in HIF-1α. While these observations establish the importance of the action of HIF-1α in oxygen-regulated degradation of reductase, they raise questions as to the HIF-target genes that mediate the response. In several DNA microarray analyses, Insig-1 and Insig-2 transcripts have been identified among those increased by either DMOG or hypoxia treatment 72-74. This observation led to the subsequent discovery that DMOG and hypoxia enhances expression of both Insigs through a HIF-dependent mechanism. Considered together, these observations establish a connection cholesterol synthesis and oxygen sensing in animal cells (Figure 4). These metabolic pathways are linked by two regulatory actions: 1) hypoxia-induced accumulation of the cholesterol biosynthetic intermediates lanosterol and 24,25-dihydrolanosterol; and 2) HIF-1α mediated induction of Insigs. Convergence of these signals triggers rapid degradation of reductase, which ultimately limits synthesis of cholesterol and helps to guard against the wasting of cellular oxygen in the face of hypoxia.
Figure 4.

Mechanism for Oxygen Sensing in the Cholesterol Synthetic Pathway
The link between synthesis of cholesterol and oxygen sensing in animal cells is provided by hypoxia induced accumulation of lanosterol and 24,25-dihydrolanosterol and HIF-1a-mediated induction of Insig-1 and Insig-2. Convergence of these responses leads to rapid degradation of HMG CoA reductase, thereby limiting synthesis of cholesterol.
Unanswered questions and future directions
Despite the recent advances in the understanding of molecular mechanisms underlying sterol-accelerated degradation of reductase, much remains to be determined. For instance, what is the mechanism by which lanosterol and 24,25-dihydrolanosterol trigger binding of reductase to Insigs? Do these methylated sterols directly bind the membrane domain of reductase in a reaction analogous to that of cholesterol and Scap? Unfortunately, attempts to demonstrate direct binding of Insigs to methylated sterols to the membrane domain of reductase have been unsuccessful. Moreover, addition of lanosterol or 24,25-dihydrolanosterol to reductase-containing membranes in vitro fails to alter the tryptic pattern of the enzyme. Thus, the possibility exists that a distinct ER membrane protein binds to methylated sterols and in turn, trigger binding of reductase to Insigs, thereby initiating reductase ubiquitination. Reductase is the target of statins, which are the most widely prescribed cholesterol-lowering drugs in humans. Interest in developing additional strategies that inhibit reductase has led to the discovery of nonsterol compounds, such as vitamin E (tocotrienols) and the bisphosphonate SR-12813, that mimic sterols in accelerating reductase degradation 75, 76. The availability of such reagents may prove useful in the on-going quest to define the molecular mechanisms for the reductase sterol-sensing reaction.
Another unresolved question in reductase degradation is the mechanism for delivery of ubiquitinated forms of the enzyme from the membrane to the cytosol for proteasomal degradation. Unlike model ER-associated degradation substrates that are either completely lumenal or contain one transmembrane domain, proteasome inhibition leads to accumulation of ubiquitinated reductase on membranes, rather than in the cytosol 36. This suggests that degradation of reductase is coupled to its ubiquitination and proceeds through a membrane-bound intermediate. However, reductase must be degraded as a unit without releasing the catalytic domain into the cytosol, which would defeat the purpose of regulated degradation.
Efficient degradation of reductase requires nonsterol isoprenoids derived from mevalonate in addition to sterols. This was borne out of experiments showing that in compactin-treated cells, sterols can trigger binding of reductase to Insigs and subsequent ubiquitination of the enzyme. However, the ubiquitinated reductase is not efficiently degraded unless the cells are also treated with mevalonate. This mevalonate requirement can be bypassed by the addition of geranylgeraniol (GG-OH), a 20-carbon isoprenoid, but not by the 15-carbon farnesol 37. GGOH does not appear to trigger reductase ubiquitination, even though it augments sterol-accelerated degradation of the enzyme. This suggests the action of nonsterol isoprenoids in a post-ubiquitination step of reductase degradation.
The current view of sterol-accelerated degradation of reductase is illustrated in the model shown in Figure 5. The reaction is initiated by sensing of membrane-embedded sterols through direct or indirect interactions with the membrane domain of reductase. This interaction causes reductase to bind to a subset of Insigs that are associated with gp78, which mediates transfer of ubiquitin from the E2 Ubc7 to lysines 89 and 248 of reductase. Ubiquitination targets reductase for recognition by gp78-associated VCP, which together with its cofactors, somehow extract ubiquitinated reductase from membranes and deliver it to proteasomes for degradation. The extraction step appears to be augmented by GG-OH. It seems likely that GG-OH, after its conversion to metabolically active geranylgeranyl-pyrophosphate (GG-PP), is incorporated into a protein that enhances the effect of sterols on reductase degradation. Possible candidates include geranylgeranylated Rab proteins, which are known to play key roles in various aspects of vesicular transport 77. Thus, the possibility exists that a vesicle-mediated transport event delivers ubiquitinated reductase to a specific organelle or subdomain of the ER in which the protein is degraded. Notably, Ufd1 appears to play a key role in this pathway by enhancing gp78 ubiquitin ligase activity and modulating a post-ubiquitination step in reductase degradation. In addition, Ufd1 seems to bind to gp78 in a sterol-regulated fashion 48, but the significance of this is presently unknown. Complete elucidation of reductase degradation will likely require the reconstitution of post-ubiquitination steps of reductase degradation in a cell-free system.
Figure 5.

Pathway for Sterol-Accelerated Degradation of HMG CoA Reductase
Accumulation of 25-hydroxycholesterol, lanosterol, or 24,25-dihydrolanosterol in ER membranes triggers binding of the reductase to Insigs. A subset of Insigs is associated with the membrane-anchored ubiquitin ligase, gp78, which binds the E2 Ubc7 and VCP, an ATPase that plays a role in extraction of ubiquitinated proteins from ER membranes. Through the action of gp78 and Ubc7, reductase becomes ubiquitinated, which triggers its extraction from the membrane by VCP, and subsequent delivery to proteasomes for degradation. The post-ubiquitination step is postulated to be enhanced by geranylgeraniol through an undefined mechanism that may involve a geranylgeranylated protein, such as one of the Rab proteins.
What is the contribution of reductase degradation to overall cholesterol homeostasis in whole animals? Insigs appear to play a major role in regulation of reductase in the mouse liver. Genetic deletion of Insigs results in the accumulation of reductase to a level approximately 20-fold higher than that in wild type mice 78. This accumulation is presumably attributable to the combination of both transcriptional and post-transcriptional regulation of reductase, but the extent to which each level of regulation contributed to the massive increase in reductase is unknown. Thus, studies that directly focus on reductase degradation are required in order to determine the contribution of protein stability to overall regulation of reductase in mice in vivo under various physiologic conditions, such as hypoxia.
The significance of Insig-mediated regulation of reductase in maintenance of cholesterol homeostasis is highlighted by the effectiveness of reductase inhibition in lowering plasma LDL-cholesterol in humans 79. However, the inhibition of reductase disrupts normal feedback inhibition of the enzyme and animals respond by developing a compensatory increase in reductase levels in the liver 80, 81. Knowledge of the mechanisms for this compensatory increase, particularly the contribution of degradation, may facilitate development of novel drugs that improve the effectiveness of statins, or in some cases provide alternative treatments. Such a drug would be modeled after lanosterol and 24,25-dihydrolanosterol, which selectively stimulates reductase degradation without affecting the Scap-SREBP pathway or LDL-receptor activity. In addition, elucidation of underlying mechanisms for sterol-accelerated, ER-associated degradation of reductase may have implications for degradation of other clinically important proteins such as the cystic fibrosis transmembrane conductance regulator (CFTR). Thus, further excitement will undoubtedly ensue once questions posed in this review begin to become clear.
Acknowledgements
Work in the DeBose-Boyd laboratory is supported by grants from the National Institutes of Health (HL20948), the Perot Family Foundation, the American Heart Association (0540128N), and the W.M. Keck Foundation.
REFERENCES
- 1.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 2.Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 1980;21:505–517. [PubMed] [Google Scholar]
- 3.Endo A, Kuroda M, Tanzawa K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett. 1976;72:323–326. doi: 10.1016/0014-5793(76)80996-9. [DOI] [PubMed] [Google Scholar]
- 4.Nakanishi M, Goldstein JL, Brown MS. Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J. Biol. Chem. 1988;263:8929–8937. [PubMed] [Google Scholar]
- 5.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roitelman J, Simoni RD. Distinct sterol and nonsterol signals for the regulated degradation of 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 1992;267:25264–25273. [PubMed] [Google Scholar]
- 7.McGee TP, Cheng HH, Kumagai H, Omura S, Simoni RD. Degradation of 3-hydroxy-3-methylglutaryl-CoA reductase in endoplasmic reticulum membranes is accelerated as a result of increased susceptibility to proteolysis. J. Biol. Chem. 1996;271:25630–25638. doi: 10.1074/jbc.271.41.25630. [DOI] [PubMed] [Google Scholar]
- 8.Ravid T, Doolman R, Avner R, Harats D, Roitelman J. The ubiquitin-proteasome pathway mediates the regulated degradation of mammalian 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 2000;275:35840–35847. doi: 10.1074/jbc.M004793200. [DOI] [PubMed] [Google Scholar]
- 9.Ness GC, Spindler CD, Moffler MH. Purification of 3-hydroxy-3-methylglutaryl coenzyme A reductase from rat liver. Arch. Biochem. Biophys. 1979;197:493–499. doi: 10.1016/0003-9861(79)90272-8. [DOI] [PubMed] [Google Scholar]
- 10.Edwards PA, Lemongello D, Fogelman AM. Purification and properties of rat liver 3-hydroxy-3-methylglutaryl coenzyme A reductase. Biochim. Biophys. Acta. 1979;574:123–135. doi: 10.1016/0005-2760(79)90091-2. [DOI] [PubMed] [Google Scholar]
- 11.Brown MS, Dana SE, Dietschy JM, Siperstein MD. 3-Hydroxy-3-methylglutaryl coenzyme A reductase. Solubilization and purification of a cold-sensitive microsomal enzyme. J Biol. Chem. 1973;248:4731–4738. [PubMed] [Google Scholar]
- 12.Liscum L, et al. Domain structure of 3-hydroxy-3-methylglutaryl coenzyme A reductase, a glycoprotein of the endoplasmic reticulum. J. Biol. Chem. 1985;260:522–530. [PubMed] [Google Scholar]
- 13.Roitelman J, Olender EH, Bar-Nun S, Dunn WA, Jr., Simoni RD. Immunological evidence for eight spans in the membrane domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for enzyme degradation in the endoplasmic reticulum. J. Cell Biol. 1992;117:959–973. doi: 10.1083/jcb.117.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luskey KL, Stevens B. Human 3-hydroxy-3-methylglutaryl coenzyme A reductase. Conserved domains responsible for catalytic activity and sterol-regulated degradation. J. Biol. Chem. 1985;260:10271–10277. [PubMed] [Google Scholar]
- 15.Gertler FB, Chiu CY, Richter-Mann L, Chin DJ. Developmental and metabolic regulation of the Drosophila melanogaster 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mol. Cell. Biol. 1988;8:2713–2721. doi: 10.1128/mcb.8.7.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gil G, Faust JR, Chin DJ, Goldstein JL, Brown MS. Membrane-bound domain of HMG CoA reductase is required for sterol-enhanced degradation of the enzyme. Cell. 1985;41:249–258. doi: 10.1016/0092-8674(85)90078-9. [DOI] [PubMed] [Google Scholar]
- 17.Skalnik DG, Narita H, Kent C, Simoni RD. The membrane domain of 3-hydroxy-3-methylglutaryl-coenzyme A reductase confers endoplasmic reticulum localization and sterol-regulated degradation onto beta-galactosidase. J. Biol. Chem. 1988;263:6836–6841. [PubMed] [Google Scholar]
- 18.Inoue S, Bar-Nun S, Roitelman J, Simoni RD. Inhibition of degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase in vivo by cysteine protease inhibitors. J. Biol. Chem. 1991;266:13311–13317. [PubMed] [Google Scholar]
- 19.Hampton RY. Genetic analysis of hydroxymethylglutaryl-coenzyme A reductase regulated degradation. Curr. Opin. Lipidol. 1998;9:93–97. doi: 10.1097/00041433-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Hampton RY, Koning A, Wright R, Rine J. In vivo examination of membrane protein localization and degradation with green fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 1996;93:828–833. doi: 10.1073/pnas.93.2.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hampton RY. Proteolysis and sterol regulation. Annu. Rev. Cell Dev. Biol. 2002;18:345–378. doi: 10.1146/annurev.cellbio.18.032002.131219. [DOI] [PubMed] [Google Scholar]
- 22.Hampton RY. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002;14:476–482. doi: 10.1016/s0955-0674(02)00358-7. [DOI] [PubMed] [Google Scholar]
- 23.Nohturfft A, Brown MS, Goldstein JL. Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. J. Biol. Chem. 1998;273:17243–17250. doi: 10.1074/jbc.273.27.17243. [DOI] [PubMed] [Google Scholar]
- 24.Hua X, Nohturfft A, Goldstein JL, Brown MS. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell. 1996;87:415–426. doi: 10.1016/s0092-8674(00)81362-8. [DOI] [PubMed] [Google Scholar]
- 25.Rawson RB, DeBose-Boyd R, Goldstein JL, Brown MS. Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J. Biol. Chem. 1999;274:28549–28556. doi: 10.1074/jbc.274.40.28549. [DOI] [PubMed] [Google Scholar]
- 26.DeBose-Boyd RA, et al. Transport-dependent proteolysis of SREBP: relocation of site-1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell. 1999;99:703–712. doi: 10.1016/s0092-8674(00)81668-2. [DOI] [PubMed] [Google Scholar]
- 27.Nohturfft A, Yabe D, Goldstein JL, Brown MS, Espenshade PJ. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell. 2000;102:315–323. doi: 10.1016/s0092-8674(00)00037-4. [DOI] [PubMed] [Google Scholar]
- 28.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 29.Yang T, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 30.Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun LP, Seemann J, Goldstein JL, Brown MS. From the Cover: Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. PNAS. 2007;104:6519–6526. doi: 10.1073/pnas.0700907104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuwabara PE, Labouesse M. The sterol-sensing domain: multiple families, a unique role? Trends Genet. 2002;18:193–201. doi: 10.1016/s0168-9525(02)02640-9. [DOI] [PubMed] [Google Scholar]
- 33.Yabe D, Xia ZP, Adams CM, Rawson RB. Three mutations in sterol-sensing domain of SCAP block interaction with insig and render SREBP cleavage insensitive to sterols. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16672–16677. doi: 10.1073/pnas.262669399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nohturfft A, Hua X, Brown MS, Goldstein JL. Recurrent G-to-A substitution in a single codon of SREBP cleavage-activating protein causes sterol resistance in three mutant Chinese hamster ovary cell lines. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13709–13714. doi: 10.1073/pnas.93.24.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nohturfft A, Brown MS, Goldstein JL. Sterols regulate processing of carbohydrate chains of wild-type SREBP cleavage-activating protein (SCAP), but not sterol-resistant mutants Y298C or D443N. Proc. Natl. Acad. Sci. U. S. A. 1998;95:12848–12853. doi: 10.1073/pnas.95.22.12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol. Cell. 2003;11:25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- 37.Sever N, et al. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 2003;278:52479–52490. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- 38.Lee PC, Sever N, DeBose-Boyd RA. Isolation of sterol-resistant Chinese hamster ovary cells with genetic deficiencies in both Insig-1 and Insig-2. J Biol. Chem. 2005;280:25242–25249. doi: 10.1074/jbc.M502989200. [DOI] [PubMed] [Google Scholar]
- 39.Song BL, DeBose-Boyd RA. Ubiquitination of 3-Hydroxy-3-methylglutaryl-CoA Reductase in Permeabilized Cells Mediated by Cytosolic E1 and a Putative Membrane-bound Ubiquitin Ligase. J Biol. Chem. 2004;279:28798–28806. doi: 10.1074/jbc.M402442200. [DOI] [PubMed] [Google Scholar]
- 40.Pickart CM. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 41.Weissman AM. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2001;2:169–178. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 42.Hershko A, Heller H, Elias S, Ciechanover A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983;258:8206–8214. [PubMed] [Google Scholar]
- 43.Hershko A, Heller H, Eytan E, Reiss Y. The protein substrate binding site of the ubiquitin-protein ligase system. J. Biol. Chem. 1986;261:11992–11999. [PubMed] [Google Scholar]
- 44.Hershko A, Ciechanover A, Varshavsky A. Basic Medical Research Award. The ubiquitin system. Nat. Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- 45.Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell. 2005;19:829–840. doi: 10.1016/j.molcel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Lorick KL, et al. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. U. S. A. 1999;96:11364–11369. doi: 10.1073/pnas.96.20.11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ponting CP. Proteins of the endoplasmic-reticulum-associated degradation pathway: domain detection and function prediction. Biochem. J. 2000;351(Pt 2):527–535. [PMC free article] [PubMed] [Google Scholar]
- 48.Cao J, et al. Ufd1 is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG-CoA reductase. Cell Metab. 2007;6:115–128. doi: 10.1016/j.cmet.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 49.Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 50.Gong Y, et al. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006;3:15–24. doi: 10.1016/j.cmet.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 51.Lee JN, Song B, DeBose-Boyd RA, Ye J. Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J Biol. Chem. 2006;281:39308–39315. doi: 10.1074/jbc.M608999200. [DOI] [PubMed] [Google Scholar]
- 52.Schroepfer GJ., Jr. Oxysterols: Modulators of Cholesterol Metabolism and Other Processes. Physiol. Rev. 2000;80:361–554. doi: 10.1152/physrev.2000.80.1.361. [DOI] [PubMed] [Google Scholar]
- 53.Bjorkhem I. Do oxysterols control cholesterol homeostasis? J. Clin. Invest. 2002;110:725–730. doi: 10.1172/JCI16388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Russell DW. Oxysterol biosynthetic enzymes. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2000;1529:126–135. doi: 10.1016/s1388-1981(00)00142-6. [DOI] [PubMed] [Google Scholar]
- 55.Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Direct Binding of Cholesterol to the Purified Membrane Region of SCAP; Mechanism for a Sterol-Sensing Domain. Mol. Cell. 2004;15:259–268. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 56.Adams CM, Goldstein JL, Brown MS. Cholesterol-induced conformational change in SCAP enhanced by Insig proteins and mimicked by cationic amphiphiles. Proc. Natl. Acad. Sci U. S. A. 2003;100:10647–10652. doi: 10.1073/pnas.1534833100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell. 2002;10:237–245. doi: 10.1016/s1097-2765(02)00591-9. [DOI] [PubMed] [Google Scholar]
- 58.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. From the Cover: Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. PNAS. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen HW, Leonard DA, Fischer RT, Trzaskos JM. A mammalian mutant cell lacking detectable lanosterol 14 alpha-methyl demethylase activity. J. Biol. Chem. 1988;263:1248–1254. [PubMed] [Google Scholar]
- 60.Leonard DA, Kotarski MA, Tessiatore JE, Favata MF, Trzaskos JM. Post-transcriptional regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase by 3 beta-hydroxy-lanost-8-en-32-al, an intermediate in the conversion of lanosterol to cholesterol. Arch. Biochem. Biophys. 1994;310:152–157. doi: 10.1006/abbi.1994.1151. [DOI] [PubMed] [Google Scholar]
- 61.Song BL, Javitt NB, DeBose-Boyd RA. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metabolism. 2005;1:179–189. doi: 10.1016/j.cmet.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 62.Gaylor JL. Membrane-Bound Enzymes of Cholesterol Synthesis from Lanosterol. Biochemical and Biophysical Research Communications. 2002;292:1139–1146. doi: 10.1006/bbrc.2001.2008. [DOI] [PubMed] [Google Scholar]
- 63.Williams MT, Gaylor JL, Morris HP. Investigation of the rate-determining microsomal reaction of cholesterol biosynthesis from lanosterol in Morris hepatomas and liver. Cancer Res. 1977;37:1377–1383. [PubMed] [Google Scholar]
- 64.Xu F, et al. Dual roles for cholesterol in mammalian cells. Proc. Natl. Acad. Sci U. S. A. 2005;102:14551–14556. doi: 10.1073/pnas.0503590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen AD, McDonald JG, Bruick RK, DeBose-Boyd RA. Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor-mediated induction of insigs. J Biol. Chem. 2007;282:27436–27446. doi: 10.1074/jbc.M704976200. [DOI] [PubMed] [Google Scholar]
- 66.Maxwell PH, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 67.Epstein AC, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 68.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 69.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 70.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular leve. Physiology. (Bethesda. ) 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 71.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 72.Kim H, et al. Analysis of the effect of aging on the response to hypoxia by cDNA microarray. Mechanisms of Ageing and Development. 2003;124:941–949. doi: 10.1016/s0047-6374(03)00166-0. [DOI] [PubMed] [Google Scholar]
- 73.Mense SM, et al. Gene expression profiling reveals the profound upregulation of hypoxia-responsive genes in primary human astrocytes. Physiol. Genomics. 2006;25:435–449. doi: 10.1152/physiolgenomics.00315.2005. [DOI] [PubMed] [Google Scholar]
- 74.Sung FL, et al. Genome-wide expression analysis using microarray identified complex signaling pathways modulated by hypoxia in nasopharyngeal carcinoma. Cancer Letters. doi: 10.1016/j.canlet.2007.01.012. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 75.Song BL, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme a reductase stimulated by delta- and gamma-tocotrienols. J Biol. Chem. 2006;281:25054–25061. doi: 10.1074/jbc.M605575200. [DOI] [PubMed] [Google Scholar]
- 76.Sever N, Lee PCW, Song BL, Rawson RB, DeBose-Boyd RA. Isolation of Mutant Cells Lacking Insig-1 through Selection with SR-12813, an Agent That Stimulates Degradation of 3-Hydroxy-3-methylglutaryl-Coenzyme A Reductase. J. Biol. Chem. 2004;279:43136–43147. doi: 10.1074/jbc.M406406200. [DOI] [PubMed] [Google Scholar]
- 77.Seabra MC, Mules EH, Hume AN. Rab GTPases, intracellular traffic and disease. Trends Mol. Med. 2002;8:23–30. doi: 10.1016/s1471-4914(01)02227-4. [DOI] [PubMed] [Google Scholar]
- 78.Engelking LJ, et al. Schoenheimer effect explained - feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J Clin. Invest. 2005;115:2489–2498. doi: 10.1172/JCI25614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Illingworth DR, Tobert JA. HMG-CoA reductase inhibitors. Adv. Protein Chem. 2001;56:77–114. doi: 10.1016/s0065-3233(01)56003-9. [DOI] [PubMed] [Google Scholar]
- 80.Kita T, Brown MS, Goldstein JL. Feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in livers of mice treated with mevinolin, a competitive inhibitor of the reductase. J. Clin. Invest. 1980;66:1094–1100. doi: 10.1172/JCI109938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singer II, et al. Hydroxymethylglutaryl-coenzyme A reductase-containing hepatocytes are distributed periportally in normal and mevinolin-treated rat livers. Proc. Natl. Acad. Sci. U. S. A. 1984;81:5556–5560. doi: 10.1073/pnas.81.17.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]