

Abstract
Multibillion-clone libraries of phages displaying guest peptides fused to the major coat protein pVIII (landscape libraries) are a rich source of probes for proteinaceous and non-proteinaceous targets. As opposed to the pIII-type fusion phages, which display peptides as independent structural domains, the guest peptides in the pVIII-fusion phages can be structurally and functionally influenced by contiguous subunits. To decipher the impact of the locale of a guest peptide on its affinity characteristics, we constructed a library of phages carrying β-galactosidase-binding peptide ADTFAKSMQ at the N-terminus of the pVIII protein surrounded by random amino acids. It was found that mutagenesis of amino acids 12–19 (domain C) has polar effects on target binding affinity of the displayed peptide. The phages with highest affinity are characterized by: (i) a net electrostatic charge around −1 of domain C of the mutated phages at pH 7.0; (ii) a lower radius of cylinder coaxial to α-helix formed by domain C; (iii) a lower higher occupied molecular orbital (HOMO) of domain C leading to a decreased formation of hydrogen bonds and (iv) positively charged surface and torsion energy of domain C, which may require a conformational transition of N-terminal peptide ADTFAKSMQ for its binding with β-galactosidase. Influence of the guest peptide on the diversity of mutations in the neighboring landscape area was also observed.
Keywords: β-galactosidase, landscape phage, major coat protein pVIII, phage library
Introduction
Phage display technology emerged as a synergy of two fundamental concepts: combinatorial peptide libraries and fusion phage. The first concept replaced the traditional collections of natural or individually synthesized compounds for libraries of peptides obtained in parallel synthesis as grouped mixtures (Geysen et al., 1984; Houghten, 1985) [reviewed in (Geysen et al., 2003)]; the second—allowed displaying foreign peptides on the surface of bacterial viruses (bacteriophages) as part of their minor or major coat proteins (Smith, 1985; Ilyichev et al., 1989) [reviewed in (Smith and Petrenko, 1997; Petrenko and Smith, 2005)]. Combination of these two concepts resulted in the development of phage display libraries—multibillion-clone compositions of self-amplified and self-assembled biological particles. In particular, a paradigm of landscape phage libraries evolved, in which fusion phages serve not just as genetic carriers for foreign peptides, like in the traditional phage display approach, but are considered as nanoparticles (nanotubes) landscaped by thousands of random peptides, which determine emergent physico-chemical characteristics of these new nanomaterials (Petrenko et al., 1996). These constructs display the guest peptide on every pVIII subunit increasing the virion's total mass by 15%. Despite the extra burden, such particles can retain their infectivity and progeny-forming ability.
Interest in landscape phage as a selectable nanomaterial with many desirable properties has shown a tremendous increase over the past years with numerous applications (Brigati et al., 2008; Petrenko, 2008). Despite the intrinsic potential of the landscape phages as a novel class of bioselective nanomaterials, structural intricacies in fusion phage architecture remain unresolved, the most important being the structural and functional independence or otherwise of the guest peptide. Data generated to date generates two diametrical hypotheses. The first one assumes a close interrelation between a guest peptide and the phage capsid because of the accommodation of the peptide in the groove formed by neighboring pVIII subunits on the phage surface (Kishchenko et al., 1991; Kishchenko et al., 1994). According to this model, the conformation of the foreign peptides is heavily constrained and their length is limited by the groove size (17–20 Å, up to 6–8 amino acids). In keeping with this model, guest peptides of more than 10 amino acids are badly tolerated by phage (Iannolo et al., 1995). This ‘constrained peptide’ hypothesis may also explain why some fusion phage peptides lose their binding efficiency when they are chemically synthesized as stand-alone entities or when phages are transformed into spheroids (Petrenko and Smith, 2000).
The second hypothesis in contrast implies that foreign peptides in the landscape phages exist as independent domains with intrinsic conformations much in the same way as they operate in a synthetic form or in the type 3 phage vectors (Malik et al., 1996). The high frequency of display achieved for peptides as long as 16 residues on hybrid virions composed of a mixture of fusion and wild-type pVIII proteins (Malik et al., 1996) creates serious doubts that residence of the pVIII-displayed peptides in a groove can be obligatory. Also, a synthetic peptide corresponding to the phage-displayed streptavidin-binding peptide inhibited binding of the phage to a streptavidin-coated matrix manifesting its intrinsic functional activity in a phage-free form (Petrenko et al., 1996). Furthermore, the successful use of landscape phage libraries for epitope-mapping (Petrenko et al., 1996) supports this concept since epitopes in the antigen scaffolds are presented in their inherent (mostly looped) conformations and it is hard to believe that the phage-constrained peptides could behave as mimotopes and bind corresponding antibodies without paying a considerable entropy fee. Solid-state NMR spectroscopy of landscape phages also coincide with the ‘independent domain’ hypothesis demonstrating that small peptides adopt persistent and biologically relevant conformations, similar to that in the native parent protein, when inserted in the N-terminal region of the pVIII of filamentous bacteriophage fd (Jelinek et al., 1997; Monette et al., 2001).
In this study, we created a novel library, G-α-library, composed of phage clones with two modifications in their major coat protein. These phage clones possess a fusion phage protein which consists of a β-galactosidase-binding peptide at the N-terminus with random mutations in amino acids 12–19 adjoining the guest peptide. We investigated the effects of these mutations on the binding affinity of the peptide towards its target, β-galactosidase (hereafter designated as β-Gal). The results of this study may help reconcile these contradictory hypotheses demonstrating that a functionally significant interaction between the displayed peptides and phage capsid depends on the structure of the peptides and electrostatic charge of amino acids of the adjacent pVIII subunits.
Materials and methods
Bacterial strains, phages and general procedures
Escherichia coli strains: K91BlueKan (Kanr) {Hfr C thi lacZ ΔM15 lac Y::mkh lacIQ} and MC1061 (Strr) {F− araD139 Δ(ara-leu)7696 galE15 galK16 Δ(lac)X74 rpsL (Strr) hsdR2 (rk− mk+) mcrA mcrB1} were kindly provided by George Smith (Yu and Smith, 1996). Oligonucleotides were obtained from Integrated DNA Technologies, Inc. (Coralville, IA, USA). Phages f8-5, 1G40 and f8α-library were described previously (Petrenko et al., 1996; Petrenko and Smith, 2000; Petrenko et al., 2002). All general procedures employed for construction of the libraries, production and analysis of recombinant phages, DNA sequencing, media and buffers, selection procedures are detailed in recently published protocols (Petrenko and Brigati, 2007; Brigati et al., 2008) and previous publications.
Enzyme-linked immunosorbent assays (ELISA)
In the direct format, immobilized phage was used to detect and bind β-Gal in solution. Briefly, candidate and control phages were immobilized in the wells of a 96-well plate followed by the addition and incubation of β-Gal for 1 h at RT. After washing the plates, the binding of β-Gal to phage was quantitated by the addition of ONPG solution (o-nitrophenyl-β-D-galactopyranoside, Sigma, prod. no. 1127) and monitoring the absorbance at 490 nm.
In the competition format, adsorbed phage 1G40, a known β-Gal binder, plays the role of a detector that binds to unbound β-Gal in solution after its pre-incubation with a tested phage. Briefly, phage 1G40 was immobilized in the wells of a Costar 96 well plate. Candidate or control phage clones were mixed with β-Gal and incubated for 1 h at RT. Aliquots of the mixtures were added to 1G40 coated wells of the enzyme-linked immunosorbent assays (ELISA) plate and incubated for 1 h at RT. After washing the plates, the binding of β-Gal to phage was quantitated by the addition of ONPG solution (o-nitrophenyl-β-D-galactopyranoside, Sigma, prod. no. 1127) and monitoring the absorbance at 490 nm. The data for each clone was fitted with a logistic function to derive sigmoidal dose-response curves using Origin software (Originlab Corporation, Northampton, MA, USA) and the phage concentration causing a 50% inhibition of the normalized ELISA signal was estimated. The dissociation constants (Kd) for each of the phage clones were then estimated in comparison to the Kd of phage 1G40, 30 nM (Petrenko and Vodyanoy, 2003) based on the premise that Kd values of the phages are proportional to the phage concentrations at the point of 50% inhibition of β-Gal binding to the indicator (immobilized) 1G40 phage, as follows from the law of mass action (Duschl., 2002). For convenience, the phage clones have been designated by the amino acids they possess at positions 12–19 of pVIII protein. The concentrations of filamentous phages for use in assays were determined spectrophotometrically (physical titer) using the general formula (Barbas et al., 2001):
For the recombinant phage from the library f8/8 (9198 nucleotides), the specific formula is:
Sequencing of the mutant pVIII proteins
To confirm that characterized phages bear the intact β-Gal-binding peptide ADTFAKSMQ at the N-terminus of the fused pVIII protein, we used N-terminal amino acid sequence analysis. Portions (2 µg) of phage proteins (corresponding to 4 × 1010 phage virions) were separated by electrophoresis on 4–20% Precise Protein Gel (Pierce, Prod. no. 25204) in HEPES-SDS buffer (0.1 M Tris, 0.1 M HEPES, 0. 1% SDS, pH 8.0) for 30 min at constant current 100 mA. Proteins were electro-blotted onto PVDF membrane (Millipore Corporation, Cat No IPVH09120, 0.45 µm) in transfer buffer (25 mM Tris, 192 mM glycine, 10% MeOH, pH 8.3) for 1.5 h at constant voltage 300 V; membrane was stained (0.25% Brilliant Blue R in 40% methanol, 7% acetic acid) for 15 min, and destained in 40% methanol, 10% acetic acid. Membrane fragments with the bands corresponding to pVIII proteins were cut and sequenced in Protein Core Lab of Baylor College of Medicine (Houston, TX, USA) to confirm that all phages carried the β-Gal binding peptide ADTFAKSMQ at the N-terminus of pVIII (data not shown). These observations were reiterated by mass spectrometry of pVIII proteins from relevant phages (Midwest Bio Services, data not shown). pVIII proteins for the mass spectrometry were obtained as for sequencing with the exception that the gel fragments containing pVIII proteins were used directly for the analysis.
Statistical analysis of phage populations in the G-α- and f8α-libraries
Phage populations were characterized using the RELIC suite of bioinformatics tools (Mandava et al., 2004). The program AAFREQ was used to analyze the frequency of amino acid occurrence in the random peptides of the G-α (77 clones), f8-α (62 clones) and amplified f8-α (108 clones) libraries. The ‘Frequency Disparity Index’ of the libraries was calculated as outlined previously (Kuzmicheva et al., 2009).
Electrophoresis of whole mutant phage virions
Mobility of different phages in an electric field was characterized by a constant voltage electrophoresis of whole phage particles (3 × 1010 virions) in 0.8% agarose gel in 4× GBB buffer (168 mM Tris base, pH 8.3 adjusted with glacial acetic acid; 80 mM NaOAc, 7.2 mM Na2EDTA) for 24 h at 4°C. The gel was soaked in 0.2 N NaOH for 1 h at RT on rocker, washed once with water and neutralized with 1 M Tris–HCl, pH 7.0 for 15 min at RT on rocker. The gel was stained with SYBR Green I Nucleic Acid Gel Stain (Cambrex ScienceRockland Inc., 15 µl/40 ml in 4× GBB buffer) by incubation for 1 h at RT on rocker and washed with water. Following final rinse, DNA of the filamentous phages were visualized at 521 nm using a KODAK 290 Electrophoresis Documentation and Analysis system (Eastman Kodak Company, Rochester, NY, USA), equipped with a DR40 Kodak camera, DR40 amber camera filter (Clare Chemical Research, Denver, CO, USA) and dark reader transilluminator (Clare Chemical Research). Images (exposure = 8 s) were captured and processed electronically using Kodak 1D software (Version 3.5.3, Eastman Kodak Company).
Computational methods
The conformational search of the peptides has been performed within MulgiGen algorithm using combined MM3/MERA force field with continual account of solvent (Potemkin et al., 2002; Aladko et al., 2006). Calculation of surface has been made according to Connolly algorithm (Connolly, 1983, 1985). Energy of torsion stresses has been computed using MM3 force field. The quantum-chemical computations of atomic charges and orbital energies have been fulfilled within semi-empirical approach PM3 using GAMESS software (Schmidt et al., 1993). Also, physico-chemical properties of amino acids affecting a peptide's affinity to the target was evaluated using the programs WebProAnalyst (Ivanisenko et al., 2005) and PROANAL Version 2 (Eroshkin et al., 1995).
Results
The pVIII major coat protein units of filamentous phages of Ff class (f1, fd, M13) are assembled in a helical lattice to form a tubular capsid around the viral DNA. Half of the pVIII subunit (N-terminal amino acids 1–24) is exposed on the phage surface, while the other half (amino acids 25–50) is buried into the phage virion with the C-terminal segment exposed into the lumen occupied by viral DNA (Fig. 1) [for review of phage structure, see (Marvin, 1998)]. For explicatory convenience, we have arbitrarily divided the exposed moiety of the fusion pVIII protein in a landscape phage into four hypothetical domains A–D: domain A (shown in white ranging from Ala1 to Asp5 incorporating the guest peptide), domain B (shown in grey ranging from Pro6 to Phe11), domain C (shown in yellow ranging from Asp12 to Thr19) and domain D (shown in red ranging from Glu20 to Ala25).
Fig. 1.

Fragment of three-dimensional structure of landscape phage, pVIII protein, fragment of nucleotide sequence of pVIII gene and amino acid sequence of pVIII protein in landscape phages (A). Fragment of three-dimensional structure of landscape phage composed from subunits of major coat protein pVIII (B). White areas correspond to the position of the guest peptides (domain A), grey—to the 5–11 amino acids (domain B), yellow—to the area of 12–19 amino acids ( domain C), and red—to the area of 20–24 amino acids exposed at the phage surface ( domain D). Area corresponding to 25–50 amino acids buried inside the capsid and is not visible. (B) Fragment of three-dimensional structure of pVIII coat protein bearing the β-galactosidase binding peptide. (C) Fragment of DNA and amino acid sequence of mature pVIII in landscape phage 1G40. (D) Fragment of DNA and amino acid sequence of mature pVIII in G-α-library. Numbers correspond to the numbers of amino acids in wild-type pVIII protein.
To ascertain the effect of the capsid structure on the functional behavior of N-terminal displayed foreign peptides, we built an experimental model in which a β-Gal specific phage peptide (DTFAKSMQ) was inserted between Ala1 and Asp5 at the N-terminus of pVIII which also had random substitutions in its domain C, thereby essentially randomizing the environs of the guest peptide (Fig. 1D). We then traced the binding affinities of the β-Gal specific peptide as a function of its contiguous amino acids. Also, the question of whether the guest peptide exerts a censoring influence on its neighboring amino acids was addressed.
Construction and biopanning of G-α-library
A new library termed G-α-library was created by cloning a synthetic oligonucleotide duplex encoding the peptide DTFAKSMQ into the gpVIII gene of members of the previously designed f8α-library (Petrenko et al., 2002). The ligated RF DNAs were electroporated into competent cells E.coli MC1061 yielding 1.2 × 106 transformed primary clones of a novel library. The yield of transformed clones was unexpectedly low considering the high competence of the cells (3.3 × 108 cfu per 1 µg of RF DNA) and amount of DNA (4 µg) used for transformation imputing poor viability of a major portion of the transformed bacterial clones. The final library containing 8.4 × 1013 phage virions was obtained by overnight amplification. The library was characterized by sequencing 74 randomly picked primary clones, obtained by plating a portion of electroporated bacterial cells, before their amplification. A total of 59% (44/74) of the clones revealed frame shifts and stop codons in pVIII gene and were unable to produce phage particles. Among the remaining, seven clones had the desired characteristics of displaying the β-Gal binding peptide as well as random amino acids in positions 12–19. Nineteen clones resembled members of the parental f8α-library, whereas one clone displayed the β-Gal binding peptide but had no mutations in amino acids 12–19. Three clones were without the insert and mutations (a parental vector f8-5 used for design of the f8α-library). All seven clones of interest produced phage virions when grown individually. Based on this analysis, we estimated that the G-α-library contains ∼2.8 × 105 unique mutant clones bearing β-Gal-binding peptide.
Biopanning against β-Gal was carried out to enrich phage library with clones displaying the desired peptide as well as to identify β-Gal binders with affinities higher than that of the parent phage 1G40 (Petrenko and Smith, 2000). Accordingly, conditions for the affinity selection were gradually made more stringent across the three rounds that were performed. Phage recovery increased during each round of selection (0.3% in the first, 0.8% in the second and 2.4% in the third round), indicating the specificity of the selection procedure. Sequence analysis of randomly picked clones from each round demonstrated an increasing portion of clones bearing β-Gal-binding peptide: 23% in original G-α-library, 58% (94/162) after the first round of selection, 82% (36/44) after the second round and 93.5% (43/46) after the third round.
Analysis of the phage binding to β-galactosidase by direct and competition ELISA
Eighty-five unique phage clones isolated in affinity selection (42, 14, 21 from the I, II, III rounds of selection, respectively) carrying β-Gal-binding peptide were initially screened by direct ELISA (data not shown) and representative clones from both ends of the binding spectrum were analyzed by direct and competition ELISA (Table. I). This latter format excludes a possibility that ELISA signals of landscape phages vary because of their different adsorption to the plastic. In the competition assay, adsorbed phage 1G40 plays the role of a detector that binds to unbound β-Gal in solution after its pre-incubation with a tested phage; as opposed to direct ELISA, the signal in the competition assay is inversely proportional to the apparent affinity of the phage to β-Gal. To further estimate affinities of representative clones identified by screening tests, serial dilutions of selected phages were subjected to the competition ELISA with 1G40 as the adsorbed detector (Fig. 2). Results revealed that the mutations had a variety of effects on the phage apparent affinity ranging from no effect like in phage DSLTLQAQ (mutations are underlined) to enhanced apparent affinity (DSLHGQAM, DQLNATAL, AELTTRAE), while other mutations lowered (EDLTQRAL, SNLEMMAT, DQLTVSAQ, ADLTVQAN and DTLTHEAT), or completely nullified ability of the phages to recognize and bind β-Gal (36 clones, Table I). To confirm that the modulation of the phage apparent affinity is not related to a change in the processing of the fusion pVIII precursor, or caused by degradation of the fusion proteins in the culture, we determined N-terminal amino acid sequences of parental phage 1G40, its analogue DSLHGQAM with improved β-Gal-binding and three representative phages, which completely lost the β-Gal binding activity (DELTVAAN, QELSIQAE and EDLSAMAG). All these clones demonstrated intact N-terminal sequence ADTFAKSMQD. These data were confirmed by mass spectrometry analysis of appropriate fused pVIII proteins.
Table I.
Direct and competition ELISA of phages
| Phage | Normalized ELISA signal |
Electrostatic chargec |
|
|---|---|---|---|
| Directa | Competitionb | ||
| Strong β-Gal binders | |||
| DSLQASATd | 100 | 17 | −1.090 |
| ADLTVQAN | 97 | 2 | −1.090 |
| AELTTRAE | 94 | 3 | −1.086 |
| DQLNATAL | 113 | 1 | −1.090 |
| DQLTVSAQ | 84 | 18 | −1.090 |
| DSLHGQAM | 157 | 1 | −0.924 |
| DSLTLQAQ | 79 | 3 | −1.090 |
| DTLTHEAT | 63 | 2 | −1.921 |
| EDLTQRAL | 66 | 3 | −1.088 |
| SNLEMMAT | 100 | 1 | −1.088 |
| Clones which lost β-Gal binding ability | |||
| DDLTASAI | 1 | 130 | −2.090 |
| DELITEAH | <1 | 160 | −2.919 |
| DELTVAAN | 0 | 142 | −2.088 |
| DLLTTQAE | <1 | 94 | −2.088 |
| DNLEMMAQ | 1 | 104 | −2.088 |
| DNLITMAD | <1 | 105 | −2.090 |
| DSLNEQAV | 1 | 138 | −2.088 |
| DTLTENAV | 0 | 160 | −2.088 |
| EDLNAQAL | 1 | 113 | −2.088 |
| EDLSAMAG | 1 | 115 | −2.088 |
| EELESIAN | <1 | 129 | −3.083 |
| EELNGQAM | 1 | 130 | −2.086 |
| EELNQQAN | 1 | 104 | −2.086 |
| EELSNSAT | 2 | 123 | −2.086 |
| EELSQQAN | 1 | 153 | −2.086 |
| EELSVQAT | 1 | 107 | −2.086 |
| EELTLEAH | 1 | 146 | −2.917 |
| EELTNSAQ | <1 | 168 | −2.086 |
| ELLEAQAK | 2 | 94 | −1.087 |
| ELLTDQAS | <1 | 122 | −2.088 |
| ENLNEVAM | 1 | 130 | −2.086 |
| EQLENQAI | 2 | 123 | −2.086 |
| EQLEVQAS | 1 | 120 | −2.086 |
| EQLSSEAN | 1 | 179 | −2.086 |
| EQLSSNAE | 0 | 132 | −2.086 |
| EQLTVEAS | 2 | 113 | −2.086 |
| ESLAAEAT | 1 | 152 | −2.086 |
| ESLEAQAT | 1 | 137 | −2.086 |
| ESLTENAY | <1 | 133 | −2.087 |
| ETLENMAS | <1 | 97 | −2.086 |
| ETLEQQAT | 1 | 114 | −2.086 |
| ETLEVQAQ | 2 | 111 | −2.086 |
| ETLTESAA | 1 | 148 | −2.086 |
| LDLHEAAV | <1 | 135 | −1.921 |
| QELSIQAE | 1 | 119 | −2.086 |
| SLLSNQAE | 1 | 131 | −1.088 |
| Control phagee | <1 | 115 | −1.090 |
| No phage | 0 | 100 | – |
aELISA signals are normalized against the 1G40 phage ELISA signal. bELISA signals are normalized against the signal of the sample without phage. cThe summary electrostatic charges of 12–19 amino acids of pVIII were estimated by employing DNA Star software. dOriginal 1G40 phage with non-modified ‘wild-type’ domain C 12–19. eControl phage without β-Gal-binding peptide.
Fig. 2.

Binding of different phages with β-galactosidase as measured by inhibition ELISA. Phages displaying indicated peptides were pre-incubated at graded concentrations with β-Gal and then added to the wells of ELISA plate coated by detection phage 1G40. Normalized ELISA signals are presented as percentage of signal from the sample without phage, which was considered as 100%. The phage concentrations were determined by spectrophotometric measurement (For details, see Materials and methods).
Since phage mutants whose affinities to β-Gal appear to be modulated demonstrate neither common motifs nor distinctive structural features (Table I) in the domain C, we investigated putative physico-chemical reasons for this modulation. Of the more than 200 physico-chemical parameters of domain C, like alpha-CH chemical shifts, hydrophobicity index, conformational parameter of inner helix, beta-structure and beta-turn that were analyzed, we found a significant correlation between the net charges of amino acids 12–19 in domain C of the mutated phages at pH 7.0 and their apparent affinity towards β-Gal as demonstrated by the ELISA assays (R = 92%, P < 0.05). Most of the strong binders had an electrostatic charge around −1 (the best binder DSLHGQAM having the charge −0.924), whereas phages which lacked β-Gal binding usually displayed a charge between −2.0 and −3.0. The comprehensive relationship is demonstrated in a scatter plot in Fig. 3A. The theoretical estimate of the net electrostatic charge on the select amino acids was confirmed by measuring their mobility in an electric field. Since each analyzed landscape phage carries at its surface 4000 copies of fused pVIII protein, even a small difference in the charge of the mutated amino acids leads to a significant difference in the phage mobility in agarose gel electrophoresis. Data obtained for the phages with different charges in mutated area (−0.924, −1.090 and −2.917) are shown on Fig. 3B. As expected, the mutant phage EELTLEAH with the highest negative charge of the mutated area (−2.917) migrates faster than phage 1G40 (−1.090), and phage DSLHGQAM (−0.924). Phages with charges between −1.090 and −2.917 demonstrated intermediate mobility (data not shown).
Fig. 3.

(A) Scatter plot representation of the normalized ELISA signals correlated to the charge carried on the phage. Selected phage clones were probed for their affinity towards β-galactosidase using different formats of ELISA. The theoretical charge borne on the phage particles was estimated using the program PROTEAN of DNAStar suite. The high affinity clones demonstrating a high normalized direct ELISA signal and a low normalized competitive ELISA signal have a charge ∼−1, whereas low affinity clones demonstrating a low normalized direct ELISA signal and a high normalized competitive ELISA signal have charges clustered between −2 and −3. (B) Mobility of different phages in 0.8% agarose gel/4×GBB electrophoresis. 1, Molecular marker (DNA λ-BstE). 2, Vector phage f8-5 (amino acids EGE instead of the guest peptide). 3, Phage EELTLEAH with the charge of 12th–19th amino acids of pVIII protein −2.917. 4, Phage 1G40 (DSLQASAT) with the charge of 12th–19th amino acids of pVIII protein −1.090. 5, Phage DSLHGQAM with the charge of 12th–19th amino acids of pVIII protein −0.924. 6, G-α-library after the first round of selection.
To reveal other characteristic features of the phage mutants that may correlate with their binding activity, we used quantum mechanics (PM3) and molecular mechanics (MM3 force field) computations to determine possible conformations, electronic, energy and geometrical characteristics of the domain C in pVIII (Table II). First, we optimized the geometric parameters of the domain C in the combined force field MM3/MERA with continuous calculation of effect of the environment and observed that in the process of optimization all octapeptides constituting domain C form α-helixes, which corresponds well to previous observations (Petrenko et al., 2002). Then, we searched for conformers in the energy range of 3 kcal/mole, relative to a conformer corresponding to the global minimum of potential energy. It was found that many octapeptides are conformationally mobile systems and may be presented in the solution by a set of conformers, as shown in Table II. We calculated different characteristics of the conformers (more than 250 characteristics for each conformer of each octapeptide) using semi-empirical quantum-chemical approximation RM3, the methods of molecular mechanics (force field MM3) and model MERA, taking into account the characteristics of the most advantageous conformer for each peptide, as well as weighted mean characteristics of all its conformational states. Any weighted mean characteristics (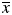 ) of the octapeptide were calculated as a sum of partial contributions of хi conformers:
) of the octapeptide were calculated as a sum of partial contributions of хi conformers:
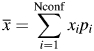 |
where Nconf is the number of conformers; Pi the probability to reveal the conformer was calculated using the formula:
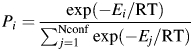 |
where Еi and Еj are the energy of conformers i and j, respectively.
Table II.
Calculated characteristics for phages with mutations in 12–19 amino acids of pVIII protein
| Structure of 12–19 amino acids | Phage concentrationa, vir ml−1 | Kd, nM | N | lg Kd | S+, Å2 | ET, kcal mol−1 | rc, Å | HOMO, eV |
|---|---|---|---|---|---|---|---|---|
| DSLQASAT | 3.2 × 1010 | 30.0 | 94 | −7.52 | 171.887 | 17.393 | 6.282 | −4.669 |
| DSLHGQAM | 1.6 × 1010 | 15.0 | 173 | −7.82 | 162.731 | 11.586 | 6.504 | −7.122 |
| DQLNATAL | 1.9 × 1010 | 17.8 | 255 | −7.75 | 180.435 | 25.520 | 6.321 | −6.297 |
| AELTTRAE | 2.5 × 1010 | 23.4 | 3404 | −7.63 | 180.415 | 23.990 | 6.472 | −3.777 |
| DSLTLQAQ | 4.0 × 1010 | 37.5 | 113 | −7.43 | 176.641 | 21.736 | 6.769 | −3.414 |
| ADLTVQAN | 5.0 × 1010 | 46.9 | 240 | −7.33 | 174.261 | 16.383 | 6.772 | −3.256 |
| SNLEMMAT | 6.0 × 1010 | 56.3 | 1043 | −7.25 | 176.313 | 16.230 | 6.710 | −2.804 |
| DQLTVSAQ | 6.0 × 1010 | 56.3 | 71 | −7.25 | 187.865 | 22.031 | 6.938 | −3.636 |
| DTLTHEAT | 8.0 × 1010 | 75.0 | 30 | −7.12 | 185.024 | 17.715 | 6.510 | −4.684 |
| EDLTQRAL | 27.0 × 1010 | 253.1 | 376 | −6.60 | 201.178 | 18.395 | 6.985 | −3.640 |
Kd, constant of dissociation. N, number of conformers. S+, area of the positively charged surface. ET, energy of torsion stresses. rc, radius of the described cylinder. HOMO, energy of the upper occupied molecular orbital. aPhage concentration (vir ml−1) estimated at the point of 50% binding of β-Gal in competition ELISA. The phage concentrations were determined by spectrophotometric measurement (for details, see Materials and methods).
The dissociation constant tended to increase with an increase in practically any extensive characteristic of the octapeptide like number of atoms, molecular weight, molecular volume to name a few. Since thermodynamic binding events depend exponentially on energy and electronic characteristics, dissociation constants were log-transformed for calculation of trends. Several important characteristics were identified through these analyses. One was the radius of the octapeptide constituting domain C which is conformationally constrained to be an α-helix. This radius was lower (6.39 ± 0.11 Å) for the more active phages when compared with the others (6.78 ± 0.17 Å) hinting at a inverse relationship between the size of the amino acid residues in domain C and phage affinity to its target. Bulkier amino acids (larger radii) tend to increase the dissociation constant (and hence lower affinity) and vice versa. However, this dependency can be considered only as a tendency, since the correlation coefficient of data is not high (∼0.76). A slightly better correlation (0.82) albeit negative is observed between phage affinity and the positive charges on the surface of octapeptide 12–19 (Fig. 4). Positive charges on domain C may favor an interaction with the terminal aspartate in the N-terminal peptide ADTFAKSMQ causing a conformational turn in the peptide precluding its binding to β-galactosidase. However, the incorporation of negatively charged amino acids into domain C also resulted in a loss of affinity of the N-terminal peptide to β-galactosidase. This apparent contradiction can be resolved if one considers the relative area of the charged surface that is exposed for interaction in the domain C. Positive charges in amino acids are generally borne by hydrogen atoms which have an extensive area of exposure (and the larger the amino acid, larger will be this area), whereas the negative charges of a peptide are localized on the carboxyl groups which occupy a relatively smaller surface. Thus, size of the amino acids and the relative area of exposure of the charges over-ride the net charge on domain C to affect affinity of the N-terminal peptide.
Fig. 4.

Correlation between the logarithm of dissociation constant (lg Kd) and octapeptide positively charged surface square (S+). The circle encloses the peptides, DQLNATAL and AELTTRAE. Without these peptides, the R would increase to ∼95%. These peptides are distinguished by high energies of torsion angle stress.
Two peptides, DQLNATAL and AELTTRAE (enclosed by circle on the graph, Fig. 4), slant from this dependence; without these points, the correlation coefficient for the positively charged surface versus the lg Kd would be 0.95. The distinguishing feature of these octapeptides is sufficiently high energies of the torsion angle stress (25.5 and 24.0 kcal/mole, accordingly); for other samples, this value is essentially less (17.7 ± 3.3 kcal/mole). High steric hindrances ensure the energy proximity for the large number of conformers, which, in turn, can increase the conformational mobility of amino acid residues and prevent possibility for the ‘internal’ association of the N-terminal peptide with the mutant region. The dependence of the logarithm of dissociation constant upon the energy of torsion stresses (force field MM3) and upon the positively charged surface is represented by the equation:
| (1) |
The experimental Log Kd values agree quite well with the calculated ones as depicted in Fig. 5.
Fig. 5.

Experimental (lg Kd, exp.) and calculated (lg Kd, calc.) values of logarithm of dissociation constant (lg Kd). Log Kd calculated =−0.7297 + 0.901*x.
Another important feature of high affinity peptides is the low energy of the higher occupied molecular orbital (HOMO) (−5.7 ± 1.7 eV) when compared with the phages with lower affinity (−3.73 ± 0.71 eV). The low energy level of the HOMO ensures that the electrons serve as weak donors of electron pairs and poor proton acceptors in the formation of hydrogen bonds. Highly active phages, with affinity higher than for the wild-type peptide DSLQASAT, and low-active phages (all other) are separated well in the factor space HOMO—Nconf (number of conformers) (Fig. 6), in which the active phages are characterized by low energy HOMO of the octapeptides 12–19. Thus, to ensure a high constant of binding (low dissociation constant), domain C must include predominantly small amino-acid residues, possess a diminutive positively charged surface and a low energy of the HOMO.
Fig. 6.

High active phages (shown by squares) and low active phages (shown by circles) in the factor space: energy of the upper occupied molecular orbital (HOMO)—number of conformers (Nconf).
Comparative analysis of parental f8α-library and G-α-library: complexity and censoring
Our data demonstrate that the binding affinity of a phage-displayed peptide towards its cognate target may be affected by the contiguous amino acids, especially if these amino acids are conformationally constrained as is the case in our model. A very interesting take on this tableau is the extent to which the N-terminal peptide controls its amino acid environs by a censoring process which may lead to exclusion of untenable amino acids. We studied this utilizing RELIC suite of programs. The theoretical frequencies at different positions of domain C of pVIII were compared with the observed frequencies at the same positions and a modified one-sample test for proportions was used to obtain a statistical indicator (Frequency Disparity Index or FDI) of over-representation or under-representation of amino acids at different positions [for details of calculation and statistical formula, refer (Kuzmicheva et al., 2009)].
Figure 7 shows the FDI profiles in positions 12, 13, 15–17 and 19 of pVIII protein in the original f8α-library (7A), amplified f8α-library (7B) and amplified G-α-library (7C). Positively charged amino acids lysine and arginine were underrepresented in all libraries while negatively charged amino acids (aspartic and glutamic acids) were overrepresented. There were no dramatic differences in the dispersion profiles between the original f8α-library and the amplified f8α-library. In contrast, the G-α-library demonstrated a dispersion of the amino acids quite different from the parental f8α-library, with an increased prevalence of negatively charged amino acids (aspartic and glutamic acids) and under-representation of phenylalanine, glycine, histidine, tyrosine and positively charged amino acids (lysine and arginine). There appeared to be an overall decrease in the diversity of amino acids occurring in domain C imputing a censoring impact of the N-terminally displayed peptide on its amino acid environment.
Fig. 7.

Frequencies of amino acids at different positions in the mutated area of 12–19 amino acids of pVIII proteins. The expected frequency of amino acids and the observed frequency calculated using AAFREQ program of the RELIC suite (Mandava et al., 2004) were used in adaptation of the one-sample test for proportion (Rosner, 2006). The results of the analysis were expressed as a frequency disparity index (FDI) which is a measure of how much the observed frequency deviates from the expected one. Amino acids at various positions of the mutated area (amino acids 12, 13, 15, 16, 17, 19) are depicted along the x-axis whereas the y-axis represents the FDI. FDIs for each amino acid are represented by clusters of columns, in which positions of each column correspond to the amino acid's position of the mutated area (see also Fig. 1). The dashed line represents statistical significance of the frequency disparity at the α-level of 0.05 (For more details see Kuzmicheva et al., 2009). (A) Parental f8α-library. (B) Amplified f8α-library. (C) G-α-library after the first round of affinity selection.
Discussion
Previous research conducted with landscape phage libraries using β-galactosidase as model target had resulted in the selection of phage clones that lost their ability to bind to the target after denaturing treatment leading to the conclusion that guest peptide has to be in complex with the surrounding phage body to exhibit target binding. As a follow-up study, we investigated in further detail the intricacies involved in this effect by creating a new library which had a β-galactosidase-specific peptide in all its members but also unique mutations in the vicinity of the foreign peptide (domain C). The present research shows that the domain C of pVIII may influence the behavior of the N-terminal displayed peptide and this effect is the culmination of numerous energy-related, charge-related and structure-related factors. Thus, the binding affinity of a peptide to its target may be positively or negatively impacted by the amino acid profile of domain C based on the manifold interactions between the foreign peptide and its environ amino acids. For example, in the case under discussion, the size of the amino acids occupying the helix of domain C: larger amino acids tend to have a deleterious effect on the guest peptide's affinity towards its target. This effect can be attributed to a number of causes; first, steric hindrances for the interaction of pVIII with its target; second, a higher degree of solvation which may interfere with the processes of peptide–peptide interaction; third, greater impedance to the dynamics of motion.
The study of the amino acid dispersion profiles of libraries demonstrated the expected absence of cysteine, proline and tryptophan in 12–19 amino acids of pVIII. Cysteine tends to form inter- and intra-molecular bridges in many random phage display libraries (Koivunen et al., 1995; Smith and Petrenko, 1997; Zwick et al., 2000; Cano et al., 2004) which combined with the dense presentation of pVIII interferes dramatically with phage assembly and survival (Petrenko et al., 1996; Petrenko et al., 2002). Proline which is responsible for the β-turn structure in proteins would not be feasible in the α-helically constrained domain C of pVIII (Petrenko et al., 2002), whereas the bulk of tryptophan precludes its fit into an α-helix. The general decrease in amino acid diversity that is observed implies that the guest peptide has a screening effect on the amino acids that could occur in its vicinity. The intricate networks of intermolecular bonds that occur in a conformationally constrained environment can be expected to mutually influence neighboring subunits affecting their physico-chemical performance. These observations revisit our earlier conclusion and suggest an interactive landscape on the phage surface with unique structural and functional characteristics.
Although, the data generated within the context of this paper agree with the constrained peptide hypothesis, this cannot be used as a foregone conclusion for all phage peptides and targets. Research involving alternative targets in our laboratory has shown that disassembled phage coat protein still possesses cognizance of its target even when instilled in a lipid bi-layer. This has shown to be true for a streptavidin-specific phage peptide (Jayanna et al., 2009) as well as several tumor-specific phage peptides (unpublished results). Thus, the effect exerted by the contiguous amino acids on the affinity of the displayed peptide has to be considered on a case-wise basis and may be an important factor to consider when harnessing landscape phage peptides for functional applications.
Phage display is becoming the technology of choice in rapid identification of high affinity ligands against a wide variety of targets. The amenities to working with phage displayed ligands have made it a mainstay in high throughput ligand screening systems with far ranging applications embracing both the biological and non-biological arenas. Furthermore, the properties of phage make it a very attractive candidate as a selective and specific homing device which expands the repertoire of applications even more (Yacoby et al., 2007). In particular, biological targets, especially cell surfaces, represent complex targets involving extensive molecular intrigues which create a need sometimes to modulate the affinity of a specific targeting ligand so as to obtain the maximum possible benefit out of the targeting. Experiments outlined within this publication represent an impetus in this direction of improving the performance of phage-derived probes by molecular evolution and affinity maturation of binding sites.
Funding
This work was supported by Army Research Office grant no. DAAD 19-01-10454 (to V.A.P.) and NIH grants no. NIH-1 R21 AI05564501 and 1 R01 CA125063-01 (to V.A.P.).
Acknowledgements
We are grateful to Dr Carpenter from Auburn University (Auburn, AL, USA) for helping us with FDI scores calculations, Dr Robert Cook from Baylor College of Medicine (Houston, TX, USA) for providing us with Erdman N-terminal amino acid sequence analysis of fused pVIII proteins from landscape phages, Dr Irina Sorokina from Midwest Bio Services (Overland Park, KS, USA) for mass spectrometry analysis of fused pVIII proteins from landscape phages and I-Hsuan Chen from Auburn University (Auburn, AL, USA) for excellent technical support.
Footnotes
Edited by Jacques Fastrez
References
- Aladko E.Y., et al. J. Phys. Chem. B. 2006;110:21371–21376. doi: 10.1021/jp061698r. [DOI] [PubMed] [Google Scholar]
- Barbas C.F., III, Burton D.R., Scott J.K., Silverman G.J. Phage Display: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. p. 736. [Google Scholar]
- Brigati J.R., Samoylova T.I., Jayanna P.K., Petrenko V.A. In: Current Protocols in Protein Science. Coligan J.E., et al., editors. New Jersey: John Wiley & Sons, Inc; 2008. pp. 1–27. [Google Scholar]
- Cano A., Viveros M., Acero G., Govezensky T., Munguia M.E., Gonzalez E., Soto L., Gevorkian G., Manoutcharian K. Immunol. Lett. 2004;95:207–212. doi: 10.1016/j.imlet.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Connolly M. J. Appl. Crystallogr. 1983;16:548–558. [Google Scholar]
- Connolly M.L. J. Am. Chem. Soc. 1985;107:1118–1124. [Google Scholar]
- Duschl C. In: Biomolecular Sensors. Gizeli E., et al., editors. London: Taylor and Francis; 2002. pp. 87–120. [Google Scholar]
- Eroshkin A.M., Fomin V.I., Zhilkin P.A., lvanisenko V.V., Kondrakhin Y.V. Bioinformatics. 1995;11:39–44. doi: 10.1093/bioinformatics/11.1.39. [DOI] [PubMed] [Google Scholar]
- Geysen H.M., Meloen R.H., Barteling S.J. Proc. Natl Acad. Sci. USA. 1984;81:3998–4002. doi: 10.1073/pnas.81.13.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geysen H.M., Schoenen F., Wagner D., Wagner R. Nat. Rev. Drug Discov. 2003;2:222–230. doi: 10.1038/nrd1035. [DOI] [PubMed] [Google Scholar]
- Houghten R.A. Proc. Natl Acad. Sci. USA. 1985;82:5131–5135. doi: 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannolo G., Minenkova O., Petruzzelli R., Cesareni G. J. Mol. Biol. 1995;248:835–844. doi: 10.1006/jmbi.1995.0264. [DOI] [PubMed] [Google Scholar]
- Ilyichev A.A., Minenkova O.O., Tatkov S.I., Karpyshev N.N., Eroshkin A.M., Petrenko V.A., Sandakhchiev L.S. Doklady Biochemistry (Proc.Acad. Sci. Ussr)-Engl.Tr. 1989;307:196–198. [Google Scholar]
- Ivanisenko V.A., Eroshkin A.M., Kolchanov N.A. Nucleic Acids Res. 2005;33:W99–W104. doi: 10.1093/nar/gki421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanna P.K., Torchilin V.P., Petrenko V.A. Nanotechnol. Biol. Med. 2009;5:83–89. doi: 10.1016/j.nano.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek R., Terry T.D., Gesell J.J., Malik P., Perham R.N., Opella S.J. J. Mol. Biol. 1997;266:649–655. doi: 10.1006/jmbi.1996.0821. [DOI] [PubMed] [Google Scholar]
- Kishchenko G.P., Minenkova O.O., Il'ichev A.I., Gruzdev A.D., Petrenko V.A. Mol. Biol. (Mosk.) 1991;25:1497–1503. [PubMed] [Google Scholar]
- Kishchenko G., Batliwala H., Makowski L. J. Mol. Biol. 1994;241:208–213. doi: 10.1006/jmbi.1994.1489. [DOI] [PubMed] [Google Scholar]
- Koivunen E., Wang B.C., Ruoslahti E. Bio-Technology. 1995;13:265–270. doi: 10.1038/nbt0395-265. [DOI] [PubMed] [Google Scholar]
- Kuzmicheva G.A., Jayanna P.K., Sorokulova I.B., Petrenko V.A. Protein Eng. Des. Sel. 2009;22:9–18. doi: 10.1093/protein/gzn060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik P., Terry T.D., Gowda L.R., Langara A., Petukhov S.A., Symmons M.F., Welsh L.C., Marvin D.A., Perham R.N. J. Mol. Biol. 1996;260:9–21. doi: 10.1006/jmbi.1996.0378. [DOI] [PubMed] [Google Scholar]
- Mandava S., Makowski L., Devarapalli S., Uzubell J., Rodi D.J. Proteomics. 2004;4:1439–1460. doi: 10.1002/pmic.200300680. [DOI] [PubMed] [Google Scholar]
- Marvin D.A. Curr. Opinion Struct. Biol. 1998;8:150–158. doi: 10.1016/s0959-440x(98)80032-8. [DOI] [PubMed] [Google Scholar]
- Monette M., Opella S.J., Greenwood J., Willis A.E., Perham R.N. Protein Sci. 2001;10:1150–1159. doi: 10.1110/ps.35901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrenko V.A. Expert Opin. Drug Deliv. 2008;5:825–836. doi: 10.1517/17425247.5.8.825. [DOI] [PubMed] [Google Scholar]
- Petrenko V.A., Brigati J.R. In: Immunoassay and other Bioanalytical Techniques. Emon J.M.V., editor. Boca Raton, FL: CRC press, Taylor & Francis Group; 2007. [Google Scholar]
- Petrenko V.A., Smith G.P. Protein Eng. 2000;13:589–592. doi: 10.1093/protein/13.8.589. [DOI] [PubMed] [Google Scholar]
- Petrenko V.A., Smith G.P. In: Phage Display in Biotechnology and Drug Discovery. Sidhu S.S., editor. Boca Raton, FL: CRC Press, Taylor & Francis Group; 2005. [Google Scholar]
- Petrenko V.A., Smith G.P., Gong X., Quinn T. Protein Eng. 1996;9:797–801. doi: 10.1093/protein/9.9.797. [DOI] [PubMed] [Google Scholar]
- Petrenko V.A., Smith G.P., Mazooji M.M., Quinn T. Protein Eng. 2002;15:943–950. doi: 10.1093/protein/15.11.943. [DOI] [PubMed] [Google Scholar]
- Petrenko V.A., Vodyanoy V.J. J. Microbiol. Methods. 2003;53:253–262. doi: 10.1016/s0167-7012(03)00029-0. [DOI] [PubMed] [Google Scholar]
- Potemkin V.A., Arslambekov R.M., Bartashevich E.V., Grishina M.A., Belik A.V., Perspicace S., Guccione S. J. Struct. Chem. 2002;43:1045–1049. [Google Scholar]
- Rosner B. Fundamentals of Biostatistics. Belmont, CA: Thomson Brooks/Cole; 2006. [Google Scholar]
- Schmidt M.W., et al. J. Comput. Chem. 1993;14:1347–1363. [Google Scholar]
- Smith G.P. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Smith G.P., Petrenko V.A. Chem. Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- Yacoby I., Bar H., Benhar I. Antimicrob. Agents Chemother. 2007;51:2156–2163. doi: 10.1128/AAC.00163-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Smith G.P. Methods Enzymol. 1996;267:3–27. doi: 10.1016/s0076-6879(96)67003-7. [DOI] [PubMed] [Google Scholar]
- Zwick M.B., Shen J.Q., Scott J.K. J. Mol. Biol. 2000;300:307–320. doi: 10.1006/jmbi.2000.3850. [DOI] [PubMed] [Google Scholar]