

Abstract
Aromatic compounds comprise a large class of natural and man-made compounds, many of which are of considerable concern for the environment and human health. In aromatic compound-degrading anaerobic bacteria the central intermediate of aromatic catabolism, benzoyl coenzyme A, is attacked by dearomatizing benzoyl-CoA reductases (BCRs). An ATP-dependent BCR has been characterized in facultative anaerobes. In contrast, a previous analysis of the soluble proteome from the obligately anaerobic model organism Geobacter metallireducens identified genes putatively coding for a completely different dearomatizing BCR. The corresponding BamBCDEFGHI complex is predicted to comprise soluble molybdenum or tungsten, selenocysteine, and FeS cluster-containing components. To elucidate key processes involved in the degradation of aromatic compounds in obligately anaerobic bacteria, differential membrane protein abundance levels from G. metallireducens grown on benzoate and acetate were determined by the MS-based spectral counting approach. A total of 931 proteins were identified by combining one-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis with liquid chromatography-tandem mass spectrometry. Several membrane-associated proteins involved in the degradation of aromatic compounds were newly identified including proteins with similarities to modules of NiFe/heme b-containing and energy-converting hydrogenases, cytochrome bd oxidases, dissimilatory nitrate reductases, and a tungstate ATP-binding cassette transporter system. The transcriptional regulation of differentially expressed genes was analyzed by quantitative reverse transcription-PCR; in addition benzoate-induced in vitro activities of hydrogenase and nitrate reductase were determined. The results obtained provide novel insights into the poorly understood degradation of aromatic compounds in obligately anaerobic bacteria.
Aromatic compounds comprise the second most abundant class of natural compounds that can be fully degraded to CO2 by aerobic and anaerobic microorganisms. In aerobic bacteria and fungi the key reactions involved in the degradation pathways of low molecular weight aromatic growth substrates are catalyzed by oxygenases that have been extensively studied in the past 50 years. In contrast, anaerobic microorganisms use a completely different enzyme inventory for the activation of chemically inert side chains or the dearomatization and cleavage of the aromatic ring. In the past 10–15 years, initial insights into the function of novel enzyme reactions involved in the catabolism of aromatic growth substrates in facultative anaerobes have been obtained (for recent reviews, see Refs. 1–4). In contrast, much less is known about the catabolism of aromatic compounds in obligate anaerobes such as Fe(III)-reducing, sulfate-reducing, or fermenting bacteria.
Benzoyl coenzyme A is a key intermediate in the anaerobic degradation of aromatic compounds in both facultative and obligate anaerobes. It serves as substrate for dearomatizing benzoyl-CoA reductases (BCRs)1 that dearomatize the aromatic ring by two-electron reduction yielding cyclohexa-1,5-diene-1-carboxyl-CoA (Fig. 1 and Refs. 5–7). A BCR enzyme has so far only been isolated and studied in the denitrifying, facultatively anaerobic Thauera aromatica (8). The extremely oxygen-sensitive enzyme has an αβγδ composition and harbors three [4Fe-4S]1+/2+ clusters (9). It couples the mechanistically difficult reduction of the aromatic ring to a stoichiometric ATP hydrolysis (10). The coupling of electron transfer to an exergonic reaction is suggested to be essential to overcome the high redox barrier for electron transfer to the aromatic ring. Initial evidence for a mechanism comprising single electron transfer and protonation steps according to the Birch reduction in chemical synthesis has been obtained (11, 12). The further conversion of the dienoyl-CoA product proceeds by enzymes of the so-called benzoyl-CoA degradation pathway yielding the CoA ester of an aliphatic dicarboxylic acid (usually 3-hydroxypimelyl-CoA), which is then further converted to three acetyl-CoA and CO2 by β-oxidation and a decarboxylating glutaryl-CoA dehydrogenase (Fig. 1). No homologues of ATP-dependent BCR enzymes are present in genomes of aromatic compound-degrading, obligately anaerobic Geobacter species or Syntrophus aciditrophicus (13, 14).
Fig. 1.

Gene products involved in the benzoyl-CoA degradation pathway in facultative and obligate anaerobes. Abbreviations for enzymes from facultative anaerobes are: Bcr, benzoyl-CoA reductase (ATP-dependent); Dch, dienoyl-CoA hydratase; Had, 6-hydroxycycloxenoyl-CoA dehydrogenase; Oah, 2-oxocyclohexonyl-CoA hydrolase. The abbreviation for an enzyme from obligate anaerobes is: Bam, benzoic acid metabolism. Additional abbreviations are: Adh, alcohol dehydrogenase (3-OH-acyl-CoA dehydrogenase); Ech, enoyl-CoA hydratase; Acd, acyl-CoA dehydrogenase; Thi, thiolase; EtfAB, electron-transferring flavoprotein subunits A and B; Oxr, oxidoreductase homologous to membrane-bound heterodisulfide reductases.
A previous analysis of the benzoate-induced soluble proteome of the Fe(III)-respiring Geobacter metallireducens separated by 2D gel electrophoresis revealed two benzoate-induced gene clusters (IA/B and II with one IA and IB separated by a transposon element (14)). They comprise genes coding for enzymes of the benzoyl-CoA degradation pathway (bam (benzoic acid metabolism) genes, clusters IA and II) and β-oxidation reactions (cluster IB). Heterologous expression and characterization of BamY (14), BamR (15), and BamA (16) from several strict anaerobes suggested that the benzoyl-CoA degradation pathways are identical in both facultative and obligate anaerobes (Fig. 1). However, a putative protein complex consisting of eight soluble protein components, BamBCDEFGHI (BamB–I), was suggested to replace the ATP-dependent BCR from facultative anaerobes. This assumption was supported by the finding that the genes coding for similar BamB–I complexes are only present in the genomes of obligately anaerobic bacteria that use aromatic growth substrates (13, 14). The individual proteins of the BamB–I complex show similarities to soluble components of NADH:quinone oxidoreductases (BamGHI), electron-transferring components of hydrogenases (BamC and selenocysteine-containing BamF), soluble heterodisulfide reductases (BamDE), and to molybdenum or tungsten-containing aldehyde:ferredoxin oxidoreductase-like proteins (BamB). In accordance, growth of G. metallireducens on benzoate depended on molybdenum/tungsten and selenium, which confirmed the essential role of the BamB–I complex in anaerobic aromatic metabolism of G. metallireducens (14).2 BamB was considered as the active site-containing component, whereas the BamC–I components were suggested to be involved in electron transfer from an unknown donor to BamB. No ATP-binding motif was found in the BamB–I complex, suggesting that electron transfer to benzoyl-CoA may be independent of ATP hydrolysis. Thus, the presence of additional, so far unknown membrane protein components putatively involved in energy conversion processes was hypothesized (14).
To obtain a more complete picture of the processes and protein components involved in the aromatic metabolism of obligately anaerobic bacteria, the membrane proteome of the model organism G. metallireducens grown on benzoate and acetate was analyzed by combining one-dimensional (1D) SDS-PAGE sample fractionation with an LC-MS/MS-based protein identification. The differential protein expression levels and the relative protein abundance levels were determined by the spectral counting approach that is based on the empirical observation that the MS/MS sampling rate of a particular peptide is directly related to the abundance of this peptide in the sample (17–22). The transcriptional regulation of selected genes with annotated function was additionally analyzed by RT-quantitative PCR (qPCR) analysis; where possible, in vitro activities of benzoate-induced enzymes were determined.
MATERIALS AND METHODS
Cultivation of G. metallireducens
G. metallireducens (German Collection of Microorganisms and Cell Cultures (DSMZ) number 7210) was cultured anaerobically with acetate (30 mm) or benzoate (5 mm) as carbon source and Fe(III)-citrate as electron acceptor as described previously (23). The growth was monitored by cell counting using a Neubauer chamber. For all experiments, cells were always harvested in the exponential growth phase at 1.5–2.5 × 108 cells ml−1 by centrifugation (20,000 × g) after which they were stored in liquid nitrogen.
Sample Preparation and 1D SDS-PAGE Separation
Frozen cells of G. metallireducens grown on either acetate or benzoate as carbon source were suspended in buffer containing 50 mm NH4HCO3, pH 7.8, and 3 mm DTT (1 g of cells in 2 ml of buffer). Cell lysates were obtained by passage through a French pressure cell at 137 megapascals. After centrifugation at 100,000 × g (1 h at 4 °C) the pellet was washed and centrifuged twice using the same buffer. The pellet was suspended in buffer containing 9 m urea, 50 mm NH4HCO3, pH 7.8, 3 mm DTT, and PMSF. After ultracentrifugation the supernatant was discarded, and the remaining pellet was suspended with 1% SDS, 50 mm Tris/HCl, pH 8, and 3 mm DTT and centrifuged again. The supernatant was used for proteome analysis. The protein content was determined using a modified Bradford protocol as described previously (24). Five hundred micrograms of protein resuspended in a solution of 1% SDS, 50 mm Tris-HCl, pH 8.3, and 3 mm DTT were diluted with an equal volume of 1D SDS-PAGE sample buffer (62.5 mm Tris/HCl, pH 8, 2% SDS, 30% glycerol, and 0.01% bromphenol blue (25)) and boiled for 5 min. The samples were cooled and vigorously shaken for 1 h at room temperature. Prior to electrophoresis the samples were centrifuged at 19,000 × g for 5 min at 20 °C. The supernatants were loaded onto a 1D SDS-PAGE gel using a 4% acrylamide stacking gel and a 15% acrylamide resolving gel. After electrophoretic separation the gels were fixed in 50% ethanol and 3% phosphoric acid for 2 h, washed three times with bidistilled water (10 min), and stained overnight with colloidal Coomassie Brilliant Blue (0.08% Coomassie Brilliant Blue G-250, 1.6% ortho-phosphoric acid, 8% ammonium sulfate, and 20% methanol (26)).
In-gel Digestion and Mass Spectrometry Analysis
After gel image analysis, the overall separating gel band of each lane containing the different protein bands was excised in 96 slices (∼1 × 10 mm) using a custom-built grid. In-gel digestion was performed with an automated protein digestion system (MassPREP Station, Waters). The gel slices were washed twice with 50 μl of 25 mm NH4HCO3 and 50 μl of acetonitrile. Cysteine residues were reduced by 50 μl of 10 mm DTT at 57 °C and alkylated by 50 μl of 55 mm iodoacetamide. After dehydration with acetonitrile, the proteins were cleaved in-gel with 10 μl of 12.5 ng μl−1 modified porcine trypsin (Promega) in 25 mm NH4HCO3 at 37 °C for 16 h. Tryptic peptides were extracted with 60% acetonitrile in 0.5% formic acid followed by a second extraction with 100% acetonitrile. The peptide extracts obtained were analyzed by micro-LC-MS/MS using an Agilent 1100 Series capillary LC system (Agilent Technologies) coupled to a High Capacity Trap Ultra ion trap (Bruker Daltonics). Peptide mixtures were loaded on a Zorbax 300SB-C18 trap column (300 μm × 5 mm; Agilent Technologies) using 0.1% trifluoroacetic acid for 3 min at 50 μl min−1. After washing, flow was reversed through the trap column, and the peptides were eluted with a gradient of 10–70% acetonitrile in 0.05% trifluoroacetic acid delivered over 120 min at a flow rate of 4 μl min−1 through a reverse phase capillary column (Zorbax 300SB-C18, 300-μm inner diameter × 15 cm; Agilent Technologies). The High Capacity Trap Ultra ion trap was externally calibrated with standard compounds. The general mass spectrometric parameters were as follows: capillary voltage, −4000 V; drying gas, 6 liters min−1; drying temperature, 300 °C. The system was operated by automatic switching between MS and MS/MS modes. MS scanning was performed in the standard-enhanced resolution mode at a scan rate of 8100 m/z s−1 with an ion charge control of 100,000 in a maximal fill time of 200 ms; five scans were averaged to obtain an MS spectrum. The three most abundant peptides and preferentially doubly charged ions were selected on each MS spectrum for further isolation and fragmentation. The MS/MS scanning was performed in the ultrascan resolution mode at a scan rate of 26,000 m/z s−1 with an ion charge control of 300,000, and a total of six scans were averaged to obtain the MS/MS spectrum. The complete system was fully controlled by ChemStation Revision B.01.03 (Agilent Technologies) and EsquireControl 6.1 Build 78 (Bruker Daltonics) software. Mass data collected during LC-MS/MS analyses were processed using the software tool DataAnalysis 3.4 Build 169 and converted into *.mgf files.
Protein Identification
The MS/MS data were analyzed using the Mascot 2.2.0 algorithm (Matrix Science, London, UK) for a search against an in-house generated protein database composed of protein sequences of G. metallireducens downloaded from NCBI (on December 20, 2007) concatenated with reversed copies of all sequences (2 × 3532 entries). Spectra were searched with a mass tolerance of 0.5 Da for MS and MS/MS data, allowing a maximum of one missed cleavage with trypsin and with carbamidomethylation of cysteines and oxidation of methionines specified as variable modifications. Protein identifications were validated when at least two peptides with high quality MS/MS spectra (less than five points below the Mascot threshold score of identity at 95% confidence level) were detected. In the case of one-peptide hits, the score of the unique peptide had to be greater (minimal “difference score” of 20) than the 95% significance Mascot threshold. The estimated false discovery rate by searching the target-decoy database (27) was found to lie below 0.8%.
Cellular component gene ontology annotation of G. metallireducens was downloaded from the Gene Ontology Annotation Database. In silico transmembrane domain prediction was performed using the Phobius Web server (28).
Differential Protein Expression and Relative Protein Abundance
To estimate the differential expression of each protein i, the differential absolute protein expression measurement scheme (21, 22, 29) was used. From all high quality MS/MS spectra (less than five points below the Mascot threshold score of identity at 95% confidence level) associated with each validated protein, we calculated the fraction fi of interpreted spectra for a given protein i in the experiment as fi = ni/N where ni is the number of spectra from protein i and N is the total number of observed MS/MS spectra in the experiment. On the basis of these measures of fi, the test statistic for differential expression of a protein was calculated as
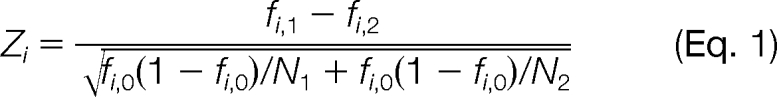 |
where the numerator represents the difference in sampled proportions of protein i in two proteomics experiments, fi,1 = ni,1/N1 and fi,2 = ni,2/N2, and the denominator represents the standard error of the difference under the null hypothesis in which the two sampled proportions were drawn from the same underlying distribution with the overall proportion fi,0 = (ni,1 + ni,2)/(N1 + N2). The abundance ratio (Fi) of a protein i between the two experiments (two growth conditions) was calculated as the ratio of the fraction of interpreted spectra Fi = fi,1/fi,2.
For proteins with no associated MS/MS spectra in one of the two cell extracts used, the abundance ratio was arbitrarily set as greater than 150 (no MS/MS spectra obtained in cells grown with acetate) or less than (no MS/MS spectra obtained from cells grown with benzoate). Proteins with |Z| > 2.58 (99% significance level) and log ratio (log2F) greater than 1 were considered significantly induced during growth on benzoate. Proteins with |Z| > 2.58 (99% significance level) and log ratio (log2F) less than −1 were considered as significantly repressed during growth on benzoate. In this analysis, the results from two biological replicates for extracts from cells grown on benzoate and acetate were pooled and the Z-score and log ratio for each validated protein were calculated.
For the comparison of protein abundances protein weights (W) were taken into consideration (18); the relative protein abundance for each of the k proteins was calculated as follows.
 |
The relative protein abundance of succinate dehydrogenase (NCBI database accession no. gi78194855) was set to 100 and used as reference for the calculation of each relative protein abundance.
Quantitative PCR Analysis
One microgram of total RNA from G. metallireducens grown on acetate or benzoate was reverse transcribed using random hexamer primers and the High Capacity® Reverse Transcription kit (Applied Biosystems, Foster City, CA). The cDNA was diluted 1:12.5 and served as the template for qPCR analysis using the TaqMan® 9700 System (Applied Biosystems) with Fast SYBR® Green Master Mix (Applied Biosystems) according to the manufacturer's protocol. Melting curve and agarose gel analyses were used to confirm the specificity of the amplification reactions. All mRNA quantification data were normalized to mRNA levels of a housekeeping enzyme (succinate dehydrogenase). As a positive control induction of bamF (coding for a putative methyl viologen-reducing hydrogenase, δ subunit) in bacteria grown on benzoate was verified (induction, ∼33-fold). Primer sequences for quantitative real time expression analysis (qRT-PCR) of mRNA levels of different enzymes are listed in supplemental Data 1. Genes were considered significantly induced or repressed during growth on benzoate if the log ratio (log2) of the mRNA abundance between the two growth conditions was greater than 1 or less than −1.
Determination of Enzyme Activities
Hydrogenase and nitrate reductase activities were analyzed at 30 °C under anaerobic conditions using oxidized benzyl viologen as artificial electron acceptor (hydrogenase) or the reduced form as electron acceptor (nitrate reductase). Substoichiometric reduction of benzyl viologen was anaerobically carried out by additions of dithionite from a 50 mm stock solution to A600 = 1.4–1.6 in the nitrate reductase and to A600 = 0.05 in the hydrogenase assay. The test buffer contained 1 mm benzyl viologen, 50 mm Tris/HCl, pH 7.8, and 100–150 μg of solubilized membrane protein fraction obtained after ultracentrifugation of crude extracts from G. metallireducens grown on acetate or benzoate. The reaction was carried out in a gas-tight sealed cuvette (400 μl) under a nitrogen atmosphere. For the hydrogenase assay, the reaction was started by injection of 100 μl of H2 via a gas-tight syringe followed by gentle shaking of the cuvette. For the nitrate reductase activity test, the reaction was started by addition of 0.5–2.5 mm sodium nitrate. As a control, the membrane-bound succinate dehydrogenase activity was determined spectrophotometrically using ferricenium as electron acceptor. Cytochrome bd oxidase activity was tested by oxygen determination using an optode (Fibox3, PreSens Precision Sensing GmbH, Regensburg, Germany) and an oxygen sensor spot in a 1.5-ml gas-tight glass vial at room temperature. Menaquinol (0.5–1.5 mm), NADH (0.5–1 mm), acetate (10–20 mm), and benzoate (1–2 mm) were tested as electron donors for cell extracts/whole cells.
RESULTS AND DISCUSSION
Differential Analysis of the Membrane Proteome of G. metallireducens
In a recent study we analyzed and compared the soluble proteomes of G. metallireducens cells grown on benzoate and acetate by 2D gel electrophoresis coupled to mass spectrometric analysis. By combining the data obtained with results from reverse transcription-PCR, 44 benzoate-induced genes were found to be organized in the gene clusters IA/B and II (14). They comprise genes for all predicted enzymes involved in the conversion of benzoyl-CoA to acetyl-CoA and CO2; an eight gene-containing cluster (bamB–I) was proposed to code for a novel BCR complex.
To reveal benzoate-induced membrane-bound/associated proteins, the spectral counting approach was performed with tryptic digests from membrane preparations obtained from cells grown on benzoate and acetate. After thorough removal of soluble or loosely membrane-attached proteins by washing/extraction steps, the membrane fractions were separated by 1D SDS-PAGE, digested in-gel, and analyzed by LC-MS/MS. Although the urea treatment step is generally accepted for membrane protein preparations, one has to consider that the chaotropic urea agent may break interactions between some non-integral membrane protein subunits, which may result in a loss of some proteins. MS/MS spectra analysis identified 931 proteins in the genome of G. metallireducens with a very low false positive rate (below 0.8% as estimated by searching a target-decoy database (27)). Among these, 804 proteins were identified with at least two peptides, and 127 proteins were identified with one peptide; for the latter the spectra associated with the best score identifications are shown in supplemental Data 2. To date, 325 of these 931 proteins have a cellular component gene ontology annotation (indicated in supplemental Data 7). 53% of them (173 proteins) are annotated as membrane-bound/associated proteins, which represents 40% of all annotated membrane-bound/associated proteins from G. metallireducens. In addition, in silico transmembrane domain prediction allowed the identification of 250 proteins with at least one transmembrane domain (supplemental Data 7). About one-third of all proteins identified in this study (303 proteins) contain at least one predicted transmembrane domain and/or are predicted as membrane-bound/associated despite the very incomplete cellular component annotation of G. metallireducens.
The differential absolute protein expression measurement method (21, 22, 29) was used to identify proteins with a significant differential expression by performing a test statistic (Z-test). To estimate abundance changes during growth on benzoate and acetate, an abundance ratio was calculated for each protein. To avoid overconsideration of small changes in protein abundances and of changes in the abundances of very minor proteins, we imposed |Z| to be greater than 2.58 (corresponding to 99% confidence) and the log2 abundance ratio to be greater than 1 to consider a protein as benzoate-induced. To estimate potential biological and analytical variabilities, preparations of the membrane fractions and analysis of the tryptic peptides were carried out with extracts from two different cell batches both harvested in the exponential growth phase. The lists of all data obtained are available in supplemental Data 2–6; the spectral counting data are shown in supplemental Data 7. Using the framework described above, spectral counting analysis revealed 130 proteins with an altered expression levels in cells grown on benzoate and acetate. From the 100 benzoate-induced proteins, 23 were identified as membrane-bound/associated proteins encoded by genes of the bam clusters. Further 18 newly identified proteins with a minimum of 4.5-fold change during growth on benzoate were selected for further investigations.
Detection of Membrane Proteins Involved in Overall Energy Metabolism
To demonstrate the efficiency and comparability of the membrane proteome analysis in cells grown on benzoate and acetate, the observed abundance ratio of components from membrane complexes involved in general energy metabolism are exemplarily discussed in the following. A non-differential abundance of the housekeeping proteins in cells grown on benzoate and acetate was expected.
All predicted components of NADH:menaquinone oxidoreductase, succinate dehydrogenase, and H+-F0F1-ATP synthase were identified in cells grown on benzoate and acetate at almost equal abundances (Table I). None of the 27 subunits of the three complexes passed the established filtering thresholds to consider a protein as differentially expressed during growth on benzoate and acetate. These results indicate that both cell types were in comparable physiological states (exponential growth phase) and that differences in abundance of other membrane proteins cannot be assigned to differences in cultivation or sample preparation and demonstrate the reliability of the spectral counting approach for differential membrane proteome analysis.
Table I. Abundance of membrane protein components involved in central energy metabolism in G. metallireducens cells grown on benzoate and acetate.
| Protein annotation | Molecular mass | NCBI database accession no. | log2 ratio of protein abundance (benzoate/acetate) | Relative protein abundance (benzoate) |
|---|---|---|---|---|
| kDa | gi | |||
| Proton-translocating NADH:quinone oxidoreductase, chain N | 52.1 | 78195787 | −0.7 | 8.4 |
| Proton-translocating NADH:quinone oxidoreductase, chain M | 57.2 | 78195788 | −0.1 | 37.4 |
| NADH:plastoquinone oxidoreductase, chain 5 | 73.3 | 78195789 | −0.4 | 26.7 |
| NADH:ubiquinone oxidoreductase, chain 4L | 11.1 | 78195790 | −0.6 | 42.3 |
| NADH:ubiquinone/plastoquinone oxidoreductase, chain 6 | 18.2 | 78195791 | −0.1 | 74.8 |
| NADH:quinone oxidoreductase, chain I | 15.1 | 78195792 | −0.1 | 51.4 |
| Respiratory chain NADH dehydrogenase, subunit 1 | 37.5 | 78195793 | −0.5 | 55.5 |
| NADH dehydrogenase I, G subunit, putative | 89.0 | 78195794 | −0.2 | 17.5 |
| NADH dehydrogenase I, F subunit | 64.3 | 78195795 | −0.3 | 21.0 |
| NADH dehydrogenase (ubiquinone), 24-kDa subunit | 19.2 | 78195796 | −0.1 | 9.3 |
| NADH dehydrogenase I, D subunit | 43.7 | 78195797 | −0.7 | 35.0 |
| NADH (or F420H2) dehydrogenase, subunit C | 18.5 | 78195798 | −0.3 | 60.6 |
| NADH dehydrogenase (ubiquinone), 20-kDa subunit | 18.3 | 78195799 | 0.0 | 34.3 |
| NADH:ubiquinone/plastoquinone oxidoreductase, chain 3 | 13.3 | 78195800 | −0.6 | 38.5 |
| Fumarate reductase, iron-sulfur protein | 27.2 | 78194853 | 1.0 | 164.4 |
| Succinate dehydrogenase or fumarate reductase, flavoprotein | 70.7 | 78194854 | 0.5 | 330.6 |
| Succinate dehydrogenase, cytochrome b558 subunit | 24.0 | 78194855 | −0.1 | 100.0 |
| H+-Transporting two-sector ATPase, A subunit | 25.5 | 78195804 | 0.6 | 4.5 |
| ATP synthase F0, C subunit | 9.4 | 78195805 | −1.0 | 73.9 |
| H+-transporting two-sector ATPase, δ/ϵ subunit | 15.2 | 78195850 | −0.1 | 17.7 |
| ATP synthase F1, β subunit | 51.0 | 78195851 | 0.4 | 87.7 |
| H+-Transporting two-sector ATPase, γ subunit | 31.9 | 78195852 | 0.1 | 38.7 |
| ATP synthase F1, α subunit | 54.6 | 78195853 | 0.2 | 66.5 |
| H+-Transporting two-sector ATPase, δ subunit | 19.4 | 78195854 | −1.0 | 45.0 |
| H+-Transporting two-sector ATPase, B/B′ subunit | 22.6 | 78195855 | −1.0 | 22.1 |
| H+-Transporting two-sector ATPase, B/B′ subunit | 15.6 | 78195856 | −0.9 | 31.5 |
| Inorganic H+-pyrophosphatase | 70.1 | 78195685 | 0.0 | 126.2 |
The genome of G. metallireducens contains three copies of a gene cluster annotated as components of (mena)quinol:cytochrome c oxidoreductases (usually termed cytochrome bc1 complexes consisting of four subunits). However, neither of the corresponding components was identified in cells grown on benzoate or acetate. This finding indicates that no such respiratory complex is involved in electron transfer from menaquinol to the terminal acceptor Fe(III) and that a so far unknown menaquinol-oxidizing membrane component should exist. Thus, the predicted cytochrome bc1 complexes are considered to exhibit other functions. Recently a special role of such a complex (gi78194581–gi78194584) in p-cresol degradation was suggested (30, 31). The complex was proposed to mediate electron transfer from the p-cresol methylhydroxylase reaction to the menaquinone pool. Accordingly the genes of this complex were only induced during growth on p-cresol but not during growth on benzoate or acetate.
Interestingly in cells grown on both acetate and benzoate the gene putatively coding for a proton-translocating pyrophosphatase was identified in equal amounts (Table I). This finding is in line with the fact that during growth on both benzoate and acetate AMP + PPi-forming carboxylic acid-CoA ligases are involved in initial ATP-dependent benzoate/acetate activations. Obviously obligate anaerobes commonly couple the exergonic hydrolysis of PPi to H+/Na+ ion translocation across the cytoplasmic membrane as it has been demonstrated for the fermenting Syntrophus gentianae (32).
Identification of Proteins Encoded by the Benzoate-induced Gene Clusters IA/B and II
A previous study revealed the benzoate-induced gene clusters IA/B and II (14). Analysis of the soluble proteomes only identified 13 of the 44 predicted benzoate-induced gene products, although RT-PCR analysis indicated that the 44 genes were all induced during growth on benzoate. The reason for this discrepancy could be due to (i) the presence of membrane/membrane-associated proteins that were not suitable for 2D gel electrophoretic analysis, (ii) very low expression level of these proteins, or (iii) posttranscriptional regulatory processes. In contrast, the membrane proteome analysis of this work identified 23 of the 44 putative gene products as benzoate-induced.
In Table II, the differential identification of individual components of the putative benzoyl-CoA dearomatizing BamB–I complex in the soluble and membrane proteome is shown. To compare abundances of the proteins in cells grown on benzoate, we additionally calculated the relative protein abundance of each protein by normalizing the spectral count by the protein molecular weight (see “Material and Methods”). BamBCDE and BamI had been identified in the soluble fraction in the previous study (14).2 These components were also identified in the membrane fraction of benzoate-grown cells albeit at low (BamB) or very low amounts (BamC–E and BamI; Table II). Although topology prediction annotated the selenocysteine-containing BamF and the BamG as soluble proteins, they were only identified at relatively high abundance in the membrane fraction, indicating that at least these components of the putative BamB–I complex were attached to the membrane. Obviously most of the BamB–E and BamHI components were removed during the multiple washing steps of sample preparation. In summary, the results obtained suggest that the predicted BamB–I complex is at least partly associated to the membrane with BamFG apparently showing a higher affinity to the membrane than the other components. As expected components of the BamB–I complex were absent in the membrane fraction of cells grown on acetate (no spectrum observed).
Table II. Differential identification of BamB–I components in the membrane and soluble protein fraction.
Semiquantitative identification in the soluble fraction was carried in a previous analysis after separation on 2D gels (14).
| NCBI database accession no. | Gene product | Amino acid similarities to | Relative abundance |
|
|---|---|---|---|---|
| Membrane fractiona | Soluble fraction | |||
| gi | ||||
| 78194549 | BamB | Aldehyde ferredoxin oxidoreductases | 1.6 | +++ |
| 78194548 | BamC | FeS subunit of hydrogenases | <1 | +++ |
| 78194547 | BamD | Soluble subunit of heterodisulfide reductases | <1 | ++ |
| 78194546 | BamE | Soluble subunit of heterodisulfide reductases | <1 | +++ |
| 78194545 | BamF | SeCys-containing subunit of hydrogenases | 19 | − |
| 78194543 | BamG | NADH:ubiquinone oxidoreductases (NuoG) | 12 | − |
| 78194542 | BamH | NADH:ubiquinone oxidoreductases (NuoF) | <1 | − |
| 78194541 | BamI | NADH:ubiquinone oxidoreductases (NuoE) | 1 | +++ |
a Arbitrary units; the abundance of the structural gene of succinate dehydrogenase (gi78194855) was set to 100.
The benzoate-induced cluster IB contains a number of genes coding for enzymes involved in β-oxidation reactions of aromatic catabolism, nine of which were identified as benzoate-induced in the membrane fraction (supplemental Data 2, 5, and 6). It has been reported that acyl-CoA dehydrogenases and their in vivo electron acceptors, electron-transferring flavoproteins (ETFs), are often found to be attached to the membrane by interaction with a membrane-bound ETF:quinone oxidoreductase (33). In accordance, two subunits of ETFs (gi78194527–8) and glutaryl-CoA dehydrogenase (BamM; gi78194537) were identified as benzoate-induced in both the soluble and the membrane proteome (14). As G. metallireducens contains no genes coding for homologues of ETF:quinone oxidoreductases, the primary electron-accepting membrane protein of reduced ETF was unclear. It has been proposed that oxr genes (gi78194527 and gi78194532), located adjacent to the genes coding for two subunits of ETF (etfAB), code for an alternative ETF:quinone oxidoreductase complex. Indeed both Oxr copies were identified as benzoate-induced membrane proteins. They show similarities to heme b/FeS cluster-containing, membrane-bound heterodisulfide reductases from Methanosarcinales species. Topology prediction revealed the presence of five to six transmembrane helices, which explains why they were not identified after 2D gel electrophoretic separation in the previous study. The role of benzoate-induced proteins, annotated as two subunits of succinyl-CoA synthetases, identified in both the soluble and membrane fractions is unknown so far.
Identification of Novel Benzoate-induced Proteins
Some of the newly identified proteins showed similarities to components of membrane-bound hydrogenases, cytochrome bd oxidase, nitrate reductase, transporter-related proteins, and a number of proteins with unclear/unknown functions (Table III and supplemental Data 2, 5, and 6). To verify the induction of the genes coding for benzoate-induced membrane proteins, the transcriptional regulation of selected genes was analyzed. For this purpose total RNA was isolated from cells grown on benzoate and acetate in the exponential growth phase and converted to cDNA by reverse transcriptase reactions, respectively. Quantitative analysis of gene expression was performed by RT-qPCR using appropriate oligonucleotide primer pairs. Primers for amplifying cDNA from the structural genes of succinate dehydrogenase and inorganic H+-pyrophosphatase served as positive controls, which are expressed in cells grown on benzoate and acetate cells at equal amounts (Table III). Primers amplifying cDNA from two benzoate genes, bamB and bamF (Table II and Ref. 14), served as positive control for benzoate-induced genes (Table III). An intergenic, non-coding region served as negative control as described previously (14).
Table III. Analysis of membrane-bound/associated proteins that are more abundant in cells grown on benzoate and transcriptional analysis of selected genes induced during growth on benzoate.
Only proteins with annotated functions are shown; proteins are grouped according the clustering of the corresponding genes. Proteins with bold data met the spectral counting criteria and are considered as induced during growth on benzoate. Gene induction was determined by RT-qPCR. Proteins marked by a check in the right column are considered as induced during growth on benzoate. The assignment is based on either protein abundance (additionally marked by checks) or by mRNA abundance (log ratio >1.1). Dashes indicate protein abundances below the criteria for unambiguous protein identification. TMH, number of transmembrane helices; ND, not determined.
| Protein annotation in genome | Molecular weight | gi | TMH | Protein abundance |
mRNA abundance |
||||
|---|---|---|---|---|---|---|---|---|---|
| Benzoatea | log ratio(benzoate/acetate) | |Z|(benzoate/acetate) | Induced in benzoate growth condition | log ratio(benzoate/acetate) | Induced in benzoate growth condition | ||||
| Succinate dehydrogenase subunit C (control 1) | 24,012 | 78194855 | 5 | 100.00 | −0.1 | 1.25 | 0.0 | ||
| Inorganic H+-pyrophosphatase (control 2) | 70,100 | 78195685 | 16 | 126.20 | 0.0 | 0.80 | 0.1 | ||
| BamB (positive control 1) | 73,802 | 78194549 | 0 | 1.65 | >7.2 | 4.14 | ✓ | >5.3 | ✓ |
| BamF (positive control 2) | 23,900 | 78194545 | 0 | 18.83 | >7.2 | 8.71 | ✓ | >5.3 | ✓ |
| Nitrate reductase, α subunit | 134,326 | 78192805 | 0 | 17.40 | 2.7 | 14.40 | ✓ | 2.9 | ✓ |
| Nitrate reductase, β subunit | 54,118 | 78192806 | 0 | 10.77 | 3.2 | 7.70 | ✓ | 1.7 | ✓ |
| Nitrate reductase, δ subunit | 22,822 | 78192807 | 0 | — | — | — | ND | ||
| Nitrate reductase, γ subunit | 26,501 | 78192808 | 5 | 3.63 | 2.2 | 2.62 | ✓ | 4.0 | ✓ |
| Complex I/energy-converting hydrogenase | 27,614 | 78192845 | 0 | 2.78 | 1.9 | 2.13 | 1.1 | ✓ | |
| Transcriptional regulator, Icl R family | 29,438 | 78192846 | 0 | 7.18 | −0.2 | 0.54 | |||
| Complex I/energy-converting hydrogenase | 54,267 | 78192847 | 0 | 1.77 | >7.2 | 3.68 | ✓ | 1.6 | ✓ |
| Complex I/energy-converting hydrogenase | 51,165 | 78192848 | 11 | 0.63 | >7.2 | 2.12 | |||
| Complex I/energy-converting hydrogenase | 22,810 | 78192849 | 7 | — | — | — | ND | 1.2 | ✓ |
| Complex I/energy-converting hydrogenase | 32,014 | 78192850 | 8 | — | — | — | ND | 2.3 | ✓ |
| Complex I/energy-converting hydrogenase | 70,567 | 78192851 | 16 | — | — | — | ND | 1.0 | |
| Complex I/energy-converting hydrogenase | 26,303 | 78195048 | 0 | 2.19 | 0.2 | 0.30 | 1.6 | ✓ | |
| Complex I/energy-converting hydrogenase | 55,236 | 78195049 | 0 | 2.55 | 0.2 | 0.48 | 2.1 | ✓ | |
| Complex I/energy-converting hydrogenase | 51,177 | 78195050 | 13 | — | — | — | ND | ||
| Complex I/energy-converting hydrogenase | 23,043 | 78195051 | 7 | 0.28 | −1.1 | 0.67 | 1.7 | ✓ | |
| Complex I/energy-converting hydrogenase | 32,263 | 78195052 | 7 | — | — | — | ND | ||
| Complex I/energy-converting hydrogenase | 70,158 | 78195053 | 14 | — | — | — | ND | 2.5 | ✓ |
| Na+/solute symporter | 61,848 | 78193214 | 13 | 50.63 | 5.2 | 20.24 | ✓ | 5.2 | ✓ |
| Protein of unknown function DUF485 | 11,918 | 78193215 | 2 | 13.43 | >7.2 | 4.75 | ✓ | ||
| Tungstate ABC transporter-related protein | 26,615 | 78193512 | 0 | 3.61 | 3.8 | 3.30 | ✓ | 1.6 | ✓ |
| Binding protein inner membrane component | 23,847 | 78193513 | 5 | 1.34 | 1.2 | 1.00 | 2.0 | ✓ | |
| Extracellular tungstate-binding protein | 29,161 | 78193514 | 0 | 9.88 | 2.8 | 5.10 | ✓ | ||
| Protein of unknown function DUF214 | 91,594 | 78193984 | 11 | 35.02 | 5.2 | 20.50 | ✓ | ||
| ABC transporter-related protein | 24,352 | 78193985 | 0 | 33.39 | 6.8 | 10.58 | ✓ | 2.2 | ✓ |
| AMP-dependent synthetase and ligase | 91,691 | 78194077 | 0 | 3.35 | 3.9 | 5.95 | ✓ | 1.3 | ✓ |
| Cytochrome bd ubiquinol oxidase, subunit II | 37,435 | 78194391 | 8 | 5.82 | >7.2 | 5.54 | ✓ | ||
| Cytochrome bd ubiquinol oxidase, subunit I | 49,956 | 78194392 | 9 | 14.23 | 3.8 | 9.04 | ✓ | −0.3 | |
| Transcriptional regulator, BadM/Rrf2 family | 14,604 | 78194393 | 0 | 6.58 | >7.2 | 3.68 | ✓ | −0.5 | |
| Rubrerythrin | 19,131 | 78195684 | 0 | 6.02 | 3.0 | 3.35 | ✓ | −1.0 | |
| NiFe hydrogenase, cytochrome b subunit | 25,101 | 78195776 | 4 | 1.79 | >7.2 | 2.51 | ✓ | 3.4 | ✓ |
| NiFe hydrogenase, large subunit | 62,264 | 78195777 | 0 | 8.12 | >7.2 | 8.45 | ✓ | 3.2 | ✓ |
| NiFe hydrogenase, small subunit | 40,167 | 78195778 | 0 | 3.83 | >7.2 | 4.66 | ✓ | 4.2 | ✓ |
a Arbitrary units; the abundance of the structural gene of succinate dehydrogenase (gi78194855) was set to 100.
Overall the proteomics and transcription analysis data are in good agreement. Of 29 benzoate-induced genes of the clusters IA/B that were previously identified by RT-PCR (14), 23 of the corresponding gene products (∼80%) were also identified in this work at higher abundance in cells grown on benzoate (supplemental Data 7). In addition, a further 19 benzoate-induced genes were newly identified by RT-qPCR analysis in the study (Table III). For 11 of these proteins, proteomics and RT-qPCR data were in full agreement. No opposite significant change between mRNA and protein levels was observed. However, a few genes showed a significant change only at the mRNA or only at the protein level. These cases are presented in Table III and are discussed in the following. In the case of NiFe/heme b hydrogenase and nitrate reductase results from in vitro enzyme activity determinations are presented (Table IV); again succinate dehydrogenase activity measurements served as a control.
Table IV. Specific activities of hydrogenase and nitrate reductase in cell extracts of G. metallireducens.
Activity was determined spectrophotometrically using reduced benzyl viologen as electron donor (nitrate reductase) or the oxidized form as electron acceptor (hydrogenase). For the succinate dehydrogenase control, ferricenium was used as electron acceptor. One milliunit is referred to as conversion of 1 nmol min−1 H2/nitrate/succinate, respectively. Mean values ± S.D. from three independent determinations are presented.
| Enzyme | Extracts |
-Fold increase benzoate/acetate (membrane fraction) | |||
|---|---|---|---|---|---|
| Benzoate-grown cells |
Acetate-grown cells |
||||
| Soluble | Membrane | Soluble | Membrane | ||
| milliunits mg−1 | |||||
| Hydrogenase | 47 ± 5 | 157 ± 12 | 10 ± 4 | 10 ± 2 | 16 |
| Nitrate reductase | 146 ± 8 | 450 ± 50 | 48 ± 12 | 125 ± 20 | 4 |
| Succinate dehydrogenase (control) | 155 ± 40 | 525 ± 70 | 270 ± 50 | 774 ± 200 | 0.7 |
NiFe/Heme b-containing Hydrogenase
All three subunits of a putative NiFe/heme B-containing hydrogenase complex (gi78195776–78) were identified at a clearly higher abundance in cells grown on benzoate (Table III). Using oligonucleotide primers deduced from the three subunits of hydrogenase, a clear induction of gene transcription was observed with cDNA from cells grown on benzoate when compared with cDNA from acetate-grown cells (Table III). Using a spectrophotometric assay monitoring the H2 and cell extract-dependent reduction of benzyl viologen, hydrogenase activities were determined mainly in the membrane protein fraction of extracts from cells grown on benzoate (Table IV). The activity was 15 times higher than in extract from cells grown on acetate; this is in good accordance with data obtained from proteome and RT-qPCR analysis. The genome of G. metallireducens contains two further gene clusters coding for putative soluble hydrogenases (e.g. gi78193586 and gi78195766 annotated as the large NiFe-containing subunit of the putative hydrogenase complexes). However, using oligonucleotide primers for amplifying DNA fragments of the genes putatively coding for the large subunits, expression was negligible and non-differential in cells grown on benzoate and acetate (not shown). Thus, only one of the three gene clusters in the genome of G. metallireducens that putatively code for hydrogenases was found to be induced during growth on benzoate.
Membrane-bound three subunit-containing NiFe/heme b hydrogenases usually catalyze the oxidation of dihydrogen to protons, and reducing equivalents are transferred to quinones (menaquinone in the case of G. metallireducens). The induction of such a hydrogenase during growth on benzoate was not expected. A possible function as a low potential electron donor for benzoyl-CoA reduction is rather unlikely as the presence of the heme b-containing transmembrane subunit rather suggests electron transfer to menaquinone. The question arises whether the enzyme uses external or endogenously produced H2 as substrate in vivo. Notably the gas phase of the culture medium did not contain dihydrogen.
Energy-converting Hydrogenases
Next to the 14 clustered genes annotated as subunits of complex I of the respiratory chain (Table I), an additional protein assigned to a putative NADH:quinone oxidoreductase subunit was identified as more abundant in the membrane fraction in cells grown on benzoate (gi78192847). The gene belongs to a cluster comprising seven open reading frames of which six are annotated as putative subunits of NADH:quinone oxidoreductases/energy-converting hydrogenases and one is annotated as a transcriptional regulator (Table III). Two additional products from this cluster were identified with an abundance ratio above the threshold (gi78192845 and gi78192848). However, the number of MS/MS spectra was too low to consider them as significantly differentially expressed using the highly stringent criteria established in this study. Topology prediction revealed that gi78192848–51 contain 42 highly hydrophobic transmembrane domains, which may explain the low recovery in the MS analysis. Highly hydrophobic proteins have been reported to escape analysis due to the lack of tryptic digestion sites close to the membrane domains (34). Interestingly an additional highly similar gene cluster is present in the genome (gi78195048–53) of which three proteins were identified (Table III). To further test the induction of both gene clusters during growth on benzoate, the induction of selected genes of each cluster was tested by RT-qPCR analysis. The results obtained indicate that transcription of genes from cluster gi78195048–53 was clearly induced during growth on benzoate, whereas expression of cluster gi78192845–51 was moderately induced (Table III). The similarities between the six genes of the two newly identified benzoate-induced gene clusters were highest with putative proteins from other Geobacter species and related Deltaproteobacteria (50–65% amino acid sequence identity). When compared with enzymes from Enterobacteriacaea or methanogens, similarities were clearly higher to components of energy converting-hydrogenases than to components of related complex I of the respiratory chain.
Energy-converting hydrogenases usually couple the exergonic reduction of protons to H2 (e.g. with formate as electron donor) to the transport of H+/Na+ across the cytoplasmic membrane (35). They usually consist of two soluble components and two or more integral membrane protein components and are homologous to the proton-translocating/energy-conserving modules of NADH:quinone oxidoreductases. Examples are hydrogenases 3 and 4 from Escherichia coli or the Ech hydrogenases from methanogens. Notably in G. metallireducens the benzoate-induced gene clusters putatively coding for energy-converting complexes cannot be assigned to “real” hydrogenases as the deduced large subunit (e.g. gi78192847) does not contain the N- and C-terminal conserved CXXC motif, which is involved in NiFe cofactor binding (35). In G. metallireducens such an energy-conserving complex would represent an attractive candidate for a benzoate-induced membrane complex that is involved in a membrane potential-driven electron transport from an unknown donor to BCR.
Cytochrome bd Oxidase and Rubrerythrin
Two membrane proteins annotated as subunits of cytochrome bd oxidases were identified at clearly higher abundance in cells grown on benzoate when compared with cells grown on acetate (Table III). In contrast, RT-qPCR analysis revealed that the levels of the corresponding mRNAs were almost equal in cells grown on benzoate and acetate (Table III). This discrepancy suggests the presence of additional posttranscriptional regulatory elements. Attempts to verify the induction of cytochrome bd oxidase by in vitro activity measurements were not feasible because of very high background reactions caused by the presence of high amounts of Fe(II), which was derived either from the growth medium or from the numerous cytochromes in the dark reddish cell extracts.
Cytochrome bd oxidases usually catalyze the reduction of dioxygen by (mena)quinol with a very high affinity to dioxygen (36, 37). When part of a respiratory chain, such enzymes serve as terminal oxidases and are usually expressed at low oxygen concentrations in some aerobic organisms. In obligate anaerobes they rather serve as powerful oxygen-scavenging enzymes for protection from oxidative stress. It is conceivable that the obligately anaerobic G. metallireducens has an increased demand for an oxygen detoxification system during growth on benzoate for protection of the assumed extremely oxygen-sensitive BCR complex. The induction of an oxygen-scavenging enzyme during anaerobic growth on an aromatic substrate has recently been demonstrated in the facultatively anaerobic T. aromatica: here a benzoate-induced dienoyl-CoA oxidase with a high affinity to dioxygen as electron acceptor was identified (38). The requirement for increased oxygen protection when an extremely oxygen-sensitive enzyme complex has to be synthesized has been studied extensively in the case of nitrogenase (39).
Next to the cytochrome bd oxidase, rubrerythrin, another protein supposed to be involved in oxygen protection (40), was identified at clearly higher abundance in cells grown on benzoate (log ratio >3; Table III). But as in the case of cytochrome bd oxidase, transcription of the gene was similarly induced in cells grown on benzoate and acetate. Thus, it appears that the regulatory pattern of both putative oxygen-scavenging proteins follows similar principles probably involving posttranscriptional regulation processes.
Dissimilatory Nitrate Reductase
Surprisingly the products of three genes putatively coding for a membrane-bound, four subunit-containing dissimilatory nitrate reductase complex were identified at clearly higher abundance in cells grown on benzoate than in cells grown on acetate (Table III). In accordance, a clear induction of three genes during growth on benzoate was verified by RT-qPCR analysis. Moreover membrane extracts from cells grown on benzoate exhibited a 4-fold higher nitrate reductase activity than similarly prepared extracts from cells grown on acetate. Together these results indicate an up-regulation on the transcriptional level, although cells were always grown with Fe(III)-citrate as terminal electron acceptor. On the first view the induction of dissimilatory nitrate reductase during growth on benzoate appears curious. A possible explanation is based on the assumption that both nitrate reductase and BamB contain a molybdo-/tungstopterin cofactor. Thus, growth on an aromatic growth substrate is expected to induce the genes coding for enzymes involved in molybdenum/tungsten uptake and molybdo-/tungstopterin cofactor synthesis. A co-induction of several molybdo-/tungstopterin cofactor-containing enzymes when molybdo-/tungstopterin cofactor synthesis is induced is conceivable. E.g. the modA gene codes for a molybdenum-sensing transcriptional regulator of genes involved in molybdenum uptake, synthesis of the molybdenum/tungsten cofactor, and molybdenum/tungsten cofactor-containing polypeptides (41).
Tungstate Uptake System
The products of the benzoate-induced genes gi78193512 and gi78193514 belong to a cluster of genes coding for a putative ABC transporter system with the former representing the gene coding for the nucleotide binding component and the latter gene coding for the periplasmic binding protein (Table III). The third permase component of a typical ABC transporter system (gi78193513) was identified with a too low number of MS/MS spectra to pass the Z-test (despite an abundance ratio above threshold) most probably because of its extreme hydrophobicity (more than 60% of the amino acids are predicted to be involved in transmembrane helices formation). RT-qPCR analysis confirmed the induction of the corresponding genes (Table III). The putative periplasmic binding protein component showed exceptionally high similarities to tungstate-binding proteins of ABC transporters referred to as TupA (42) from several bacteria (up to 87% amino acid sequence identity to other Geobacter species and up to 56% identity to annotated TupA proteins from Ralstonia species). In particular the unique typical features of TupA proteins that distinguish them from molybdate-binding proteins were identified (e.g. the conserved TTTS motif near the N terminus and the SRGDXSGT motif (43)). The genes coding for the putative tungstate uptake system are part of a cluster containing several further genes coding for annotated enzymes involved in molybdo-/tungstopterin cofactor biosynthesis (gi78193508–19). Notably both the molybdo- and tungstopterin cofactors are usually synthesized by the same set of enzymes, whereas molybdate and tungstate uptake systems are highly specific (42).
The identification of a putative tungstate uptake system is remarkable as BamB, the proposed active site-containing component of benzoyl-CoA reductase from G. metallireducens, is similar to aldehyde:ferredoxin oxidoreductases that also often contain a tungstopterin cofactor (44, 45). Consequently BamB is rather supposed to be a tungsten rather than a molybdenum cofactor-containing protein.
Further Newly Identified Membrane Proteins That Were More Abundant in Cells Grown on Benzoate
A number of further benzoate-induced proteins were identified in the membrane fraction (supplemental data). Although many of them code for unknown proteins, some of them show similarities to other proteins (Table III and supplemental data). E.g. next to the putative ABC transporter for tungstate uptake two additional benzoate-induced transporter related proteins, annotated as components of a Na+/proline transporter (gi78193214–5) and an additional ABC transporter (gi78193984–5), were identified. RT-qPCR analysis confirmed a high induction of the corresponding genes (Table III). Furthermore a benzoate-induced membrane-associated, AMP-dependent ligase was found at higher abundance in cells grown on benzoate that was also slightly induced on the transcriptional level (Table III). The specific function of these and other benzoate-induced unknown proteins remains to be elucidated.
Conclusions: Membrane Proteins Involved in Aromatic Catabolism of G. metallireducens
The integration of the data obtained from (i) membrane proteome analysis by the spectral counting approach/1D SDS-PAGE fractionation, (ii) quantitative gene expression determinations, and (iii) enzyme activity measurements enabled a number of novel insights into key processes of aromatic degradation in obligately anaerobic bacteria. The so far non-characterized dearomatizing benzoyl-CoA reductase complex is proposed to be membrane-associated possibly by interaction with components similar to those of energy-converting hydrogenases. This suggestion is first based on the presence of the BamFG components of the putative dearomatizing BamB–I complex in the membrane fraction and the inability to identify them in the soluble proteome. Second two benzoate-induced gene clusters putatively coding for proteins with high similarities to components of energy-converting hydrogenases were identified. This finding implies that electron transfer to the aromatic ring is not coupled to a stoichiometric ATP hydrolysis but may rather be driven by a membrane potential involving the energy-conserving hydrogenase modules. The identification of benzoate-induced genes coding for an ABC transporter that contains all characteristic and unique features of tungstate uptake systems indicates that the putative active site-containing component of benzoyl-CoA reductase, BamB, contains rather a tungsten- than molybdenum-containing cofactor. The induction of a dissimilatory nitrate reductase by benzoate can be explained by common regulatory circuits of molybdo-/tungstopterin-containing proteins, whereas the role of benzoate-induced NiFe/heme b-containing uptake hydrogenase remains to be studied. Growth on benzoate also increased the level of oxygen-scavenging proteins most probably by posttranscriptional regulation. Thus, anaerobic aromatic degradation appears to be associated with a higher demand for protection from oxidative stress, a phenomenon that is probably common for all anaerobic bacteria using aromatic growth substrates.
Supplementary Material
Footnotes
* This work was funded by the Deutsche Forschungsgemeinschaft Grant BO 1565/6-1.
 The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
2 D. Heintz, S. Gallien, S. Wischgoll, A. K. Ullmann, C. Schaeffer, A. K. Kretzschmar, A. van Dorsselaer, and M. Boll, unpublished results.
1 The abbreviations used are:
- BCR
- benzoyl-CoA reductase
- ABC
- ATP-binding cassette
- 2D
- two-dimensional
- 1D
- one-dimensional
- ETF
- electron-transferring flavoprotein
- qPCR
- quantitative PCR
- Bam
- benzoic acid metabolism.
REFERENCES
- 1.Boll M., Fuchs G., Heider J. (2002) Anaerobic oxidation of aromatic compounds and hydrocarbons. Curr. Opin. Chem. Biol 6, 604–611 [DOI] [PubMed] [Google Scholar]
- 2.Díaz E. (2004) Bacterial degradation of aromatic pollutants: a paradigm of metabolic versatility. Int. Microbiol 7, 173–180 [PubMed] [Google Scholar]
- 3.Fuchs G. (2008) Anaerobic metabolism of aromatic compounds. Ann. N.Y. Acad. Sci 1125, 82–99 [DOI] [PubMed] [Google Scholar]
- 4.Gibson J., Harwood C. S. (2002) Metabolic diversity in aromatic compound utilization by anaerobic microbes. Annu. Rev. Microbiol 56, 345–369 [DOI] [PubMed] [Google Scholar]
- 5.Boll M. (2005) Dearomatizing benzene ring reductases. J. Mol. Microbiol. Biotechnol 10, 132–142 [DOI] [PubMed] [Google Scholar]
- 6.Boll M. (2005) Key enzymes in the anaerobic aromatic metabolism catalysing Birch-like reductions. Biochim. Biophys. Acta 1707, 34–50 [DOI] [PubMed] [Google Scholar]
- 7.Boll M., Fuchs G. (2005) Unusual reactions involved in anaerobic metabolism of phenolic compounds. Biol. Chem 386, 989–997 [DOI] [PubMed] [Google Scholar]
- 8.Boll M., Fuchs G. (1995) Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur. J. Biochem 234, 921–933 [DOI] [PubMed] [Google Scholar]
- 9.Boll M., Fuchs G., Meier C., Trautwein A., Lowe D. J. (2000) EPR and Mossbauer studies of benzoyl-CoA reductase. J. Biol. Chem 275, 31857–31868 [DOI] [PubMed] [Google Scholar]
- 10.Unciuleac M., Boll M. (2001) Mechanism of ATP-driven electron transfer catalyzed by the benzene ring-reducing enzyme benzoyl-CoA reductase. Proc. Natl. Acad. Sci 98, 13619–13624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Möbitz H., Boll M. (2002) A Birch-like mechanism in enzymatic benzoyl-CoA reduction: a kinetic study of substrate analogues combined with an ab initio model. Biochemistry 41, 1752–1758 [DOI] [PubMed] [Google Scholar]
- 12.Thiele B., Rieder O., Golding B. T., Müller M., Boll M. (2008) Mechanism of enzymatic Birch reduction: stereochemical course and exchange reactions of benzoyl-CoA reductase. J. Am. Chem. Soc 130, 14050–14051 [DOI] [PubMed] [Google Scholar]
- 13.McInerney M. J., Rohlin L., Mouttaki H., Kim U., Krupp R. S., Rios-Hernandez L., Sieber J., Struchtemeyer C. G., Bhattacharyya A., Campbell J. W., Gunsalus R. P. (2007) The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc. Natl. Acad. Sci. U.S.A 104, 7600–7605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wischgoll S., Heintz D., Peters F., Erxleben A., Sarnighausen E., Reski R., Van Dorsselaer A., Boll M. (2005) Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol. Microbiol 58, 1238–1252 [DOI] [PubMed] [Google Scholar]
- 15.Peters F., Shinoda Y., McInerney M. J., Boll M. (2007) Cyclohexa-1,5-diene-1-carbonyl-coenzyme A (CoA) hydratases of Geobacter metallireducens and Syntrophus aciditrophicus: evidence for a common benzoyl-CoA degradation pathway in facultative and strict anaerobes. J. Bacteriol 189, 1055–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuntze K., Shinoda Y., Moutakki H., McInerney M. J., Vogt C., Richnow H. H., Boll M. (2008) 6-Oxocyclohex-1-ene-1-carbonyl-coenzyme A hydrolases from obligately anaerobic bacteria: characterization and identification of its gene as a functional marker for aromatic compounds degrading anaerobes. Environ. Microbiol 10, 1547–1556 [DOI] [PubMed] [Google Scholar]
- 17.Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., Mann M. (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 [DOI] [PubMed] [Google Scholar]
- 18.Blondeau F., Ritter B., Allaire P. D., Wasiak S., Girard M., Hussain N. K., Angers A., Legendre-Guillemin V., Roy L., Boismenu D., Kearney R. E., Bell A. W., Bergeron J. J., McPherson P. S. (2004) Tandem MS analysis of brain clathrin-coated vesicles reveals their critical involvement in synaptic vesicle recycling. Proc. Natl. Acad. Sci. U.S.A 101, 3833–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao B. B., Clermont A., Rook S., Fonda S. J., Srinivasan V. J., Wojtkowski M., Fujimoto J. G., Avery R. L., Arrigg P. G., Bursell S. E., Aiello L. P., Feener E. P. (2007) Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat. Med 13, 181–188 [DOI] [PubMed] [Google Scholar]
- 20.Gilchrist A., Au C. E., Hiding J., Bell A. W., Fernandez-Rodriguez J., Lesimple S., Nagaya H., Roy L., Gosline S. J., Hallett M., Paiement J., Kearney R. E., Nilsson T., Bergeron J. J. (2006) Quantitative proteomics analysis of the secretory pathway. Cell 127, 1265–1281 [DOI] [PubMed] [Google Scholar]
- 21.Lu P., Rangan A., Chan S. Y., Appling D. R., Hoffman D. W., Marcotte E. M. (2007) Global metabolic changes following loss of a feedback loop reveal dynamic steady states of the yeast metabolome. Metab. Eng 9, 8–20 [DOI] [PubMed] [Google Scholar]
- 22.Wang R., Marcotte E. M. (2008) The proteomic response of Mycobacterium smegmatis to anti-tuberculosis drugs suggests targeted pathways. J. Proteome Res 7, 855–865 [DOI] [PubMed] [Google Scholar]
- 23.Lovley D. R., Phillips E. J. (1988) Novel mode of microbial energy metabolism: organic carbon oxidation coupled to dissimilatory reduction of iron or manganese. Appl. Environ. Microbiol 54, 1472–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramagli L. S., Rodriguez L. V. (1985) Quantitation of microgram amounts of protein in two-dimensional polyacrylamide gel electrophoresis sample buffer. Electrophoresis 559–563 [Google Scholar]
- 25.Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 26.Neuhoff V., Stamm R., Pardowitz I., Arold N., Ehrhardt W., Taube D. (1990) Essential problems in quantification of proteins following colloidal staining with Coomassie brilliant blue dyes in polyacrylamide gels, and their solution. Electrophoresis 11, 101–117 [DOI] [PubMed] [Google Scholar]
- 27.Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 28.Käll L., Krogh A., Sonnhammer E. L. (2007) Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res 35, W429–W432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu P., Vogel C., Wang R., Yao X., Marcotte E. M. (2007) Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol 25, 117–124 [DOI] [PubMed] [Google Scholar]
- 30.Johannes J., Bluschke A., Jehmlich N., von Bergen M., Boll M. (2008) Purification and characterization of active-site components of the putative p-cresol methylhydroxylase membrane complex from Geobacter metallireducens. J. Bacteriol 190, 6493–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters F., Heintz D., Johannes J., van Dorsselaer A., Boll M. (2007) Genes, enzymes, and regulation of para-cresol metabolism in Geobacter metallireducens. J. Bacteriol 189, 4729–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schöcke L., Schink B. (1998) Membrane-bound proton-translocating pyrophosphatase of Syntrophus gentianae, a syntrophically benzoate-degrading fermenting bacterium. Eur. J. Biochem 256, 589–594 [DOI] [PubMed] [Google Scholar]
- 33.Parker A., Engel P. C. (2000) Preliminary evidence for the existence of specific functional assemblies between enzymes of the beta-oxidation pathway and the respiratory chain. Biochem. J 345, 429–435 [PMC free article] [PubMed] [Google Scholar]
- 34.Jansson M., Wårell K., Levander F., James P. (2008) Membrane protein identification: N-terminal labeling of nontryptic membrane protein peptides facilitates database searching. J. Proteome Res 7, 659–665 [DOI] [PubMed] [Google Scholar]
- 35.Hedderich R. (2004) Energy-converting [NiFe] hydrogenases from archaea and extremophiles: ancestors of complex I. J. Bioenerg. Biomembr 36, 65–75 [DOI] [PubMed] [Google Scholar]
- 36.Borisov V. B. (1996) Cytochrome bd: structure and properties. Biokhimiia 61, 786–799 [PubMed] [Google Scholar]
- 37.Jünemann S. (1997) Cytochrome bd terminal oxidase. Biochim. Biophys. Acta 1321, 107–127 [DOI] [PubMed] [Google Scholar]
- 38.Thiele B., Rieder O., Jehmlich N., von Bergen M., Müller M., Boll M. (2008) Aromatizing cyclohexa-1,5-diene-1-carbonyl-coenzyme A oxidase. Characterization and its role in anaerobic aromatic metabolism. J. Biol. Chem 283, 20713–20721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fay P. (1992) Oxygen relations of nitrogen fixation in cyanobacteria. Microbiol. Rev 56, 340–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann Y., Meile L., Teuber M. (1996) Rubrerythrin from Clostridium perfringens: cloning of the gene, purification of the protein, and characterization of its superoxide dismutase function. J. Bacteriol 178, 7152–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grunden A. M., Shanmugam K. T. (1997) Molybdate transport and regulation in bacteria. Arch. Microbiol 168, 345–354 [DOI] [PubMed] [Google Scholar]
- 42.Andreesen J. R., Makdessi K. (2008) Tungsten, the surprisingly positively acting heavy metal element for prokaryotes. Ann. N.Y. Acad. Sci 1125, 215–229 [DOI] [PubMed] [Google Scholar]
- 43.Makdessi K., Andreesen J. R., Pich A. (2001) Tungstate Uptake by a highly specific ABC transporter in Eubacterium acidaminophilum. J. Biol. Chem 276, 24557–24564 [DOI] [PubMed] [Google Scholar]
- 44.Roy R., Menon A. L., Adams M. W. (2001) Aldehyde oxidoreductases from Pyrococcus furiosus. Methods Enzymol 331, 132–144 [DOI] [PubMed] [Google Scholar]
- 45.Roy R., Adams M. W. (2002) Characterization of a fourth tungsten-containing enzyme from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol 184, 6952–6956 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.