

Abstract
Reactive oxygen species can cause widespread cellular damage, including base alterations and strand breaks in DNA. An array of DNA-repair enzymes constitutes an essential part of the line of defense that cells use against oxidative damage to the genome. A DNA glycosylase/β-lyase enzyme, Ogg1, scavenges the genome for 8-oxoguanine, a major mutagenic DNA adduct induced by reactive oxygen species, and catalyzes its excision and subsequent cleavage of the DNA phosphate backbone. Several polymorphisms of Ogg1, including the single amino-acid substitutions R46Q, R131Q and R154H, are associated with a variety of human cancers. These three mutations have previously been characterized experimentally but no structural data have been published. We have performed multiple molecular dynamics simulations of R46Q, R131Q and R154H human Ogg1 to predict the structural and dynamical effects of the substitutions throughout the protein and specifically within the active site and substrate recognition site. None of the substitutions induced unfolding or global structural changes, instead their effects were confined principally to the active and recognition sites. Although the enzyme active site is located 18-21 Å from the three investigated mutation sites, these mutations' structural effects propagate through space and cause a major change in the orientation and chemical environment of the active site side chains. This change appears likely to compromise the ability of the Lys 249 side chain to undergo a necessary deprotonation step prior to its nucleophilic attack of the DNA. The mutations also cause an expansion of the active site cavity, which may explain the experimentally observed decreases in substrate specificity.
Introduction
Aerobic respiration is the process by which nearly all higher organisms generate energy. Aerobic respiration involves the four-electron reduction of molecular oxygen to water. Partially reduced molecular species that are generated during this reduction process, including O2•-, OH• and H2O2, are strong electrophilic oxidants that can escape from the mitochondria and cause cellular damage. Such reactive oxygen species (ROS) can also be generated by ionizing radiation and organic free radicals. ROS frequently attack the cellular genome, proteins and lipids, with the genome being a particularly frequent target. Attack of the genome by ROS generates numerous genotoxic adducts and DNA strand breaks.1 It is believed that these adducts play an essential role in pathophysiologic processes such as aging and cancer, and consequently considerable research effort has been devoted to understanding the mechanisms of oxidative DNA damage and repair.2
The principal ROS-induced adduct in DNA is 7,8-dihydro-8-oxoguanine (Figure 1a).3,4 Among the most toxic of all adducts3, 7,8-dihydro-8-oxoguanine (also known as 8-oxoguanine or oxoG) is formed by the oxidation of the guanine base in DNA.3,4 Although guanine strongly prefers to pair with cytosine, oxoG pairs with both cytosine and adenine. The ability of oxoG to pair with adenine allows frequent mispairing of oxoG residues with adenine during DNA replication in vivo and in vitro, generating G·C → T·A transversion mutations, the second most common somatic mutation in human cancers that frequently affect the tumor suppressor gene p53 (ref. 5). OxoG is repaired by the base excision DNA repair pathway.5-7 Lesion-specific DNA glycosylases, the principal components of this pathway, scan the genome for damaged bases and catalyze scission of their glycosidic bond (Figure 1b). DNA glycosylase/β-lyases, one class of DNA-repair enzymes, employ an amine-containing residue in their active sites as a catalytic nucleophile and create a covalently linked enzyme-DNA adduct. This adduct subsequently undergoes a set of transformations to produce a DNA strand scission at a site on the 3′-side of the lesion.8 Verdine and co-workers have identified a structural superfamily of DNA glycosylase/β-lyases.9,10 Enzymes in this superfamily are characterized by an active site HhH-GPD motif that contains a helix-hairpin-helix (HhH) element followed by a Gly/Pro-rich loop and ending in a conserved Asp residue.11 Members of this superfamily use an active site Lys located within the HhH-GPD motif as the catalytic nucleophile. Substitution of a highly conserved Asp residue in the active site by Asn disrupts the catalytic activity of the DNA glycosylase/β-lyases, demonstrating a key role for the Asp residue in catalysis.10,11
Figure 1.
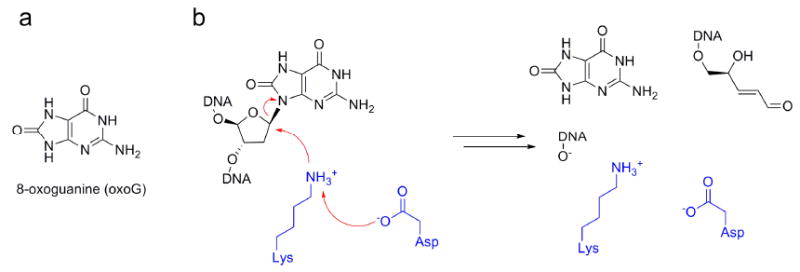
DNA glycosylase substrate and mechanism. (a) Chemical structure of 8-oxoguanine (oxoG), generated by oxidative damage of DNA by reactive oxygen species. (b) Overview of Schiff-base enzymatic mechanism of DNA glycosylases responsible for excision of oxoG from oxidatively damaged DNA. An Asp side chain is proposed to deprotonate a nearby Lys side chain, which proceeds to undergo a nucleophilic attack on the C1′ atom of the oxoG nucleotide, ultimately leading to strand cleavage at the 3′ phosphate group.
The DNA glycosylase/β-lyases of the HhH-GPD superfamily are critical for the recognition and removal of a large number of DNA lesions arising from alkylation, oxidation, deamination and other types of genetic damage. The enzyme responsible for repair of oxoG in eukaryotes is 8-oxoguanine glycosylase (Ogg1), a DNA glycosylase/β-lyase.12 The base-excision activity of Ogg1 is specific for oxoG and is context dependent, requiring the presence of cytosine on the complementary strand.13,14 Similar to many other DNA glycosylases2, Ogg1, after recognizing oxoG, flips out the oxoG residue from the DNA double strand to bind the altered base in a specific recognition pocket, catalyzes excision of oxoG, and then mediates cleavage of the phosphate backbone.
Wild-type human Ogg1 (hOgg1) consists of three domains assigned by CATH15 (Figure 2a). Residues 12-100 form a α/β domain with a 2-layer sandwich architecture. Residues 104-124 and 265-323 form a mainly-α domain with an orthogonal bundle architecture. The third domain, consisting of residues 136-261, is also mainly α and has an orthogonal bundle architecture. As shown in Figure 2b, the overall hOgg1 secondary structure is 43% helical (16 helices; 149 residues) and 10% β-sheet (7 strands; 35 residues). According to SCOP16 classification, hOgg1 residues 136-323 form a DNA-glycosylase fold. This fold is common to other members of the superfamily of DNA glycosylase/β-lyases, including E. coli endonuclease III, E. coli alkylation repair DNA glycosylase AlkA and E. coli DNA glycosylase MutY (Figure 2c). As members of the DNA-glycosylase/β-lyase superfamily, these proteins are typified by the presence of a HhH-GPD motif. In addition to the two α-helices conserved in the HhH-GPD motif, hOgg1 contains a third antiparallel β-sheet domain found in few other DNA glycosylases.
Figure 2.
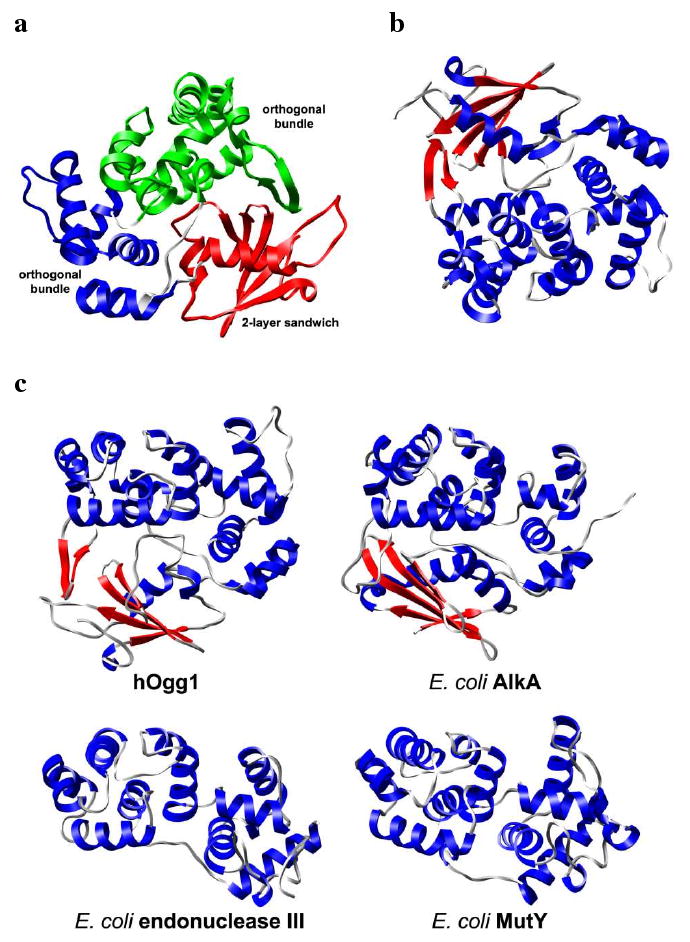
Structure of hOgg1 bound to oxoG-containing DNA (DNA not shown). (a) CATH-designated hOgg1 domains (colored separately). (b) Alternate view of protein colored by secondary structural elements. α-helices and β-strands are colored blue and red, respectively. (c) Representative structures from the DNA glycosylase/β-lyase superfamily with a HhH-GPD motif sharing a DNA-glycosylase fold. Secondary structural coloring is the same as that for part b. Structures were aligned to human hOgg1 by Cα-RMSD fitting. All hOgg1 structures are from PDB code 1ebm.
Several mutations in hOgg1 are associated with human cancers. Chevillard and co-workers have reported that homozygous mutations in OGG1 are frequently found in human lung and kidney tumors.17 One identified single nucleotide polymorphism (SNP) leads to a R131Q substitution. Despite the 20 Å distance between Arg 131 and the Lys 249 nucleophile (Figure 3a), R131Q completely abolishes hOgg1 activity.17 No experimental structure of R131Q hOgg1 has been published. It has been speculated that the R131Q substitution has an adverse effect on protein folding, as the guanidinium group of Arg 131 is located in the interior of the structure and is involved in an extensive network of hydrogen bonds (Figure 3b).18 A R46Q substitution is observed in many human kidney tumors.19 Structures of R46Q hOgg1 are not available. Arg 46 in wild-type (wt) hOgg1 is buried in the interior of the protein and its guanidinium group forms two hydrogen bonds with the carbonyl oxygen atom of His 179 (Figure 3c). Experiments show that, despite the 21 Å separation between Arg 46 and Lys 249, the R46Q substitution diminishes hOgg1 activity and decreases hOgg1 selectivity for the base opposite oxoG.20 Furthermore, a R154H substitution has been linked to gastric cancer.20 Although the side chain of Arg 154 is located on the exterior of the protein 18 Å from Lys 249 (Figure 3d), R154H causes a greater decrease in the enzyme activity and selectivity for oxoG than does R46Q.20
Figure 3.
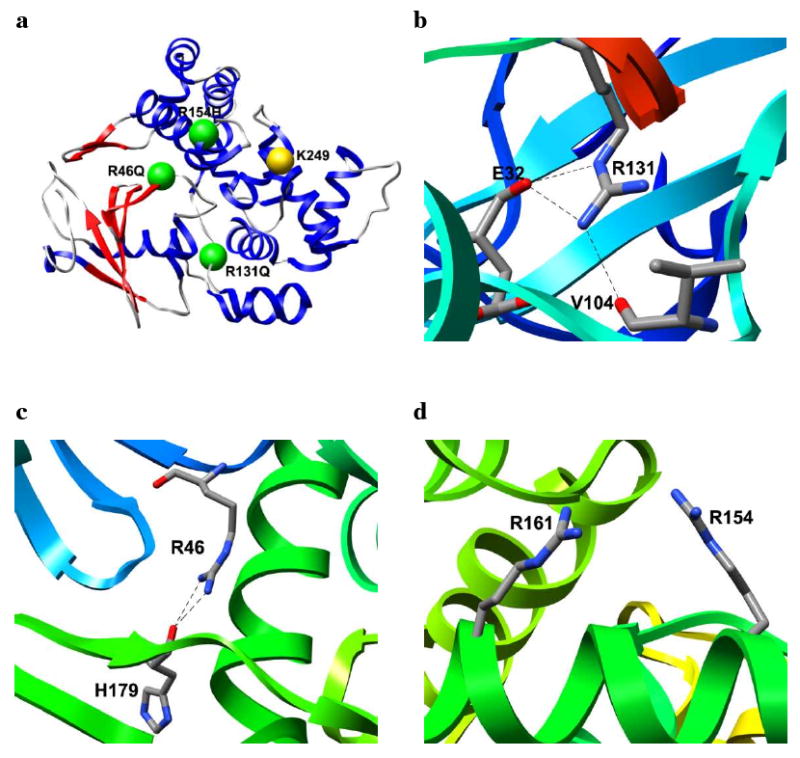
Locations and local environments of hOgg1 polymorphisms. (a) Positions of polymorphic residues (green) relative to the active site Lys 249 residue (yellow). Protein structure is colored blue for α-helices and red for β-strands. Local environments of (b) Arg 131, (c) Arg 46 and (d) Arg 154 in hOgg1 wt. Hydrogen bonds are denoted by dashed lines.
Given the lack of experimental structural data for these particular hOgg1 polymorphisms, we have predicted their structural and functional effects via multiple molecular dynamics (MD) simulations. These simulations are part of our Dynameomics effort.21-23 The goal of our Dynameomics effort is to create a repository for molecular-dynamics data to supplement static structural information. We are currently expanding the scope of Dynameomics to develop a database for molecular dynamics data of proteins encoded by non-synonymous SNPs. For example, in addition to hOgg1, simulations of mutations of human catechol O-methyltransferase24, thiopurine S-methyltransferase25, histamine N-methyltransferase26, and DJ-127 have been added to our data repository.
The hOgg1 simulations presented here reveal that the R46Q, R131Q and R154H mutations induce structural changes that propagate through space (∼18-21 Å) to distort the orientation of the distant active site and oxoG-recognition site residues while leaving the overall fold unchanged. Side chain reorientations in the active site occurred that are predicted to jeopardize the ability of the catalytic Lys 249 to be deprotonated, a prerequisite for nucleophilic attack of the C1′ atom in the oxoG residue.28 Likewise, in contrast to the wt protein, the mutants all formed active-site cavities large enough to accommodate oxoG without first having to undergo reorganization, which may further reduce the impetus for Lys 249 to become deprotonated. Expansion and distortion of the recognition pocket was also observed for the R46Q and R131Q mutations while interruption of important hydrogen bonding interactions occurred in the R154H structure. Taken together, these results are consistent with, and provide explanations for, the decreased catalytic activity and oxoG selectivity of hOgg1 observed experimentally.
Methods
Molecular Dynamics Simulations and Analysis
All wt simulations were based on the 2.15 Å resolution crystal structure28 of human hOgg1 as solved at 4 °C and pH 4.6 (PDB code 1ko9). The structure includes residues 12-323 of the wt protein. Based on apparent hydrogen bonding in the structure, His 237 was protonated at its Nε atom. Starting structures of the R46Q, R131Q and R154H polymorphs were prepared by making the amino acid substitutions to the wt protein and minimizing the torsional, electrostatic and van der Waals interactions of the resulting mutant structure in vacuo using the ENCAD simulation package29.
Atomic partial charges and the potential energy function for all proteins were taken from Levitt et al30. Starting protein structures were minimized in vacuo for 1000 steps of steepest descent minimization. The minimized structures were subsequently solvated in a rectangular box of flexible three-center (F3C) waters31 with walls located ≥ 10 Å from any protein atom. The solvent density of the water box was preequilibrated to a density of 0.993 g/mL, the experimental water density at 310 K and 1 atm pressure32. The solvent was minimized for 1000 steps and this minimization was followed by 1 ps of dynamics of the solvent only and then by an additional 500 steps of solvent minimization. The entire system was subsequently minimized for 500 steps and then heated to 310 K.
MD simulations were performed at 310 K in the microcanonical (NVE) ensemble using the in lucem molecular mechanics (ilmm) program33. Three simulations ranging from 22 ns to 24 ns were performed for wt hOgg1 and for each of the three polymorphs. Simulations performed in triplicate differed by using different random number seeds to initiate assignment of the initial velocity distributions. Protocols and the MD potential energy function have been described elsewhere.30,34 The simulations included all hydrogen atoms, which were added by ilmm. A force-shifted nonbonded cutoff of 10 Å was used and the nonbonded interaction pair list was updated every three steps. A time step of 2 fs was applied in all simulations and structures were saved every 1 ps for analysis.
Analysis of MD simulations was performed using ilmm. Cα root-mean-square deviations (Cα-RMSD), Cα root-mean-square fluctuations (Cα-RMSF) about the mean, contact distances, solvent-accessible surface areas (SASA), and secondary structure assignments were calculated. An atomic contact was defined as occurring when an interresidue C-C atom distance was ≤ 5.4 Å or a heavy-atom (C, O, N, S) distance was ≤ 4.6 Å in two nonadjacent residues. Residues were considered to be in contact if any interresidue pair of atoms formed a contact. A contact was counted as a hydrogen bond if the donor-acceptor angle was >135° and the hydrogen-acceptor distance was ≤ 2.6 Å. SASAs were calculated using the NACCESS algorithm35 and secondary structures were assigned by the DSSP algorithm36. Protein images were produced using Chimera37.
Results and Discussion
Effects of R46Q, R131Q and R154H Mutations on hOgg1 Global Structure
Table 1 lists several overall structural parameters from the simulations for wt hOgg1 and the R46Q, R131Q and R154H polymorphs at 310 K. The amino acid substitutions do not induce a notable increase in the average Cα-RMS deviations from the appropriate starting structure relative to the wt simulations. Each polymorphism's average Cα-RMSD is within the range of wt behavior. Similarly, the polymorphic structures do not display an increase in total solvent-accessible surface area (SASA) relative to wt. The failure to increase SASA is consistent with the overall structural stability implied by the Cα-RMS deviations of the polymorph structures and indicates that the structures do not expand appreciably during the simulation. It is interesting to note that the average number of total intramolecular contacts is well conserved between the wt and polymorph simulations at 310 K, with alternative, nonnative contacts compensating for lost native contacts. Such behavior contrasts with that of DJ-1, where a single amino acid substitution is associated with a significant reduction in the number of total intramolecular residue-residue contacts.27
Table 1.
General properties of hOgg1 simulations. Properties at 310 K represent averages for 3 simulations over the last 5 ns of each simulation.
| Temp (K) | Cα-RMSD (Å) | Total SASA (Å2) | Intramolecular residue contacts | |
|---|---|---|---|---|
| wt | initial | 0.0 | 13730 | 1317 |
| 310 | 3.5 ± 0.7 | 16320 ± 506 | 1199 ± 13 | |
| R46Q | initial | 0.0 | 13733 | 1315 |
| 310 | 3.7 ± 0.3 | 16399 ± 277 | 1198 ± 16 | |
| R131Q | initial | 0.0 | 13718 | 1318 |
| 310 | 3.5 ± 0.4 | 16322 ± 356 | 1196 ± 13 | |
| R154H | initial | 0.0 | 13705 | 1315 |
| 310 | 3.8 ± 0.8 | 16223 ± 312 | 1203 ± 16 |
Figure 4 shows the average structures over the last 5 ns of the simulations. No one of the simulations displays unfolding or significant loss of secondary structural elements. The R46Q and R131Q simulations, however, show some disruption of the four-residue β1 strand. In R154H, β2 is lost and a α-helix forms over residues 14-19, which immediately precede α1. The native α-helices are stable in all simulations.
Figure 4.
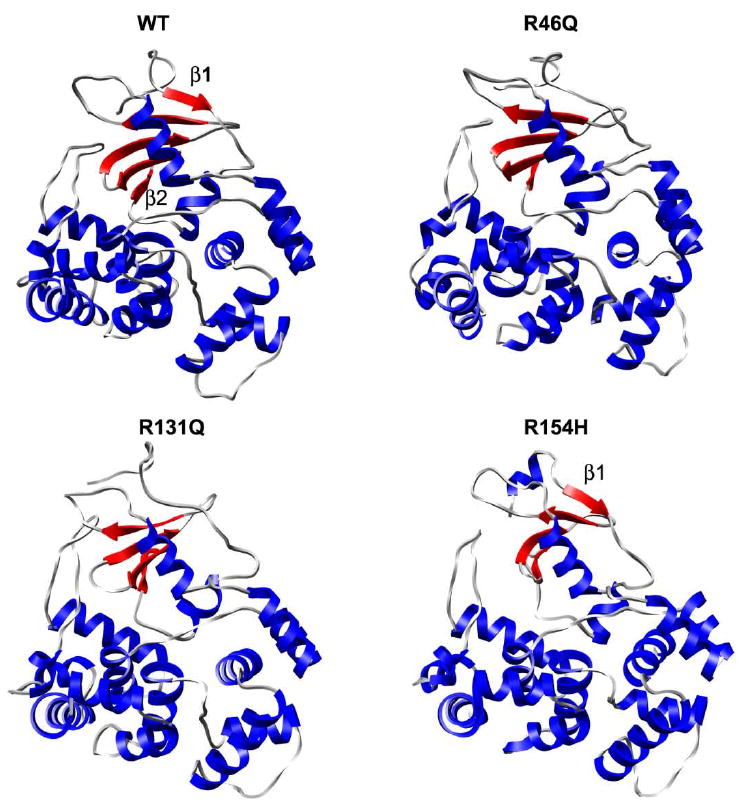
Average structures from last 5 ns of hOgg1 simulations at 310 K. α-helices and β-sheets are colored blue and red, respectively.
As shown in Figure 5, despite the similar average Cα-RMS deviations in the wt and mutant simulations, there is a broad distribution of Cα-RMSDs over the last 10 ns of the simulations, particularly for the wt protein and the R154H polymorph. The R154H polymorph occurs predominantly in either a state similar to the starting structure or in a structurally distorted state, with 26% and 40% of its sampled conformations having Cα-RMS deviations <3 Å and >4 Å, respectively. The α/β domain of R154H (Figure 6) has the largest average per-residue Cα-RMS deviations, whereas the two mainly-α domains show considerably less deviation from the starting structure. In contrast to the R154H polymorph, the wt has only 13% of its sampled conformations with Cα-RMSD >4 Å, and two peaks in its Cα-RMSD distribution occur at 2.4 Å and 3.5 Å. The R46Q and R131Q polymorphs have single, wider distribution peaks centered at 3.9 Å and 3.5 Å, respectively.
Figure 5.
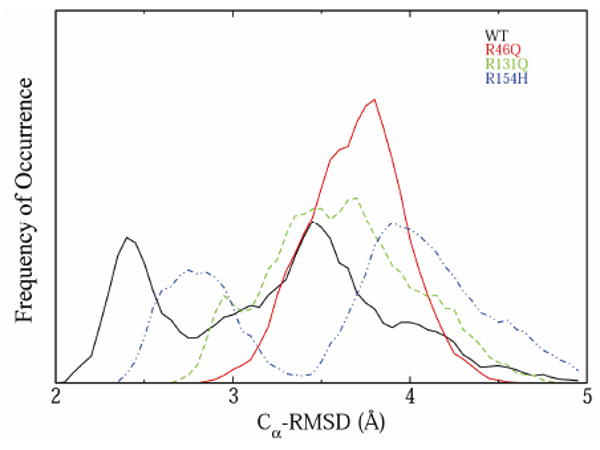
Cα-RMSD distributions during the last 10 ns of simulations of hOgg1 wt structure and polymorphs at 310 K. Plots are solid black (wt), solid red (R46Q), dotted green (R131Q) and dotted blue (R154H).
Figure 6.
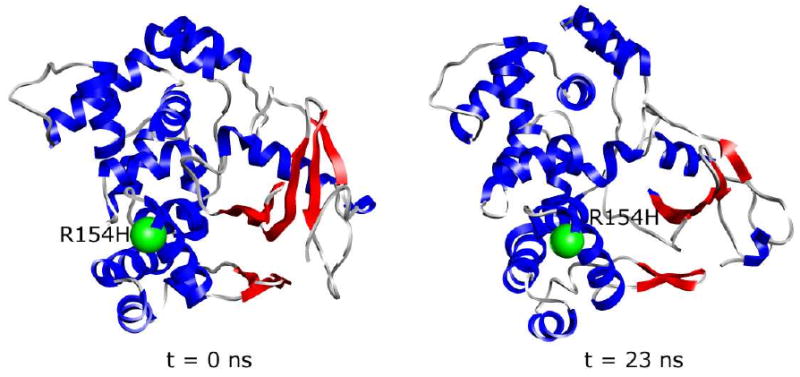
Distortion of the α/β domain of R154H polymorph at 310 K. The β-sheets of the α/β domain are colored red. The site of the R154H substitution is represented as a green sphere. The starting structure (left) is compared with the structure at 23 ns of one of the three R154H hOgg1 simulations (right).
The average per-residue Cα-RMS fluctuations are nearly equal in magnitude for the wt and all polymorph simulations. Residues 287-290, located in the loop connecting α14 and α15 within the mainly-α domain with orthogonal bundle architecture, show the largest Cα-RMSF (∼1.5 Å) in all simulations. Residues of the active site that are involved in the catalytic mechanism (Lys 249, Asp 268) and that recognize the flipped out oxoG (Gly 42, Cys 253, His 270, Gln 315, Phe 319) have average or below-average Cα-RMS fluctuations (0.5-0.8 Å) both in the wt and polymorph simulations. These small fluctuations in the hOgg1 active site indicate that structural distortions induced by the amino acid substitutions do not lead to increased dynamic motions in the active site. Nevertheless, these mutations disrupt the relative orientations of the active site residues that are necessary for effective catalysis and oxoG recognition. The resulting structural alterations remain stable throughout the simulations.
Structural Distortion of Active Site in hOgg1 Polymorphisms
A proposed mechanism10,38 for hOgg1 and other glycosylases and β-lyases involves Lys 249 acting to displace the oxoG base and to promote elimination of the DNA 3′-phosphodiester via a Schiff base mechanism (Figure 1b). Asp 268, a conserved feature of the HhH-GPD motif, may also assist the catalytic mechanism by transferring protons to and from Lys 249.9,10,39Figure 7 shows the effects of the side chains of the residues involved in molecular recognition and catalysis in the active site (Lys 249, Cys 253, Asp 268, His 270, Gln 315, Phe 319). Although the simulations did not include the oxoG-containing DNA, the active site perturbations that they reveal provide insight into the structural basis for the decreased hOgg1 activity and selectivity caused by the R46Q, R131Q and R154H mutations.
Figure 7.
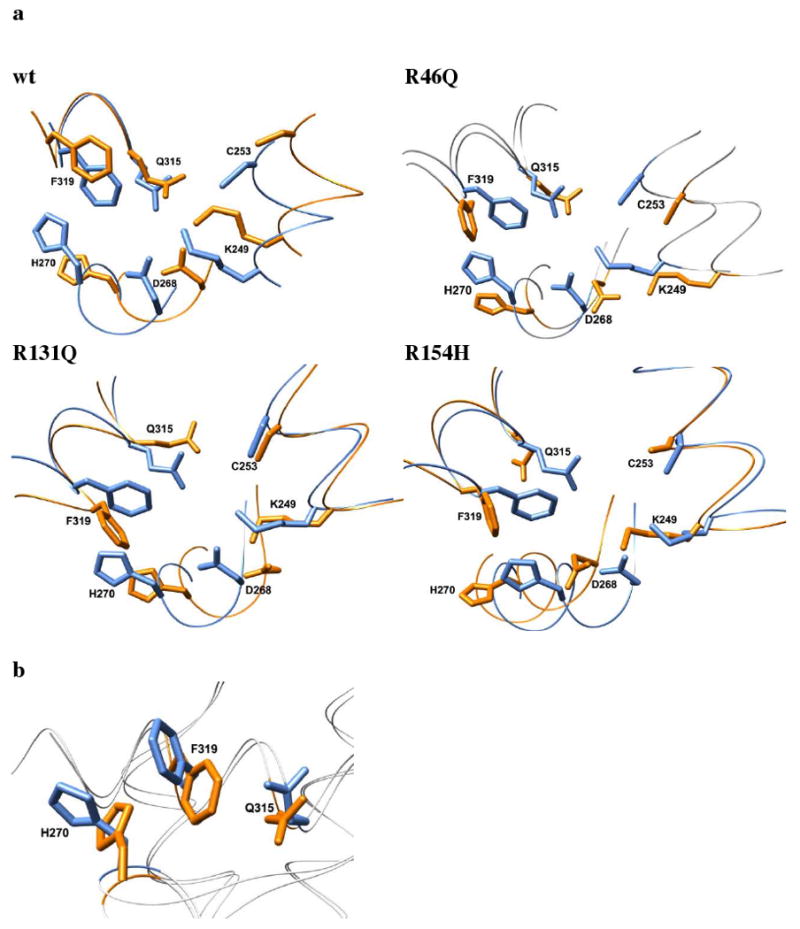
Structural effects of mutations on hOgg1 active site at 310 K from molecular dynamics simulations. (a) The wt hOgg1 crystal structure (blue) is shown overlapped with the average structures (orange) from the last 5 ns of one simulation of each mutated protein. The depicted structural changes were observed in all simulations for each mutation. (b) Experimentally observed changes in hOgg1 oxoG binding pocket between free and DNA-bound states. Blue and orange side chains represent the DNA-bound and free states of hOgg1, respectively. DNA is not shown. Alignment generated by Cα-RMSD fit of X-ray crystal structures of the hOgg1-oxoG DNA complex (PDB code: 1ebm) and the unbound protein (PDB code: 1ko9).
The orientations of the active site side chains relative to each other are conserved in all wt simulations at 310 K, and the top two layers (Phe 319 and Gln 315) of the three-layer sandwich formed in the crystal structure by these residues and His 270 remain intact. The His 270 side chain undergoes a dynamic swinging motion between the solvent and a stacked position with Phe 319 and Gln 315, completing the sandwich. This conformational change of His 270 is the only significant conformational change observed in the active site in the wt hOgg1 simulations and appears to be intrinsic to the dynamics of hOgg1.
Although the active site is situated ∼21 Å from residue 46, the R46Q mutation destroys the active site three-layer sandwich by reversing the orientation of the Phe 319 side chain such that it moves to the surface of the protein from its initial buried position in each simulation (Figure 7). The Phe 319 phenyl group becomes approximately perpendicular to the average plane of the imidazole ring of the His 270 side chain, which itself undergoes the same dynamic swinging motion as that in the wt simulations. The Phe 319 conformational change, which is never observed in the wt simulations, greatly increases the distance between the Phe 319 and Gln 315 side chains. Moreover, the orientation of Asp 268 also changes such that the torsion angle about its Cα-Cβ bond is ∼180° relative to that of the wt crystal structure. This reorientation occurs at the same time that the Lys 249 side chain moves slightly outward from its initial buried position, such that the salt bridge between Lys 249 and Asp 268 is maintained throughout the simulations.
The hOgg1 active site is ∼20 Å from residue 131, and the R131Q mutation likewise disrupts the active site's three-layer sandwich in each simulation (Figure 7). The Phe 319 side chain becomes roughly perpendicular to the imidazole ring of His 270, increasing its distance from the Gln 315 side chain. As in the other simulations, His 270 is dynamic throughout the simulation, consistently flipping outward into the solvent. In contrast to the R46Q simulations, the orientation of Asp 268 is not significantly altered relative to that of the wt crystal structure. The salt bridge formed between Lys 249 and Asp 268 remains intact throughout all simulations.
The site of the R154H mutation is located ∼18 Å from the active site and also perturbs the active site structure. Like the other two mutations, R154H disrupts the three-layer sandwich in all three simulations (Figure 7). The Phe 319 side chain changes orientation so that it is ∼90° from its original orientation and becomes roughly perpendicular to the plane of the His 270 ring. Despite this change, its position is still compatible with forming contacts with oxoG, as it does in the crystal structure of the DNA-bound protein. Similar to the R46Q mutation, R154H is associated with a reorientation of Asp 268 such that the torsion angle about the Cα-Cβ bond is ∼180° relative to that of the wt crystal structure. However, its carboxylate group remains in the proper orientation to accept a hydrogen bond from the N3 atom of oxoG. As in simulations of the other mutations, the salt bridge between Lys 249 and Asp 268 is maintained during the simulations. Moreover, nonnative contacts are formed between Lys 249 and Ser 148, and a hydrogen bond is formed between these two residues' side chains.
A major difference between R154H and the other mutations is its drastic effect on Gln 315. In the simulated R154H structures, Gln 315 points away from the active site, forming contacts with the side chains of Val 265, Met 271, Phe 303, Trp 307 and Leu 318. Gln 315 is oriented such that it loses the two hydrogen bonds with the N1 and N2 atoms of the oxoG base that it forms in the crystal structure of the wt protein. An inability of oxoG to form these two hydrogen bonds with the hOgg1 active site would be expected to reduce the binding affinity of oxoG-containing DNA for hOgg1.
Implications of Structural Effects for Catalysis
A common observation across the different mutant simulations is that the three-layer sandwich formed by the side chains of His 270, Phe 319 and Gln 315 is disrupted. The mutations consistently cause the Phe 319 side chain to adopt a conformation roughly perpendicular to that in the wt simulations and in the wt crystal structure. As shown in Figure 7b, this perpendicular orientation of the Phe 319 side chain is also adopted in the wt structure upon complex formation with oxoG-containing DNA. Thus, the R46Q, R131Q and R154H mutations induce a Phe 319 conformation in the unbound hOgg1 protein that mimics that in the wt oxoG-bound state.
The similarity in the Phe 319 side chain conformation between the unbound mutants and the bound wt protein has important implications for hOgg1 catalytic activity. In order to perform a proposed nucleophilic addition to the C1′ carbon atom of the oxoG sugar ring, the amino group of Lys 249 needs to be deprotonated beforehand.28 This deprotonation step is unlikely to occur when Asp 268 forms a stabilizing salt bridge with Lys 249. A mechanism has been suggested by which movements in Phe 319 and His 270 induced by oxoG binding change the position of Asp 268, allowing its stabilizing interactions with Lys 249 to be broken.28 In the case of the mutants studied here, however, it appears that little or no movement of Phe 319 would occur upon DNA binding, as the Phe 319 side chain is already in a position similar to that of the hOgg1-oxoG complex. Moreover, unlike the wt unbound structure, the unbound mutants already have a cavity of sufficient volume to accommodate an oxoG residue (Figure 8), thereby eliminating major reorganization of the active site as a prerequisite for binding. Additionally, the stabilizing interactions between Lys 249 and Asp 268 are maintained over the entirety of every mutant simulation, despite the reorientation of other side chains in the catalytic site. For these reasons, it is likely that these stabilizing interactions would not be interrupted by oxoG binding. The Lys 249 amino group may then be prevented from undergoing deprotonation and would not become armed for nucleophilic catalytic activity.
Figure 8.
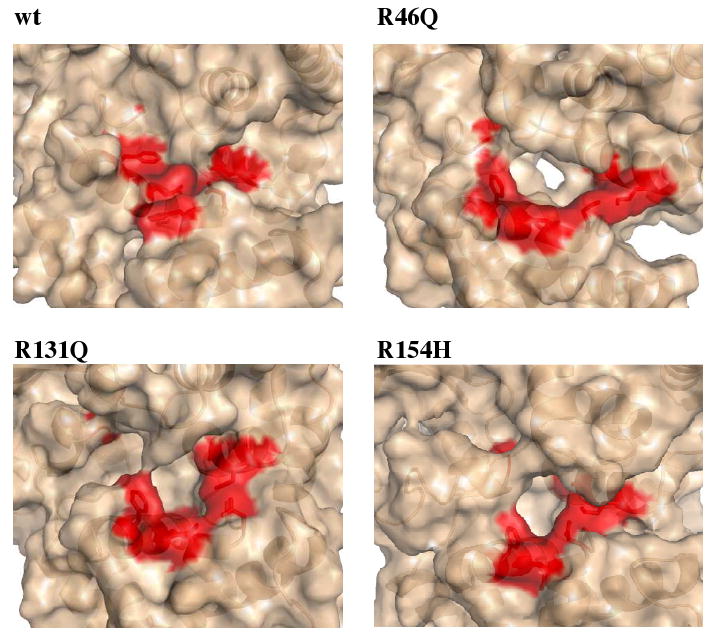
Molecular surface in the vicinity of the hOgg1 active site at 310 K. The surface portions corresponding to residues involved in catalysis and oxoG base recognition (Gly 42, Lys 249, Cys 253, Asp 268, His 270, Gln 315, Phe 319) are colored red. A cavity sufficiently large to accommodate the oxoG base is formed in the mutant structures but not in the unbound wt protein. Structures are taken from snapshots at 23 ns.
Structural Distortion of Recognition Pocket for Unpaired Cytosine in Mutant hOgg1
The hOgg1 specificity pocket comprises Asn 149, Arg 154, Tyr 203 and Arg 204. The crystal structure of hOgg1 bound to DNA containing oxoG shows that the protein binds to the backbone of the DNA strand containing oxoG but does not contact the backbone of the complementary strand. hOgg1 extrudes oxoG, the substrate, from its intrahelical position within the DNA, and oxoG becomes deeply inserted into an extrahelical active-site cleft on the enzyme (Figure 9a). The protein forms an extensive network of stabilizing contacts with the backbone of the oxoG-containing DNA strand (Figure 9b). The Asn 149 side chain, which is buried in the protein in its unbound state, rotates outward away from the protein upon DNA binding in order to form a stabilizing interaction with the N4 atom of the unpaired cytosine base. In the simulations, the effects of the mutations R46Q, R131Q and R154H propagate not only to the active site but also to the recognition pocket for the unpaired cytosine base opposite the flipped-out oxoG residue in the DNA-hOgg1 complex (Figure 9c).
Figure 9.
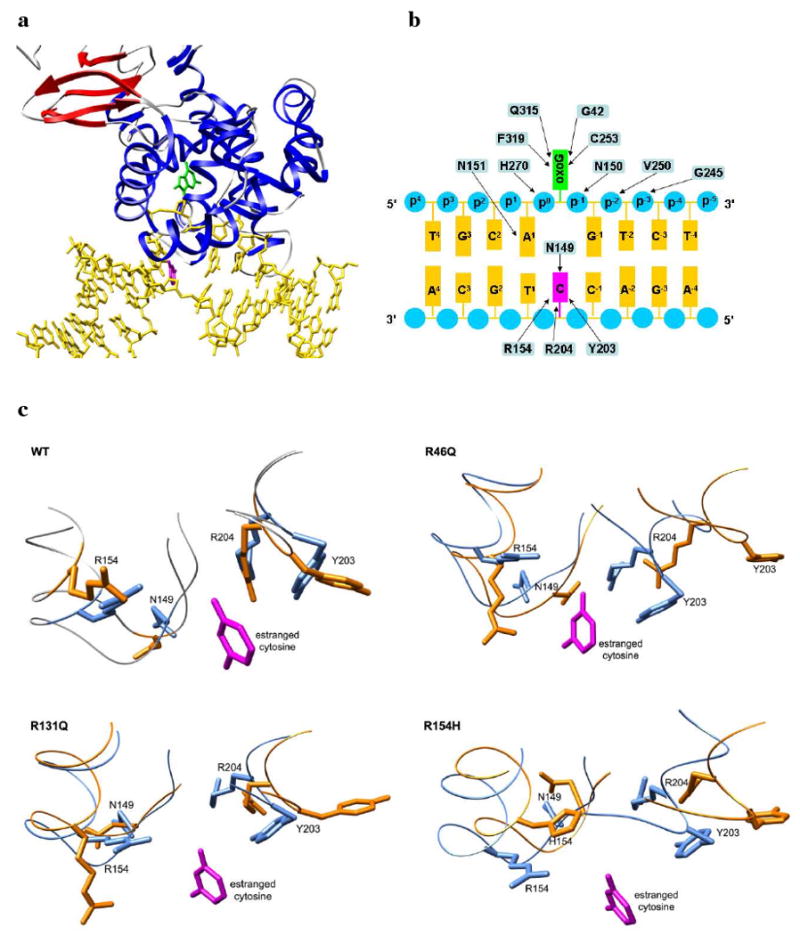
Recognition of oxoG-containing DNA by hOgg1 and structural disruption of binding pocket by mutations. (a) Experimental structure of DNA (yellow) bound to hOgg1 (PDB code: 1ebm). The extruded oxoG residue and the resulting unpaired cytosine residue of the DNA are colored green and magenta, respectively. Protein structure is colored blue for α-helices and red for β-strands. (b) Diagram of contact interface between hOgg1 and oxoG-containing DNA observed in experimental structure. Arrows indicate interactions between amino acid residues (blue rectangles) and oxoG (green rectangle), DNA backbone phosphate groups (blue circles), DNA bases (orange rectangles) and the unpaired cytosine opposite oxoG (magenta rectangle). Figure adapted from ref. 18. (c) Structural effects of mutations on hOgg1 recognition pocket for the unpaired cytosine residue at 310 K from molecular dynamics simulations. The average structures from the last 5 ns of one simulation of each mutant (orange) are shown overlapped with the wt unbound crystal structure (blue). The cytosine base (magenta) is shown by overlapping the crystal structure of the oxoG DNA-bound protein (PDB code: 1ebm) with the other two structures.
In each of the wt hOgg1 simulations the orientations and relative positions of the recognition pocket residues are reasonably well conserved. Tyr 203 is flexible about its Cα-Cβ bond and fluctuates between having its hydroxyl group pointing outward into solution and pointing inward toward the position that would be occupied by the cytosine base. Despite this flexibility, the positions of the residues do not change notably relative to their positions in the unbound wt crystal structure.
In contrast to the wt simulations, each of the R46Q simulations shows a major widening of the pocket between the α5-loop-α6 motif and the α8-loop-α9 motif, which recognizes the unpaired cytosine base. The Cα atom of Tyr 203, whose side chain forms a critical interaction with the cytosine base, moves ∼7 Å away from its position in wt and the side chain points outward into solution for the majority of the simulation. Similarly, the Arg 204 Cα atom moves ∼5 Å from its wt position. The side chain of Arg 154 is likewise reoriented, changing by ∼90° relative to wt, but the positions of its backbone atoms do not change significantly. The large displacement of Tyr 203 and Arg 204, along with the widening of the pocket, would likely prevent these residues from forming their specific interactions with the unpaired cytosine base, reducing or eliminating the selectivity for oxoG-containing DNA.
In two of the three R131Q simulations the mutation does not cause widening of the pocket between the α6-loop-α7 motif and the α9-loop-α10 motif, as occurs for the R46Q mutation. The Cα positions of the residues comprising the pocket are not notably altered in these two simulations, and Tyr 203 spends roughly equal amounts of time pointing outward into solution and inward toward the position of cytosine binding. In one of the simulations, however, the pocket does widen drastically, with the Tyr 203 main chain moving ∼8 Å from its initial location. In all three simulations the side chain orientation of Arg 154 also periodically changes but still spends a considerable amount of time in its native orientation. Although the pocket widening is observed in only one of the three R131Q simulations, it is likely that the displacement of Tyr 203 and Arg 204 would interrupt the interactions that these residues form with the unpaired cytosine. This interruption may reduce the overall hOgg1 binding selectivity for oxoG relative to wt.
The R154H substitution does not alter the width of the pocket appreciably. However, replacement of Arg 154 by His, whose side chain normally forms three hydrogen bonds with the O2 and N3 atoms of the unpaired cytosine, interrupts these hydrogen bonds. Since the lost hydrogen bonds represent half of the total hydrogen bonds formed by hOgg1 with the cytosine base, the R154H substitution is expected to lower the selectivity of hOgg1 for oxoG-containing DNA.
Effects of Mutations on hOgg1 Residue Contacts
The structural effects of the mutations on the hOgg1 active site and recognition pocket are presumably caused by interruptions of the residue-residue contact networks that link these regions of the protein to the mutation sites. In order to evaluate this proposition, residue-residue contacts that were either diminished or accentuated in the mutant hOgg1 simulations relative to the wt simulations were determined (Figure 10). Contacts that are present for an average occupancy time percentage that is 50% lower or higher across all three simulations of a mutant relative to that for wt are counted as diminished or accentuated, respectively.
Figure 10.
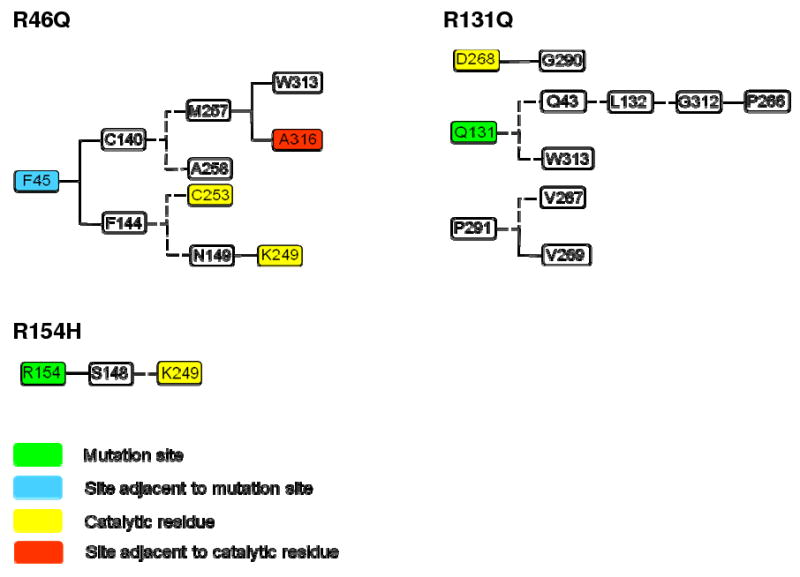
Networks of altered residue-residue contacts for mutant hOgg1 simulations that link mutation sites to active site. Solid and dashed lines represent diminished and accentuated contacts, respectively. A contact is considered diminished or accentuated if the average value of its percentage occupancy time over all three simulations for a particular mutant is 50% smaller or larger than the corresponding average of all three wt simulations, respectively.
The R46Q mutation involves contact perturbations that span the whole width of the protein on both sides of the mutation site. The network of diminished and accentuated contacts links the vicinity of the mutation site to the active site (Figure 10). Replacement of Arg 46 by a Gln induces an adjacent residue, Phe 45, to have diminished contacts with both Cys 140 and Phe 144. The diminution of contacts between Cys 140 and Phe 45 is followed by formation of nonnative contacts between Cys 140 and Met 257. Met 257, located in the vicinity of the active site, has in turn diminished contacts with Ala 316, which is adjacent to Gln 315, a member of the active site three-layer sandwich. The disruption of contacts between Phe 45 and Phe 144 likewise links the vicinity of the mutation site to the protein active site. After losing contact with Phe 45, Phe 144 forms contacts with Cys 253, which is part of the active site. In addition, Ser 143 moves near Asn 149, which has diminished contacts with Lys 249, another active-site residue. Lys 249 does not compensate for these diminished contacts by interacting with alternative residues. Furthermore, Val 267, which is adjacent to Asp 268, a proposed catalytic residue, forms a contact with Pro 291 that is not observed in the wt simulations. This contact may stabilize the alternate Asp 268 side chain conformation that is observed in the R46Q simulations. For the wt simulations the standard deviation of percentage occupancy time of most of the contacts plotted in Figure 10 is similar to the standard deviation for the R46Q simulations. However, the Phe 144-Asn 149, Asn 149-Lys 249, and Met 257-Ala 316 contacts have greater standard deviations of percentage occupancy time for the wt simulations than for the R46Q simulations.
The altered contacts in the R131Q simulations are similarly located throughout the protein. As shown in Figure 10, the proposed catalytic residue Asp 268 and residues immediately adjacent to it are involved in altered contacts. Asp 268 has a diminished contact with Gly 290 and does not have any compensatory contacts. Val 267 and Val 269 have accentuated and diminished contacts with Pro 291, respectively. Gln 131 forms new contacts with Gln 43, which in turn makes novel contacts with Leu 132. Leu 132 likewise forms new contacts with Gly 312, and the latter has diminished contact with Pro 266. A position change in Pro 266 accompanying its loss of contacts with Gly 312 is associated with a position change of Asp 268, as well. Moreover, not only does R131Q break a contact of Asp 268 but it also breaks a contact of Ala 314, which is positioned next to the critical Gln 315 residue of the active site three-layer sandwich. The standard deviation of occupancy time percentage among the three wt simulations exceeds that among the three R131Q mutations for the Arg/Gln 131-Gln 43, Arg/Gln 131-Trp 313, Gln 43-Leu 132, and Leu 132-Gly 312 contacts, whereas the other contacts have similar standard deviations among wt and mutant simulations.
R154H is associated with the largest total number of residues involved in contact alterations among the simulated mutants, with 13 diminished contacts and 20 accentuated contacts. The catalytic Asp 268 residue does not have appreciably altered contacts and only one of its neighbors, Val 267, is involved in contact changes, diminishing contacts with Pro 291 and Gly 300 while having an accentuated contact with Trp 272. However, the R154H substitution is involved in a network of contact changes that links residue 154 to the active-site Lys 249 residue (Figure 10). His 154 has diminished contacts with Ser 148 that Arg 154 forms in the wt simulations. In turn, Ser 148 forms nonnative contacts, including a hydrogen bond, with Lys 249. The standard deviations of occupancy time percentage for the Ser 148-Lys 249 contact are similar among the three wt and three R154H simulations, while that of the Arg/His 154-Ser 148 contact is greater among the three wt simulations than among the mutant simulations.
In all simulations of mutant hOgg1, the structure of hOgg1 at or adjacent to the mutation sites is perturbed by the relative disruption or formation of at least one residue contact. R131Q, however, is the only mutation for which there is a disruption or formation of more than a single adjacent residue contact. This finding implies that the immediate environment of the region of the mutation does not necessarily need to be greatly disturbed in order for structural disruptions elsewhere in the protein to occur. Analysis of contact changes reveals a plausible contact pathway for active site disruption for each of the three mutations, demonstrating how single amino acid mutations may induce distant structural changes. In the case of R154H the network of altered contacts is simple, with a single amino acid, Ser 148, mediating contact changes that link the mutation site to Lys 249 of the active site. The R46Q and R131Q mutations, in contrast, have more complicated contact networks, but the networks nevertheless suggest a possible mechanism for structural disruptions that travel 18-21 Å through space. These observations indicate that networks of sequentially changed residue contacts may provide a general explanation for local and global structural changes in mutant proteins.
Conclusions
DNA glycosylase/β-lyase hOgg1 is a crucial enzyme for the repair of oxidatively damaged DNA containing 8-oxoguanine residues in humans. The ability of hOgg1 to perform its catalytic function hinges on both its proper recognition of DNA containing 8-oxoguanine and the proper positioning and protonation state of residues within its catalytic site. R46Q, R131Q and R154H compromise the ability of hOgg1 to recognize oxoG-containing DNA and its catalytic activity, despite the fact that the mutation sites are positioned 18-21 Å from the active site. However, experimental structures of these mutant proteins are not available.
Simulations performed in triplicate for each mutation at physiological temperature show that the mutations do not cause unfolding of hOgg1 within the time limits sampled. However, the mutations induce often drastic conformational changes in both the active site and the unpaired-cytosine recognition pocket. These changes appear sufficient to diminish or abolish proper recognition of the oxoG base and the cytosine base. Moreover, side chain reorientations in the active site were revealed that could jeopardize the ability of the catalytic Lys 249 residue to be deprotonated, a prerequisite for nucleophilic attack on the C1′ atom of the oxoG residue. These predicted structural and functional effects are consistent with the experimentally characterized disruption of hOgg1 activity. The sequences of contact changes spatially linking the mutation sites and the active site are clearly suggested for each mutation.
This work builds on the case of catechol O-methyltransferase24, thiopurine S-methyltransferase25, histamine N-methyltransferase26 and DJ-127, for which it has been observed that single amino acid mutations cause major structural perturbations in protein functional sites situated ∼16 Å from mutation sites. The ability of local structural effects upon mutation to propagate to distant regions of proteins may be a general trait of proteins that are mutated in human disease. We are investigating this possibility as part of our SNP database in Dynameomics.
Abbreviations
- ROS
reactive oxygen species
- oxoG
7,8-dihydro-8-oxoguanine
- Ogg
8-oxoguanine glycosylase
- wt
wild-type
- MD
molecular dynamics
- ilmm
in lucem Molecular Mechanics
- RMSD
root-mean-square deviation
- RMSF
root-mean-square fluctuation
- SASA
solvent-accessible surface area
- SCOP
structural classification of proteins
Footnotes
Financial support for this work was provided by National Institutes of Health Grant NLM #T15 LM07442 (to P.A.) and GM 50789 (to V.D.), and the Microsoft External Research Program (www.microsoft.com/science). This study is a part of our ongoing Dynameomics project (www.dynameomics.org).
References
- 1.Lindahl T. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T, Wood RD. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 3.Grollman AP, Moriya M. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 4.Michaels ML, Miller JH. J Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollis T, Lau A, Ellenberger T. Prog Nucleic Acid Res Mol Biol. 2001;68:305–314. doi: 10.1016/s0079-6603(01)68109-1. [DOI] [PubMed] [Google Scholar]
- 6.McCullough AK, Dodson ML, Lloyd RS. Annu Rev Biochem. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 7.Scharer OD, Jiricny J. Bioessays. 2001;23:270–281. doi: 10.1002/1521-1878(200103)23:3<270::AID-BIES1037>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 8.Norman DPG, Chung SJ, Verdine GL. Biochemistry. 2003;42:1564–1572. doi: 10.1021/bi026823d. [DOI] [PubMed] [Google Scholar]
- 9.Labahn J, Scharer OD, Long A, Ezaz-Nikpay K, Verdine GL. Cell. 1996;86:321–329. doi: 10.1016/s0092-8674(00)80103-8. [DOI] [PubMed] [Google Scholar]
- 10.Nash HM, Bruner SD, Scharer OD, Kawate T, Addona TA, Spooner E, Lane WS, Verdine GL. Curr Biol. 1996;6:968–980. doi: 10.1016/s0960-9822(02)00641-3. [DOI] [PubMed] [Google Scholar]
- 11.Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Kamp PA, Thomas D, Barbey R, de Oliveira R, Boiteux S. Proc Natl Acad Sci USA. 1996;93:5197–5202. doi: 10.1073/pnas.93.11.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjoras M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T, Seeberg E. EMBO J. 1997;16:6314–6322. doi: 10.1093/emboj/16.20.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girard PM, D'Ham C, Cadet J, Boiteux S. Carcinogenesis. 1998;19:1299–1305. doi: 10.1093/carcin/19.7.1299. [DOI] [PubMed] [Google Scholar]
- 15.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 16.Murzin AG, Brenner SE, Hubbard T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 17.Chevillard S, Radicella JP, Levalois C, Lebeau J, Poupon MF, Oudard S, Dutrillaux B, Boiteux S. Oncogene. 1998;16:3083–3086. doi: 10.1038/sj.onc.1202096. [DOI] [PubMed] [Google Scholar]
- 18.Bruner SD, Norman DPG, Verdine GL. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 19.Audebert M, Chevillard S, Levalois C, Gyapay G, Vieillefond A, Klijanienko J, Vielh P, El Naggar AK, Oudard S, Boiteux S, Radicella JP. Cancer Res. 2000;60:4740–4744. [PubMed] [Google Scholar]
- 20.Audebert M, Radicella JP, Dizdaroglu M. Nucleic Acids Res. 2000;28:2672–2678. doi: 10.1093/nar/28.14.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck DAC, Jonsson AL, Schaeffer RD, Scott KA, Day R, Toofanny RD, Alonso DOV, Daggett V. Protein Eng Des Sel. 2008;21:353–368. doi: 10.1093/protein/gzn011. [DOI] [PubMed] [Google Scholar]
- 22.Kehl C, Simms AM, Toofanny RD, Daggett V. Protein Eng Des Sel. 2008;21:379–386. doi: 10.1093/protein/gzn015. [DOI] [PubMed] [Google Scholar]
- 23.Simms AM, Toofanny RD, Kehl C, Benson NC, Daggett V. Protein Eng Des Sel. 2008;21:369–377. doi: 10.1093/protein/gzn012. [DOI] [PubMed] [Google Scholar]
- 24.Rutherford K, Bennion BJ, Parson WW, Daggett V. Biochemistry. 2006;45:2178–2188. doi: 10.1021/bi051988i. [DOI] [PubMed] [Google Scholar]
- 25.Rutherford K, Daggett V. J Mol Biol. 2008;379:803–814. doi: 10.1016/j.jmb.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rutherford K, Parson WW, Daggett V. Biochemistry. 2008;47:893–901. doi: 10.1021/bi701737f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson PC, Daggett V. Biochemistry. 2008;47:9380–9393. doi: 10.1021/bi800677k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bjørås M, Seeberg E, Luna L, Pearl LH, Barrett TE. J Mol Biol. 2002;317:171–177. doi: 10.1006/jmbi.2002.5400. [DOI] [PubMed] [Google Scholar]
- 29.Levitt M. ENCAD, Energy Calculation and Dynamics. Stanford University; Palo Alto, CA: 1990. [Google Scholar]
- 30.Levitt M, Hirshberg R, Sharon R, Daggett V. Comput Phys Commun. 1995;91:215–231. [Google Scholar]
- 31.Levitt M, Hirshberg R, Sharon R, Laidig KE, Daggett V. J Phys Chem <y> 1997. 101:5051–5061. [Google Scholar]
- 32.Kell GS. J Chem Eng Data. 1967;12:66–69. [Google Scholar]
- 33.Beck DAC, Alonso DOV, Daggett V. ilmm, in lucem Molecular Mechanics. University of Washington; Seattle, WA: pp. 2000–2008. [Google Scholar]
- 34.Beck DAC, Daggett V. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Hubbard SJ, Thornton JM. NACCESS, Department of Biochemistry and Molecular Biology. University College; London: 1993. [Google Scholar]
- 36.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 37.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 38.Dodson ML, Michaels ML, Lloyd RS. J Biol Chem. 1994;269:32709–32712. [PubMed] [Google Scholar]
- 39.Nash HM, Lu R, Lane WS, Verdine GL. Chem Biol. 1997;4:693–702. doi: 10.1016/s1074-5521(97)90225-8. [DOI] [PubMed] [Google Scholar]