

Abstract
Hepatitis C virus (HCV) infection frequently persists despite eliciting substantial virus-specific immune responses. Thus, HCV infection provides a setting in which to investigate mechanisms of immune escape that allow for viral persistence. Viral amino acid substitutions resulting in decreased MHC binding or impaired antigen processing of T cell epitopes reduce antigen density on the cell surface, permitting evasion of T cell responses in chronic viral infection. Substitutions in viral epitopes that alter TCR contact residues frequently result in escape, but via unclear mechanisms since such substitutions do not reduce surface presentation of peptide-MHC complexes and would be expected to prime T cells with new specificities. We demonstrate here that a known in vivo HCV mutation involving a TCR contact residue significantly diminishes T cell recognition and, in contrast to the original sequence, fails to effectively prime naïve T cells. This mutant epitope thus escapes de novo immune recognition because there are few highly specific cognate TCR among the primary human T cell repertoire. This is the first example of viral immune escape via exploitation of a “hole” in the T cell repertoire, and may represent an important general mechanism of viral persistence.
Keywords: Human, T cells, Viral, T cell receptors
INTRODUCTION
Chronic viral infections, such as those caused by HIV, hepatitis C virus (HCV), and hepatitis B virus (HBV) are among the leading causes of death in the world.1 HCV infection is a particularly important model for elucidating mechanisms of viral persistence since most HCV infections persist but some spontaneously resolve in the first year. This permits comparison of host responses to viral evasion tactics between those who control infection and those who do not.
Acute HCV infection usually elicits a CD8 T cell response yet evades clearance in approximately 75% of infected individuals.2–4 If infection persists, the T cell responses generated during acute infection typically decline, and chronic infection is characterized by low frequencies of virus-reactive CD8 T cells in peripheral blood.2,3,5 The mechanisms contributing to decline of the immune response during chronic infection have not been fully elucidated, but viral evolution over the course of infection can contribute to persistence, enabling escape by mutation of key epitopes targeted by T-lymphocytes. In the chimpanzee model of HCV infection as well as in humans, a strong association between viral persistence and the development of escape mutations has been demonstrated.6–8
Evasion of the immune response via substitution within or near T cell epitopes occurs during HCV, HBV, SIV, and HIV infections via decreased MHC binding and impaired antigen processing for presentation, both of which decrease epitope density on the cell surface.7–16 Substitutions in TCR contact residues could also affect T cell recognition, but these substitutions would be expected to lead to only transient immune escape since the new epitope is still present on the cell surface and should be recognized by a distinct repertoire of T cells. De novo generation of CD8 T cell responses specific for some but not all escape variants has been demonstrated for HIV.17 Model systems have suggested mechanisms by which substitutions affecting TCR contact residues might prevent efficient de novo recognition by T cells bearing high affinity TCR in the context of a competent immune system. One possibility is that the substituted epitope is less well recognized because the initial antigen has already elicited a subset of T cells cross-reactive with but of lower affinity for the new antigen that dominate the response to the substituted epitope. Such a mechanism, which we term “repertoire fixation”, is analogous to the phenomenon of original antigenic sin that has been applied to T cell responses to reinfection with a microbial variant of the pathogen to which an individual was previously exposed.18,19 An alternative mechanism involves generation of an epitope for which the T cell repertoire contains a paucity of high affinity TCRs, a so called relative “hole” in the TCR repertoire. This may occur if, for example, the peptide epitope or the peptide/MHC complex mimics self. This mechanism was proposed to explain differential responsiveness to the synthetic antigen GT by different strains of MHC congenic mice.20 Escape through impaired T cell recognition via this mechanism has not been demonstrated for infection by any pathogenic organism.
We report here evidence for escape by a naturally occurring mutation in a T cell epitope that only affects TCR contact without altering peptide processing or MHC affinity. We show that the naive T cell repertoire in most individuals lacks clonotypes capable of high-avidity cognate recognition of the escape epitope sequence. Our results provide evidence in humans that HCV can evade the immune response by exploiting a relative “hole” in the T cell repertoire.
MATERIALS AND METHODS
Participants
The Risk Evaluation Assessment of Community Health (REACH) prospective study of young IDUs in Baltimore, MD examined the incidence and risk factors for HCV infection, as described previously.21 Participants eligible for the study were anti-HCV antibody negative, between 15 to 30 years of age, and acknowledged use of injection drugs. Participants were invited to co-enroll in a substudy of acute hepatitis C and those who consented had blood drawn for isolation of serum, plasma, and peripheral blood mononuclear cells in a protocol designed for monthly follow up. At each visit, participants were provided counseling to reduce the risks of drug use. The REACH protocol and the HCV substudy protocols were approved by the institutional review boards of the Johns Hopkins Schools of Medicine and Hygiene and Public Health. In addition, peripheral blood leukocytes were obtained from HLA-A2+ healthy volunteers from the general population either by leukapheresis or venipuncture according to protocols approved by the University of Washington Institutional Review Board. All subjects gave written informed consent. Donors were initially screened for HLA-A2 expression by flow cytometry of peripheral blood mononuclear cells (PBMC) using an A2-specific monoclonal antibody (mAb; BB7.2). The expression of HLA-A2 was subsequently confirmed by molecular typing in the Clinical Immunogenetics Laboratory at the Puget Sound Blood Center (Seattle, WA). Markers for infectious agent exposure (HBsAg, anti-HBc, anti-HCV, anti-HTLV-I/-II, anti-HIV-1/-2, HCV-, HBV-, HIV-PCR) were assessed by the Puget Sound Blood Center (Seattle, WA). Only HCV/HBV/HIV negative donors were included for the naïve population.
Hemigenomic HCV sequencing and analysis
From 140–280 µL of serum or plasma, the 5.2 kb region from the 5’UTR to the NS3/NS4A junction was cloned as previously described.22 For each specimen, thirty-three clones were assigned to clonotypes by using a previously-described gel shift assay, and 2 clones representing the modal clonotype were sequenced, with a third clone used as needed to resolve discrepancies.23 Sequences were assembled into contigs using Aligner (CodonCode). Sequence data were obtained at the point of initial viremia and approximately six months later, as previously described.2
IFN-γ ELISPOT assay
HCV-specific CD8+ T-cell responses were quantified by ELISPOT assay as previously described24 with the following modifications. Ninety-six well polyvinylidene plates (Millipore, Billerica, MA) were coated with 2.5 µg/ml recombinant human anti-IFN-gamma antibody (Endogen, Pierce Biotechnology, Rockford, IL) in PBS at 4°C overnight. Previously frozen PBMC were added at 200,000 cells/well or 30,000 T cells from lines in 140 µl R10 media (RPMI 1640 [Sigma-Aldrich Corp., St. Louis, MO,], 10% FCS [Sigma-Aldrich], and 10mM Hepes buffer [Sigma-Aldrich] with 2 mM glutamine and antibiotics [50 U/ml penicillin-streptomycin]). The PBMC obtained six months after initial viremia were tested for IFN-γ production in response to serial dilutions of synthetic peptides representing the viral sequence present at initial viremia (LP5) or at six months following initial viremia (MP5). Comparison of the LP5 and MP5 epitopes was performed using 10 fold dilutions of those peptides from 10 µM to 0.001µM. The plates were incubated for 20 hours (PBMC) or five hours (T cell lines) at 37°C, 5% CO2. Plates were then washed, labeled with 0.25 µg/ml biotin-labeled anti-IFN-γ (Endogen), and developed by incubation with streptavidin-alkaline phophatase (Bio-Rad Lab., Hercules, CA) followed by incubation with BCIP/NBT (Bio-Rad) in Tris-buffer (pH 9.5). The reaction was stopped by washing with tap water and the plates were dried, prior to counting on an ELISPOT reader (Cellular Technology Ltd, Cleveland, Ohio). The assay was performed at least in duplicate and background was not more than 15 SFC per million PBMC. Responses were considered positive if the number of spots per well minus the background was at least 25 SFC per million PBMC.24A control of pooled cytomegalovirus, Epstein-Barr virus, and influenza antigens (CEF control peptide pool) and phytohemmaglutinin (PHA) were used as positive controls.25 Responses to the CEF control peptide pool were quantifiable and varied by less than 10% over time. Responses to PHA were uniformly positive.
Intracellular cytokine staining (ICS) and Degranulation Assay
The assay was essentially performed as previously described.26,27 Briefly, 2×105 T-cells were stimulated for 5 hours in 96-well plates using 4×105 peptide-pulsed T2 cells. CD107a–FITC (Becton Dickinson (BD)) and 10ug/ml brefeldin A (Sigma) was added at the beginning of the stimulation period. After 5 hours, cells were stained for CD107a and co-stained for CD8. Cells were then fixed, permeabilized, and stained with antibodies against IFNγ, TNFα or IL-2 (BD), using FIX/PERM and PERM/Wash solution (BD).Cells were then washed and analyzed on a FACS-Calibur flow cytometer using CellQuest software (BD).
MHC-peptide binding assays
EBV transformed cell lines were used as the primary sources of HLA molecules. Cells were maintained in vitro and HLA molecules purified by affinity chromatography as previously described.28Quantitative assays to measure the binding of peptides to purified HLA A*0201 molecules are based on the inhibition of binding of a radiolabeled standard peptide.28 Briefly, 1–10 nM of radiolabeled peptide was co-incubated at room temperature with 1 µM to 1 nM of purified HLA A*0201 in the presence of 1 µM human β2-microglubulin (Scripps Laboratories, San Diego, CA) and a cocktail of protease inhibitors. After a two-day incubation, binding of the radiolabeled peptide to the corresponding HLA*A0201 molecule was determined by capturing HLA*A0201/peptide complexes on Greiner Lumitrac 600 microplates (Greiner Bio-one, Longwood, FL) coated with the W6/32 antibody, and measuring bound cpm using the TopCount microscintillation counter (Packard Instrument Co. Meriden, CT). A five fold difference in binding is considered significant in this assay.
mRNA electroporation
mRNA constructs containing the epitope region derived from autologous virus of Subject 28 at initial viremia (KLVALGINAV, Lp5) or the KLVAMGINAV sequence obtained 6 months after infection (Mp5) were designed. Clones with either the KLVALGINAV or KLVAMGINAV sequence served as a template. The precise composition of these PCR products was confirmed by sequencing. Optimization of viability and transfection efficiency was performed by electroporation of EBV transformed B cells with GFP mRNA using the Amaxa electroporation system (Amaxa GmbH, Cologne, Germany). Cultured B cells (5–10 × 106) were washed with PBS and resuspended at 5–10 × 106/100µl in Human B cell nucleofector solution. The cells were then electroporated (pulse program P016), with 5µg of either Lp5 or Mp5 mRNA derived from Subject 28. The B cells were incubated for 16 hours at 37C/5%CO2 in 20% FBS prior to use in intracellular cytokine secretion assays.
Bulk stimulation of peripheral blood mononuclear cells
To establish CD8 T cell lines, cryopreserved or fresh PBMC (4–10 × 106) were stimulated with 10 µg/ml of synthetic HCV peptide and 0.5µg/ml of the costimulatory antibodies anti-CD28 and anti-CD49d (Becton Dickinson) in R20 media. Recombinant interleukin-2 (IL-2, 25 IU/ml) was added on day 2 and every other day thereafter. T cells were counted and restimulated with an equal number of irradiated allogeneic PBMC and 10 µg/ml of synthetic HCV peptide after ten and 20 days in culture.
Induction of T cell lines from healthy, HCV-seronegative individuals and DC priming of CD8+ T cells from HCV exposed subjects
T cell lines were generated as previously described with some minor modifications.29 29Briefly DC were derived from adherent monocytes, cultured for 5 days in Cellgenix DC Medium (Cellgenix, Freiburg, Germany), supplemented with 1% human serum, 800 IU/ml GM-CSF (Chiron, Emeryville,CA) and 1000 IU/ml IL-4 (R&D Systems, Minneapolis). On day 3, 1.5ml of fresh medium, supplemented with 1600 IU/ml GM-CSF and 1000 IU/ml IL-4, was added to each well. On day 5 of the DC culture, cells were harvested, resuspended for maturation in fresh DC medium and supplemented with a cytokine cocktail of 800 IU/ml GM-CSF 1000 IU/ml IL-4, 10ng/ml LPS from E.coli 055:B5 (Sigma, St. Louis, MO) and 50IU/ml IFN-γ (Intermune, Brisbane, CA). Parallel to the maturation phase, DC were also incubated with 10µg/ml of peptide. DC were incubated in 6-well-plates and used for T cell stimulation the following day.
On day 0 of the T cell culture, naïve CD8 T cells were obtained by first depleting CD45RO+ cells using anti-CD45RO microbeads (Miltenyi Biotec, Auburn, CA) and LD columns, followed by a positive selection step using anti-CD8 microbeads and LS columns. This approach resulted in a >95% pure CD45RO-/CD8+ population and CD8+ population, respectively. For subject 28, CD8+ T cells were isolated using anti-CD8 microbeads (Miltenyi Biotec, Auburn, CA) and LS columns. Mature, peptide pulsed DC were washed and incubated with the CD8 cells (1×106/ml) at a ratio of 1:2 – 1:10 in complete T cell medium (RPMI 1640 medium supplemented with 12.5 mM HEPES, 4 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin (Invitrogen, Carlsbad, CA), 50 µM β-mercaptoethanol (Sigma, St. Louis, MO), and 10% human serum) supplemented with 100µM 1-methyltryptophane (Sigma). Each group consisted of a minimal concentration of 1×106 T cells, distributed equally either on 96-well-v-bottom plates or 24-well-plates (2×105 to 5×105 T cells/well, respectively).
On day 4 of culture, IL-7 and IL-15 (R&D Systems, Minneapolis) was added with fresh medium at a final concentration of 5ng/ml. Cell lines were fed every 2–3 days with fresh medium and cytokines until harvest or restimulation. For restimulation, autologous PBMC were pulsed with peptide (10µg/ml) for 2h and irradiated with 30Gy. Each T cell line was harvested, washed, resuspended in fresh medium without cytokines, and mixed with 1×107 irradiated PBMC in a 6-well plate (final volume 2ml). 24h later 2ml of fresh medium was added containing cytokines (IL-2 (50IU/ml, Chiron, Emeryville,CA), IL-7 (5ng/ml), and IL-15 (5ng/ml). 7–9 days after the last restimulation the T cell lines were evaluated by tetramer staining or restimulated again as above.
Tetramer staining, titration and dissociation
Tetramer staining was routinely performed using previously optimized concentrations of MHC tetramer –PE (Beckman Coulter). Both tetramers for the Lp5 and Mp5 variant were synthesized using the same batch of MHC monomers and controlled for equivalent labeling with PE. Approximately 1 ×105 T cells per sample were stained for 30min at RT, followed by incubation with CD8 antibody (BD). For tetramer titration, T cells were stained with graded concentrations of tetramer (25min, 37°C), followed by staining with ant-CD3 and anti-CD8-antibody on ice. To analyze the tetramer dissociation rate, cells stained with tetramer and then were incubated for various times at 37C with an HLA-A2 blocking antibody (clone BB7.2; BD Biosciences, 275µg/ml) was added to prevent tetramer rebinding. At each time point an aliquot was immediately fixed using cold PBS/1%PFA. Serial dilution of tetramer was performed using half log dilutions.
Antagonism Studies
A T-cell line was generated by stimulating naïve T-cells with the Lp5 peptide and restimulated using the Lp5, Mp5 and the control peptide as indicated n the text. As control peptide the sequence FVFDRPLPV, which is derived from the Human Proteasome component C2 97 and has an HLA A*0201 binding capacity of 1.0 nM.30 For antagonism studies using CFSE, T cells were labeled with the optimal concentration of CFSE (Invitrogen) and stimulated and restimulated as indicated in the text. FACS-analysis for CFSE-dilution was performed 5 days later.
Statistical analysis
The percent of Lp5 or Mp5 tetramer positive T cells was analyzed for each antigen utilizing generalized estimating equations (GEE) with an exchangeable correlation structure.31Comparisons of naïve and memory T-cells were made using unpaired T and Fisher’s exact tests. Differences were considered significant if p-values were <0.05.
RESULTS
The HLA A*0201-restricted T cell epitope spanning positions 1406 to 1415 in the HCV NS3 protein (KLVALGINAV, NS31406) was frequently recognized in our cohort of HCV infected patients identified prospectively, with 10 of 19 (53%) HLA A*0201 positive subjects with acute HCV infection bearing T cells specific for this epitope as measured by IFN-γ ELISpot assay (Data not shown). Viral sequencing for one of our subjects who had T cells specific for NS31406 (Subject 28) indicated that over time this epitope had been substituted at the leucine at position five to methionine to become KLVAMGINAV. This substitution has been observed frequently in HLA A*0201 positive subjects with chronic HCV infection.32,33 To assess the potential impact of the methionine substitution on T cell recognition, bulk peripheral blood mononuclear cells (PBMC) obtained from Subject 28 at approximately one year after initial viremia, after the methionine-substituted variant had become the dominant viral sequence, were tested for IFN-γ production by ELISpot in response to serial dilutions of the KLVALGINAV (Lp5) peptide or the KLVAMGINAV (Mp5) peptide.(Figure 1a) A 1 to 2 log left shift in the dose-response curve was observed, suggesting the substitution of leucine with methionine represented an escape mutation. Similar data were observed using PBMC obtained from Subject 28 at approximately 6 months after initial viremia, another point when the methionine-substituted variant was the dominant viral sequence.7
Figure 1.
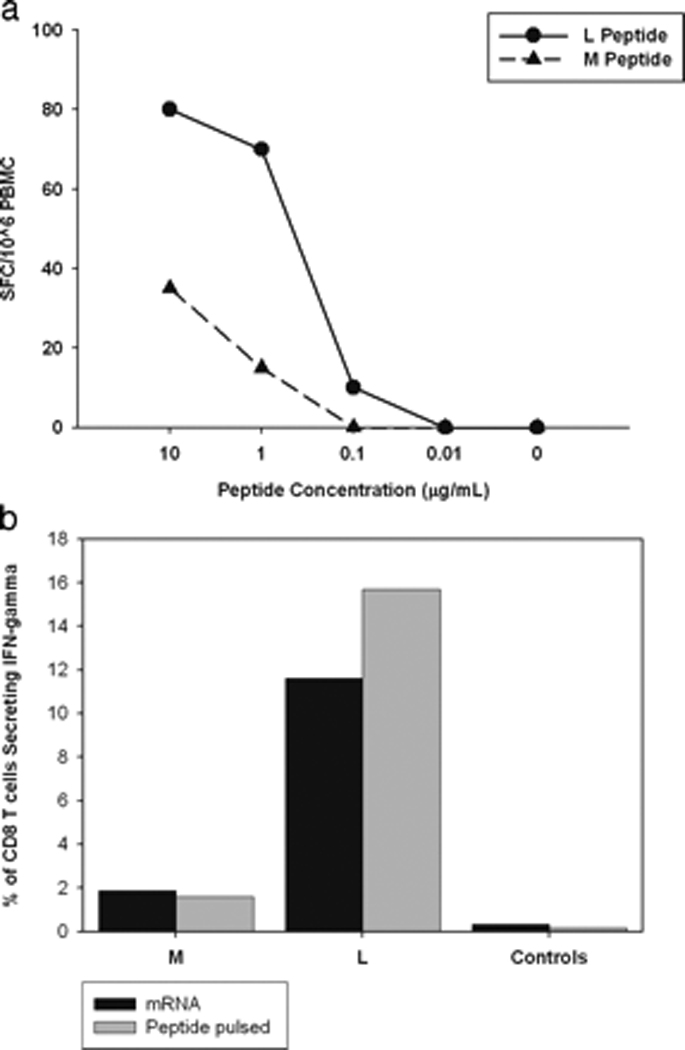
Decreased recognition of the methionine variant (Mp5) of the HLA A*0201 restricted T-cell epitope NS31406 is not due to loss of antigen processing and presentation or masking of low level responses. A. IFN-γ ELISpot was performed using PBMC obtained from Subject 28 one year following HCV infection when Mp5 was the dominant form of virus. Responses for PBMC incubated with the LP5 peptide (circles), which was present upon initial viremia, and Mp5 peptide (triangles), which was dominant six months following infection demonstrate decreased recognition of the Mp5 variant. B. Constructs containing either the Lp5 or Mp5 sequence were introduced via electroporation into EBV transformed cells from Subject 28. Immunogenicity of the construct-containing EBV cells (Black bars labeled mRNA) was assessed via intracellular cytokine staining (ICS) for IFN-γ using NS31406 specific-T cell lines generated from Subject 28. As a positive control, the same T cell line was tested for recognition of Lp5 and Mp5 peptides pulsed onto the surface of the autologous EBV transformed cells (Gray bars labeled peptide pulsed). There was no recognition of mock transfected EBV transformed cells (Black Control) or EBV transformed cells not pulsed with peptide (Grey Control). Recognition of the constructs as well as control constructs known not to represent processing mutations were similar to recognition when the peptides were pulsed onto the cell surface at a concentration of 0.001M, indicating that the Mp5 substitution did not prevent processing.
To determine if the decreased recognition reflected lower affinity binding by Mp5 to HLA A*0201, quantitative assays of binding of the Lp5 and Mp5 variant peptides to purified HLA A*0201 were performed. These direct binding assays are based on the inhibition of binding of a radiolabeled standard peptide and, as described previously, a five-fold or greater difference in binding capacity is significant.28 The assay demonstrated that the Lp5 and Mp5 peptides had comparable A*0201 binding capacity with inhibitory concentrations of 50% of 5.0 nM and 2.3 nM, respectively. Therefore, the markedly decreased recognition of the Mp5 could not be explained by decreased HLA A*0201 binding. Indeed, the substitution at position five is predicted to interact with the TCR more than the MHC based on both algorithms and the crystal structure analysis of other HLA-A2-peptide complexes.34
Impaired antigen processing can not explain the observed decreased recognition of peptide pulsed onto the cell surface since pulsing bypasses the requirement for intracellular processing. However, it is possible that the Mp5 substitution completely prevents antigen processing and presentation, thereby mitigating the development of a strong Mp5-specific response in vivo following the Lp5 to Mp5 epitope conversion in the patient. To determine if the Mp5 substitution prevents presentation, mRNA encoding either the Lp5 or Mp5 variant of NS31406 was transfected into autologous EBV–transformed B cells to serve as antigen presenting cells (APC) in an intracellular cytokine secretion (ICS) assay.(Figure 1b) B cells transfected with the Lp5 or Mp5 variant mRNA both stimulated IFN-γ secretion from an NS31406–specific T cell line recognizing both the Lp5 and Mp5 variants of NS31406 similarly to B cells pulsed directly with peptide, indicating that the reduced response to the methionine substitution in vivo did not reflect the inability of the Mp5 containing NS31406 epitope to reach the cell surface and be presented to the T cell.
Since decreased MHC binding or failure to undergo processing and presentation failed to explain the reduced recognition of the methionine-substituted peptide, we investigated other mechanisms that could result in impaired T cell recognition of Mp5. To determine if a minor population of T cells with greater specificity for Mp5 were being masked by the more abundant Lp5-specific T cells when tested directly ex-vivo, we performed in vitro restimulation of PBMC obtained from Subject 28 at approximately six months of viremia, which was after he developed Mp5 as the dominant form of the virus. The PBMC were incubated with the Mp5 or Lp5-peptide for two 10 day cycles and subsequently tested for recognition of the Mp5 or Lp5-peptide in an ELISpot assay for IFN-γ (Figure 2a). Independent of the peptide used for restimulation, both T cell lines recognized targets pulsed with the Lp5 peptide better than the Mp5 peptide. To determine if the preference for Lp5 could be overcome by using a more potent APC, either Lp5 or Mp5 –pulsed mature dendritic cells (DC) were used to restimulate and expand CD8 T cells isolated from PBMC obtained from Subject 28 after the Mp5 sequence became dominant in vivo (Figure 2b). Stimulation with Lp5-peptide-pulsed DC and subsequently PBMC resulted in a robust expansion of antigen-specific T cells from 0.6% of CD8+ T cells specific for Lp5 directly ex-vivo to 60% of the cells specific after three rounds of stimulation. However Mp5 -pulsed DC did not induce a response that demonstrated greater specificity for Mp5 than Lp5, despite the T cells having been exposed in vivo to Mp5 and the magnitude of the resulting response (12%) was clearly reduced compared to the 60% of CD8 T cells specific for the antigen after Lp5 peptide stimulation (Figure 2b). Thus, regardless of the peptide used for expansion, the mode of stimulation, or the assay used to assess IFN-γ production, T cells derived from Subject 28 at a time when the Mp5 mutation dominated his HCV sequences none-the-less consistently displayed better recognition of the Lp5 peptide than of the Mp5 peptide. Despite even longer in vivo stimulation with the M variant and very low precursor frequency of T cells specific for NS31406, similar recognition patterns were observed when PBMC from the one year time point were used. For the Lp5 expanded T cell line, the pattern of reduced cytokine production in response to Mp5 versus Lp5 held for T cell degranulation as well with reduced CD107a expression on the T cell surface in response to Mp5 versus Lp5. (Figure 2c.) In contrast, the Mp5 expanded line from PBMC at the one year time point displayed no significant degranulation in response to cells pulsed with either antigen. Given the low precursor frequency, confirmation of the poor capacity of Mp5 to induce T cell degranulation was obtained using T cell lines derived from another host where there was no exposure to Mp5 in vivo and the lines could be expanded well using Lp5. (Figure 2d) Mp5 induced poor degranulation versus Lp5 of T cells derived from this subjects as well.
Figure 2.
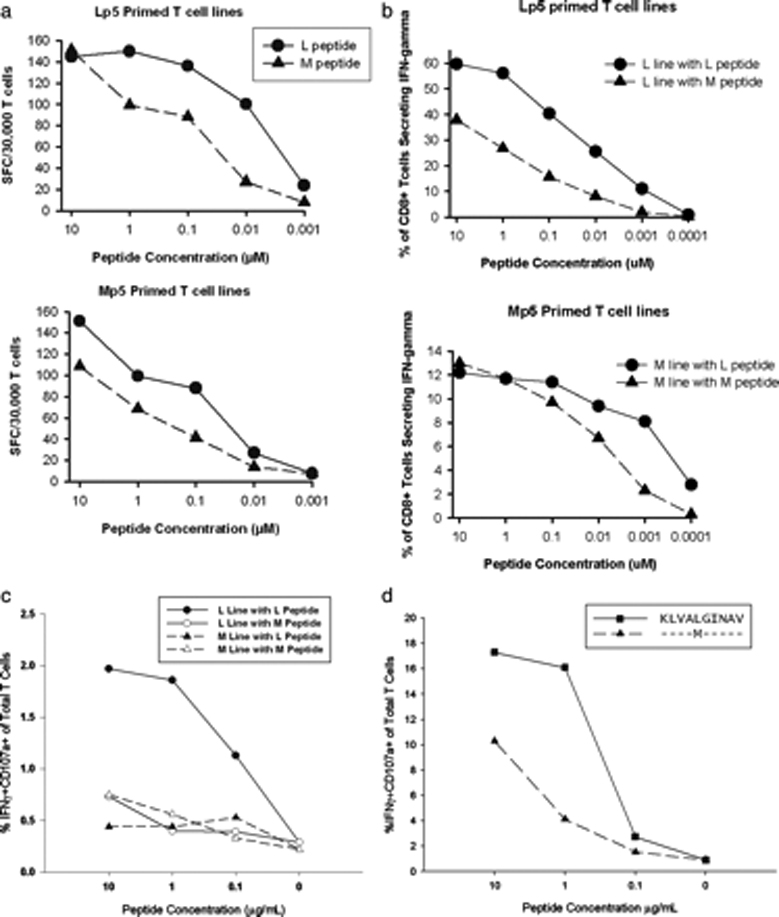
Prolonged CD8 T cell restimulation with Mp5 peptide failed to select a T cell line which recognized Mp5 better than Lp5A. PBMC were obtained from Subject 28 approximately six months after initial viremia when the Mp5 form of the virus was dominant. Those PBMC were stimulated in vitro with either the Lp5 (top panel) or Mp5 -peptide (lower panel) and supplemental interleukin-2 for two 10 day cycles and were subsequently tested for recognition of the Mp5 or Lp5-peptide. Independent of the peptide used for stimulation, both T cell lines recognized targets pulsed with the Lp5 peptide better than the Mp5 peptide in an ELISpot assay for IFN-γ. B. CD8 T cells isolated from Subject 28 after he developed Mp5 as the dominant form of the virus were restimulated with either Lp5 (top panel) or Mp5 pulsed (lower panel) DC followed by two 7 day rounds of stimulation with autologous PBMC pulsed with the same peptides as were pulsed on the DC. The response of both the Lp5 and Mp5 peptide stimulated CD8 T cells was subsequently assessed via IFN-γ ICS using LP5 (circles) or Mp5 (triangles) as antigens. Lp5 remained better recognized. C. PBMC form Subject 28 taken from the one-year time point were cultured in the presence of either Lp5 or Mp5 peptide. The precursor frequency of T cell specific for Lp5 or Mp5 was extremely low at this time point, however, after two rounds of stimulation with the peptide or the variant peptide, cells were tested for recognition of both peptides as demonstrated by IFN-gamma production and CD107a degranulation, in serial peptide dilutions in an intracellular cytokine assay. D. PBMC from an additional subject were used to generate an Lp5 specific T cell line as described in 2c. The T cell line recognized Lp5 peptide better than the Mp5 peptide, as measured by IFN-gamma production and degranulation.
To determine if the weaker recognition of Mp5 observed were due to impaired engagement of TCR by the Mp5/A*0201 complex, the avidity of TCR’s in T cell lines from Subject 28 for the Lp5 or Mp5/A*0201 complex was analyzed. Recent analyses indicate that the dissociation rate best correlates with ‘functional avidity’ as defined by the cytokine response of a T cell.35 Therefore, we analyzed tetramer off-rate by performing dissociation experiments in the presence of an HLA-blocking antibody to prevent rebinding of the tetramer. For cells expanded with Lp5, the rate of dissociation of Lp5- tetramer from the TCR’s was 90 fold slower than that of the Mp5-tetramer. The dissociation rate of the Lp5- tetramer from cells expanded with Mp5 remained slower with a 7 fold reduction in off-rate versus the Mp5-tetramer.(Figure 3a). Regardless of the peptide used for restimulation to expand the reactive cells, the TCR’s of the resulting cells always bound Lp5 -tetramer better than Mp5-tetramer. Serial dilution of tetramers also demonstrated uniformly better binding of Lp5- tetramer.(Figure 3b.) Thus, T cells derived from a chronically infected individual that were primed against the Lp5-variant in vivo demonstrated a higher avidity for Lp5, even after the Mp5 variant became dominant in vivo and after repeated in vitro stimulations with Mp5.
Figure 3.
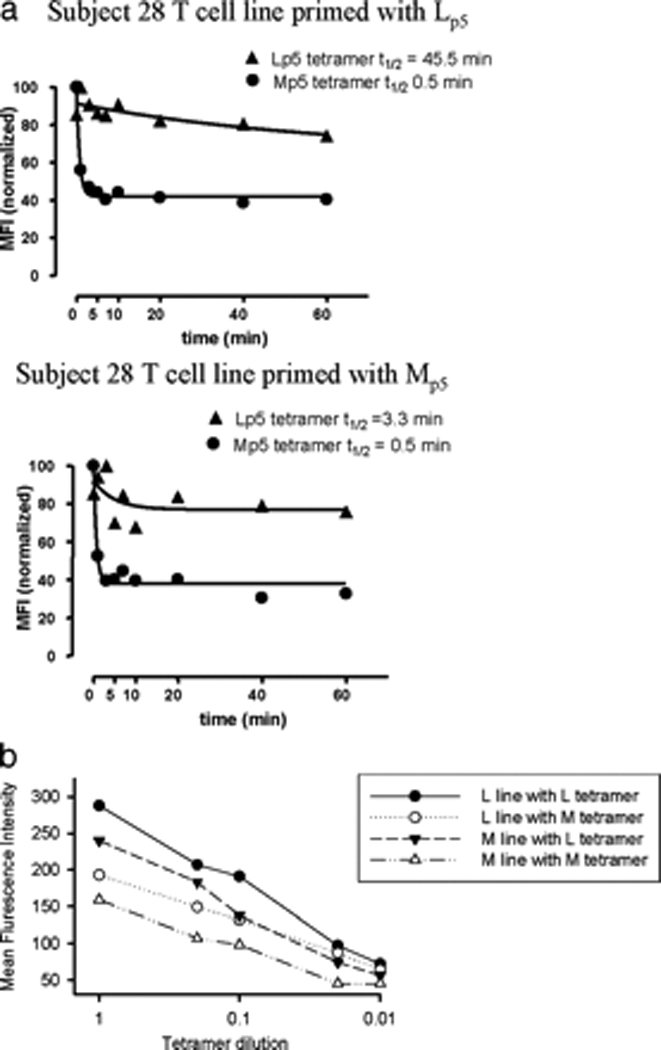
TCR from Subject 28 have decreased avidity for the Mp5/HLA-A*0201 complex. A. Binding of TCR from Subject 28 to Lp5 and Mp5 on HLA A*0201 was assessed by dissociation rates using the T cell lines described in Figure 2. Tetramer off-rate experiments were performed by staining Lp5- and Mp5- peptide primed T cell lines with Lp5- or Mp5- tetramer, washing, then incubating the cells with excess HLA-A2 antibody that prevented rebinding of any tetramer that had fallen off. An aliquot of each sample was removed following 0, 1, 2, 5, 7, 10, 20, 40, and 60 minutes of incubation and the MFI of Lp5- and Mp5- tetramer bound to the TCR for each line was recorded. The data were used to determine the time it took for half the tetramer to dissociate (t½) and demonstrated higher avidity of both Lp5 and Mp5 primed T cell lines for the Lp5-tetramer than the Mp5-tetramer. B. Serial dilution of tetramers were performed using half-log dilutions of tetramer and again demonstrated higher avidity of both Lp5 and Mp5 primed T cell lines for the Lp5-tetramer than the Mp5-tetramer.
We next considered two distinct mechanisms that could account for these unexpected observations. Mp5 could potentially be less well recognized because Lp5 had already elicited a subset of T cells cross-reactive with but of lower affinity for the new antigen that dominate the response to the substituted epitope. Such a mechanism, which we term “repertoire fixation”, is analogous to the phenomenon of original antigenic sin that has been applied to T cell responses to reinfection with a microbial variant of the pathogen to which an individual was previously exposed.18,19 Alternatively, the failure to mount a better response to Mp5 could represent poor intrinsic immunogenicity of this variant. This intrinsically reduced capacity to stimulate could result from a ‘hole in the repertoire’, defined as an absence or paucity within the primary repertoire of T cells bearing high affinity TCR with specificity for that variant.
To investigate these potential mechanisms, we evaluated recognition of Lp5 and Mp5 by the primary naïve T cell repertoires in HLA A*0201+ individuals never exposed to HCV. In such HCV-naïve individuals, a similar response to both Lp5 and Mp5 epitopes would be expected if both peptides had similar immunogenicity, since repertoire fixation resulting in a preference for the Lp5 epitope could not have occurred. In contrast, a hole in the T cell repertoire for Mp5 would be manifest by diminished Mp5 recognition by the HCV-naïve T cell repertoire. We have recently defined optimal in vitro conditions for induction of antigen-specific T cell responses from the naïve repertoire.29 CD45RO−CD8+ T cells from HCV-negative individuals were used as the responding population to the Lp5 or Mp5 peptides. In seven independent experiments, the outcome of priming of T cells from five different HCV unexposed donors was assessed. Assays were set-up in multiple parallel wells and T cells were incubated with DC pulsed either with the Lp5 or the Mp5 peptide followed by one or two rounds of stimulation with peptide-pulsed PBMC. Peptide concentrations ranging from 100ng/ml to 10ug/ml were used. The percentage of tetramer positive cells after two cycles of stimulation resulting from each of 38 attempts to prime responses from naïve T cells is indicated in Figure 4. Because we sampled the different naïve hosts different numbers of times, we used a statistical method called the generalized estimating equations (GEE). This statistical method determines the statistical significance based on the number of subjects sampled and the number of times each subject is sampled. The results demonstrate a dramatic difference between the ability of the HCV-naïve T cell population to respond to Lp5 versus Mp5. An Lp5-specific response was obtained in 36 out of 38 T cell lines, with the majority of responses vigorous. In contrast, stimulation with the Mp5 variant did not result in any detectable response in three of five donors. A response was detected by MHC tetramer staining in 14 out of the 38 attempts to generate T cell lines using Mp5 peptide for stimulation, but these responses were nearly all of very low magnitude, residing at the limit of detection of the assay. Additionally, the Mp5 and Lp5 peptide batches used to stimulate in the priming experiments were both re-evaluated for maintenance of integrity via mass spectrometry after completion of the majority of experiments, which demonstrated that Mp5 had not degraded and, importantly, that the failure to prime a response to Mp5 could not be explained by Mp5 oxidation or another chemical modification. This diminished ability of Mp5 to prime naïve T cells is unusual among peptides with maintained HLA binding capacity as demonstrated by our previous work with this assay with Melan-A, Wilms Tumor antigens of multiple HLA restrictions, and an HIV gag epitope.29,36, and M. Wolfl, unpublished data. T cells capable of recognizing the heteroclitic HLA-A0201-restricted Melan-A epitope (L26–35) are relatively frequent, which has allowed us to assess the robustness and reproducibility of the culture system.37 Effective use of this priming system has also been described for the Wilms tumor antigen 1, a transcription factor, which is being explored as an immunological target for leukemic cells.38 Proliferation of antigen-specific T-cells specific for several WT-1 epitopes with different HLA-restrictions has been detected.36 We have also evaluated the induction of T cell responses against the HLA-A0201 restricted epitope HIVgagp17 76–84 in healthy donors and were able to induce T-cell responses against this epitope (M. Wolfl, unpublished data). Thus, this method has been highly successful in priming naïve T cells specific for other antigens and the lack of priming with Mp5 is unusual.
Figure 4.
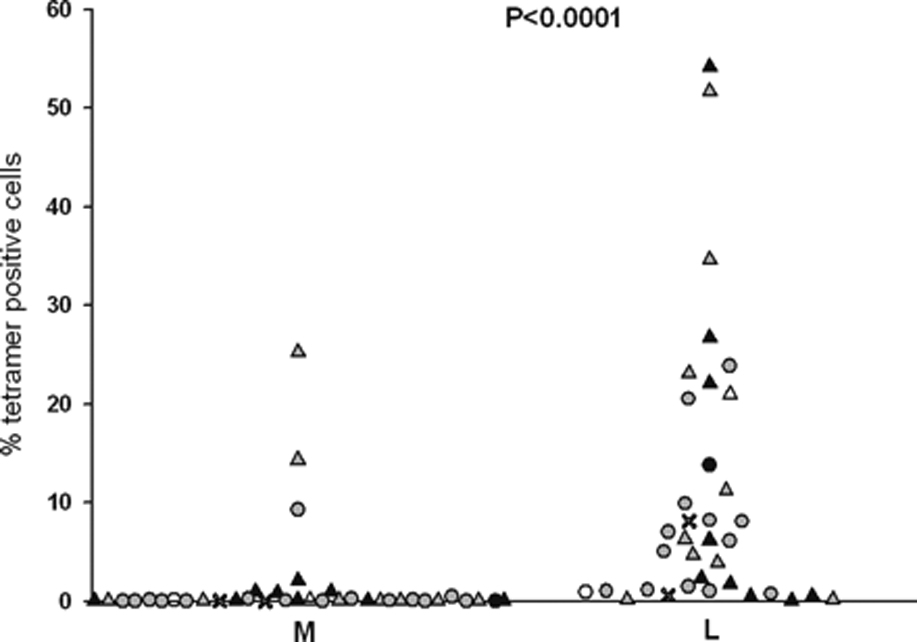
Mp5 ineffectively primes naïve T cells.
CD45RO−/CD8+ (naïve) T cells from HCV-negative individuals were primed under optimal conditions for generation of an antigen specific response with Lp5 or Mp5 and the generation of antigen specific responses assessed by tetramer staining. Priming was performed with five different donors in multiple experiments and in parallel groups for Lp5 and Mp5. An Lp5-specific response was obtained in 36 out of 38 lines and the majority of responses were vigorous. In contrast, a total of 14 out of 38 lines generated using Mp5 had a specific response for that variant as detected by tetramer staining, but nearly all were very low level responses and at the limit of detection in the assay. The Lp5 and Mp5 specific responses were compared via GEE analysis and the Lp5-specific responses were significantly more frequent and of higher magnitude than Mp5-specific responses. (p<.0001)
Since the CD45RO-CD8+ population contains not only naïve T-cells but also CD45RA+CCR7- TEMRA, a population of late effector memory cells, we performed additional purifications to confirm that the observed responses truly arose from the naïve T cell population. Additional experiments using CD45RO, CD45RA, CD62L, and CCR7 as selective markers were performed. Immunophenotyping of CD45RA+CCR7+ sorted cells showed a high purity of CD45RA+CCR7+CD28+CD62L+ naïve T cells (Figure 5a, left panel), whereas a CD45RA-CCR7- control population could be characterized as a CD45RO+ memory subpopulation (Figure 5a, right panel). When these two populations of cells were expanded and restimulated for 2 cycles with Lp5, only the naïve CD45RA+CCR7+ population yielded a response to the Lp5 HCV epitope, which was statistically significant regardless whether the magnitude (Figure 5b, top) or percent positive responses (Figure 5b, bottom) was taken into account. Additionally, CD8+ cells were first isolated by negative selection and then positively selected for CD62L or CD45RO expression. The CD62L+ fraction contains both naïve T-cells (CD62L+CD45RO-) and central memory cells (CD62L+CD45RO+) while the CD45RO+ fraction contains effector and central memory cells. As demonstrated in Figure 5c, it was not possible to generate an Lp5-specific response from the CD45RO+ population, whereas a statistically significant number of wells were positive in the CD62L+ group.(Fisher’s exact test, p=0.007) Finally, cells isolated by CD45RO depletion followed by CD8 positive selection were compared with CD8 cells obtained from PBMC by negative selection, depleted of CD45RO+ cells, and then positively selected for CD62L expression, yielding essentially completely naïve cells. When these two populations were expanded and restimulated for 2 cycles, similar numbers of tetramer+ wells were detected from both populations of naïve T cells (Figure 5d, Fisher’s exact test p=0.63.). In summary, stimulation of flow sorted naïve CD45RA+CCR7+ T cells yielded similar results to those obtained with CD45RO-CD8+ T cells, whereas no responses were detected from the CD45RO+ memory population of these HCV-negative donors, confirming that the responses were elicited from priming of naïve T cells.
Figure 5.
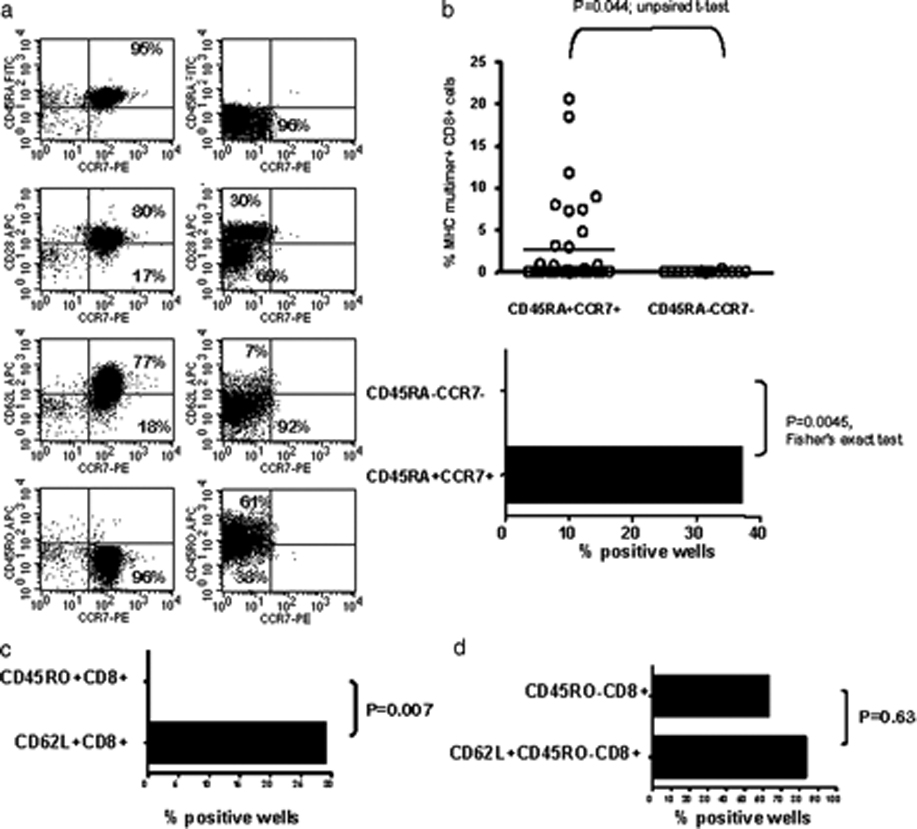
Responses to Lp5 in HCV naïve individuals derive from the naïve T cell population. A. Immunophenotyping of CD45RA+CCR7+ T-cells sorted cells showed a high purity of CD45RA+CCR7+CD28+CD62L+ naïve T cells (left panel), whereas a CD45RA-CCR7- control population was a CD45RO+ memory subpopulation (right panel). B. CD45RA+CCR7+ and CD45RA−CCR7− sorted T cells were expanded and restimulated for 2 cycles. Only the naïve CD45RA+CCR7+ population yielded a response to the Lp5 HCV epitope, as measured by magnitude (top) or percent positive (bottom). C. CD8+ cells were isolated by negative selection and then positively selected for CD62L or CD45RO expression. No Lp5-specific response was obtained from the CD45RO+ memory population, whereas a statistically significant number of wells were positive in the CD62L+ naïve group. D. Cells isolated by CD45RO depletion followed by CD8 positive selection were compared with CD8 cells obtained from PBMC by negative selection, depleted of CD45RO+ cells, then positively selected for CD62L expression. Following expansion and restimulation for 2 cycles, similar numbers of tetramer+ wells were detected from both populations containing naïve T cells.
We then assessed whether any T cells responding to Mp5 reflect the existence of a potential Mp5-specific repertoire rather than just cross-reactivity to Mp5. Priming naïve cells with Lp5 peptide resulted in Lp5-specific responses that generally included a population of cells cross-reactive for the Mp5-epitope as shown for three representative lines (Figure 6a). While cross-reactive, recognition of Lp5 was better than of Mp5 as reflected by tetramer binding and functional analysis by intracellular IFN-γ (Figure 6a). For the few T cells lines successfully generated with the Mp5 peptide, parallel staining with either Mp5 or Lp5/A*0201-MHC tetramers demonstrated that 8 of the14 lines were cross-reactive to the Lp5 epitope (three of which are shown in Figure 6b) while only six demonstrated a higher MFI after staining with the Mp5-multimer than the Lp5-multimer and one a much larger percent of Mp5 specific cells (25 vs. 0.1%), indicating that these cells were more specific for Mp5. Assessment of the functional response (IFNγ-production) of the one T cell line displaying a high percentage (25%) of Mp5-tetramer+ cells, demonstrated that these cells exclusively recognized the Mp5-variant, revealing the existence of very rare truly Mp5-specific T cells.(Figure 6c)
Figure 6.
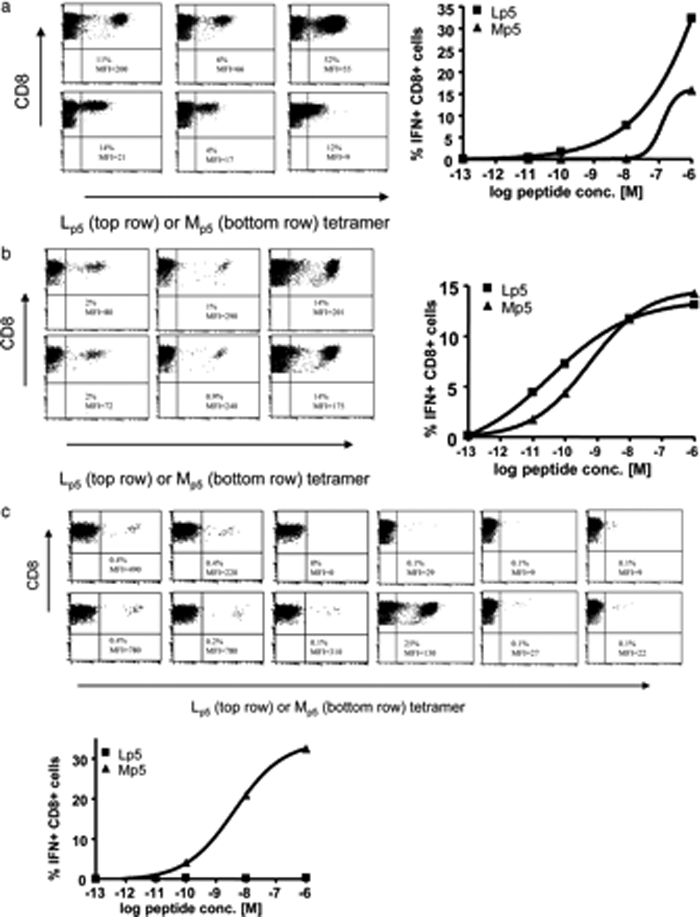
Mp5 primes naïve T cells that are primarily cross-reactive. In the right lower quadrant for each T cell line, numbers separated by a slash indicate the percent of T cells specific for NS31406/MFI. The percent of tetramer bound cells is shown using Lp5 (top row) or Mp5 (bottom row). Representative ICS data are shown for one line to the right of the FACS plots in each panel. A. Priming naïve cells with Lp5 peptide resulted in Lp5-specific responses that were cross-reactive for the Mp5-epitope as shown for three representative lines. The percent of T cells specific for Lp5 was usually higher and the MFI was always higher for Lp5 than Mp5 tetramer. Although cross-reactive, better IFN-γ production was observed in response to Lp5 versus Mp5B. For the T cells lines generated with Mp5 peptide, parallel staining with either Mp5 or Lp5/A*0201-MHC tetramers demonstrated that 8 of these lines were cross-reactive to the Lp5 epitope, displaying a similar or higher MFI, when stained with the Lp5 multimer, as shown for three representative lines. The percent of T cells specific for Lp5 and Mp5 and the MFI were usually equivalent. Responses vigorous enough to assess for cytokine production were very rare, but better or equivalent IFN-γ production was observed in response to Mp5 as compared to Lp5 for the lines that recognized both. C. Six of the 14 lines generated by Mp5 priming demonstrated a higher MFI after staining with the Mp5-multimer than the Lp5-multimer and one a much larger percent of Mp5 specific cells (25 vs. 0.1%), suggesting they were more specific for Mp5 and where assessed, they also produced more IFN-γ in response to Mp5.
That priming of Mp5-specific cells was detectable as a rare event demonstrates that the experimental system is sensitive enough to detect such rare responses in the few instances when such cells are present. The highly significant difference (p<.0001, Figure 4) in the capacity to prime naïve T-cells from HCV unexposed individuals with the Mp5 peptide compared to the Lp5 peptide indicates that variant virus containing Mp5 is highly inefficient at priming naïve T cells relative to the virus with the Lp5 epitope. Thus, in the setting of viral infection, the variant virus containing the Mp5 mutation can exploit this relative hole in the T cell repertoire in most individuals as an escape mechanism.
After demonstrating that naïve T cells were poorly primed by the Mp5 variant, we next asked whether the Mp5 variant also failed to boost previously primed responses, There was evidence of poor capacity of Mp5 to expand preexisting responses to Lp5 in vivo because viral replacement of leucine with methionine at position 1410 in Subject 28 was associated with eventual loss of T cell responses to Lp5 and Mp5. Responses to both peptides declined after the replacement and became undetectable by approximately 460 days following infection. This might at least in part be explained by the possibility that the methionine substitution fails to boost an Lp5-specific response as well as failing to prime cells with greater Mp5 specificity. Indeed, T cells from naïve individuals primed and then boosted in vitro with the Lp5 peptide completely failed to be boosted by the Mp5 peptide (Figure 7a). In addition to a failure to expand tetramer+ T cells, Mp5 boosting resulted in reduced functional activation in response as assessed by intracellular IFN-γ and TNF-α staining, CD107a mobilization shift assay for degranulation and upregulation of the activation markers CD137 and CD25.(Figure 8)
Figure 7.
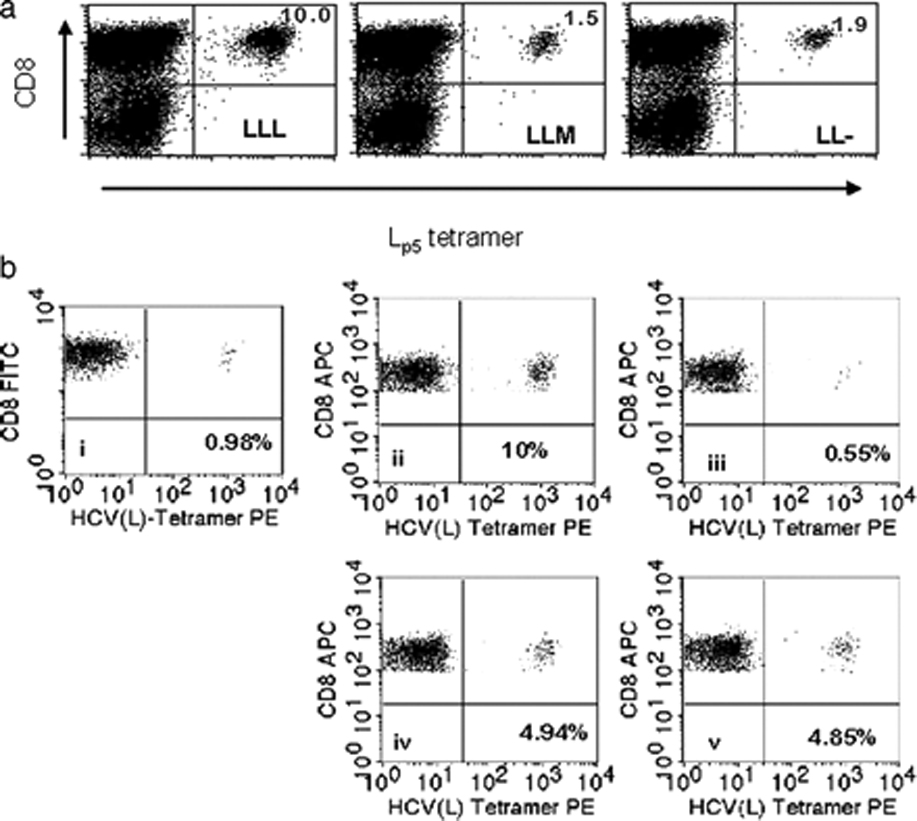
Mp5 acts as a partial agonist and not as an antagonist.
A. Mp5 fails to boost an Lp5-specific response. Using naïve T cells that had been primed with Lp5 -pulsed DC and expanded with Lp5-pulsed PBMC, a third round of stimulation was performed using either Lp5 (LLL) or Mp5 (LLM) and compared to a third round of culture with media alone (LL-). Both Lp5 and Mp5 bearing tetramers were used to assess the percent of cells specific for each antigen with no significant differences in recognition pattern. The percent of CD8+/Lp5 tetramer positive T cells did not differ significantly between LLM and LL-, demonstrating that Mp5 expanded specific T cells no better than media alone. B. The Mp5 peptide did not boost the Lp5-specific response, but does not have an inhibitory effect relative to a control peptide of comparable HLA-A*0201 binding either. A T-cell line was generated by stimulating naïve T-cells with the Lp5 peptide and the percent of Lp5 variant-specific T cell is shown in the FACS plot labeled (i) as 0.98%. This line was divided and restimulated with equal numbers of autologous PBMC pulsed with one of four peptide combinations; Lp5 only (ii), Mp5 only (iii), an equal mixture of Lp5 and control peptide (iv) or an equal mixture of Lp5 and Mp5 (v). Evaluation by tetramer staining 1 week after stimulation showed that Mp5 peptide did not boost or inhibit the Lp5-specific response relative to control peptide (Lp5 specific T cells 4.94% versus 4.85%).
Figure 8.
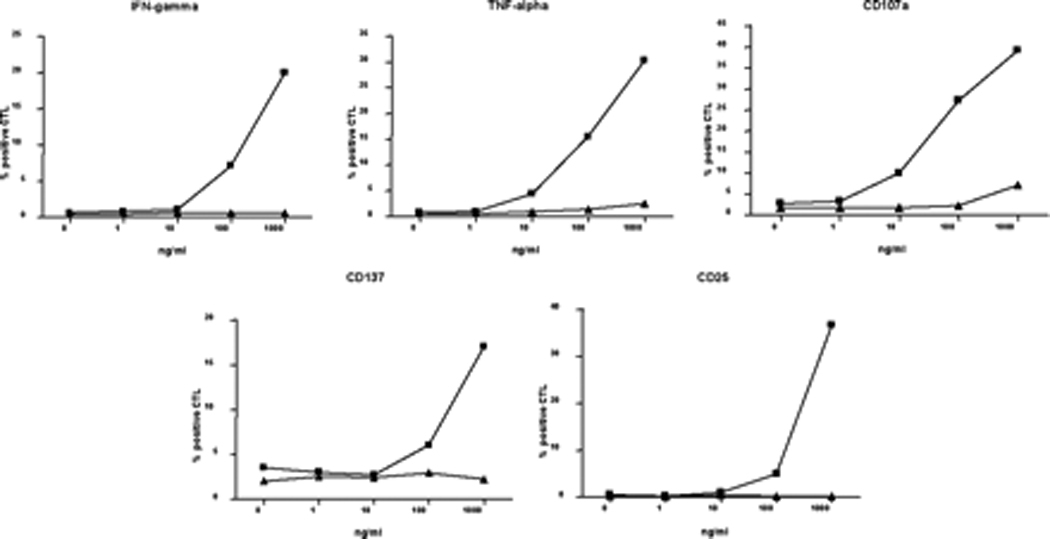
Priming naïve cells with Lp5 peptide resulted in Lp5-specific responses that were very weakly cross-reactive for the Mp5-epitope. Data are shown for three representative lines in Figure 6a. Although cross-reactive, better IFN-γ (Figure 6a) and TNF-α production, degranulation (CD107a), and upregulation of activation markers (CD137, CD25) was observed in response to the Lp5 as compared to Mp5.
Since the Mp5 variant had poor capacity to activate and boost, we next asked whether TCR engagement of Mp5 on HLA A*0201 is not only less stimulatory but actually inhibitory, a mechanism that has been described as antagonism for altered peptide ligands of other viral epitopes.32 Since the Lp5 HCV variant was present first in vivo, we assessed the effects of Mp5 on expansion of a previously Lp5 peptide primed T cell line. We found that Lp5 specific lines were not inhibited at all when the Mp5 peptide was mixed with Lp5 in restimulation assays relative to mixture with control peptide (Figure 7b). The failure of the Mp5 peptide to inhibit stimulation with Lp5 was observed in multiple formats of in vitro restimulation using either expansion of tetramer+ cells or CFSE dilution as readouts (Figure 7, data not shown). Based on these findings, we conclude that there is no clear evidence for an antagonistic function of the Mp5 peptide. However, altered peptide ligands can act as partial agonists in some settings and as antagonists in others so the possibility that Mp5 may have antagonistic function under certain conditions cannot be excluded.39
DISCUSSION
We identified an escape mutation that arose in vivo in a commonly recognized HCV peptide epitope involving a primary TCR contact residue that could not be explained by impaired HLA binding or loss of processing. A paucity of reactivity against the substituted Mp5 epitope was consistently observed in HCV naïve individuals, all of whom mounted robust responses to the original Lp5 epitope, suggesting that immune escape via conversion to Mp5 is exploiting a relative hole in the T cell repertoire. This represents the first example of a viral escape substitution resulting in evasion of the T cell response via exploitation of a hole in the T cell repertoire. Although the hole in the repertoire is not absolute, the paucity of T cell lines with even modest recognition of the Mp5 peptide that could be generated from five HCV-naïve individuals using the Mp5 peptide suggests the frequency of cells bearing a TCR that can react with this peptide is exceedingly small. Given the diversity and complexity of the TCR repertoire, “absence” of reactivity among the total T cell population is never absolute but rather a spectrum of frequencies and affinities, and will differ between individuals despite sharing the same restricting allele because of different genetic backgrounds as the source for self-peptides. Numerous examples exist of residual T cells with low affinity and frequency that are specific for self antigens, even for those antigens shown to be expressed in the thymus. The overall difference in reactivity between the L and M peptides among the HCV naïve subjects is dramatic and highly statistically significant. The host TCR repertoire would be expected to include only a few T cells specific for the M variant if the peptide mimics self or has a conformation that prevents TCR interaction. No self-peptide homologues to Mp5 were detected in a search of the nr, RefSeq and SwissProt databases, but self-mimicry is often determined by the three dimensional structure of the peptide/MHC complex and thus cannot generally be predicted based on the primary sequence of the peptide.40,41 Thus, we could not determine from these studies if the relative hole in the repertoire is the result of self-mimicry (with resultant tolerance induction) or the failure to generate a primary TCR repertoire specific for Mp5 because of its conformation.
Despite having assessed and not detected antagonism in a number of in vitro formats, antagonism may be operative in vivo as there is no definitive assay that defines the absence of antagonism. However, the results from our analysis of responses in the HCV naïve subjects clearly demonstrate a significant hole in the repertoire independent of any antagonistic effects of the M variant peptide for L specific responses. Similarly, the results in naïve hosts suggest that a hole in the repertoire is exploited for escape rather than that repertoire fixation limits the immune response. Nonetheless, the repertoire fixation and repertoire hole mechanisms are likely not mutually exclusive in vivo and may operate simultaneously. Preexisting repertoire fixation may further decrease the response to an epitope that has relatively few TCR with specificity for it and a paucity of TCR specific for an epitope may enhance repertoire fixation by decreasing competition for expansion with T cells primed to an epitope present earlier.
T cell escape mutations resulting in complete abrogation of HLA binding or disruption of processing will not be present on the surface of the cell and are thus invisible to the immune system. In contrast, substitutions that result in decreased recognition but the continued presence of the viral peptide on the surface of the infected cell, such as those that affect TCR recognition or those that decrease but do not abrogate HLA binding, remain immunologically important because they still represent targets against which host responses can be generated and could be targeted by vaccines or that could be potentially amplified via manipulations that block T cell inhibitory pathways.42,43 Understanding mechanisms for escape via mutation of TCR contact residues will facilitate development of vaccines containing antigens that stimulate cross recognition of antigens still present on the cell surface while avoiding amino acid substitutions in the vaccine strain that reduce T cell priming and recognition.
Supplementary Material
Effects of proteinase K in the generation of AVDCs. Three surface markers were studied, CD86, MHC-I and MHC-II. Red: effects of supernatant conditioned by NDV-infected DCs in one trans-well-chamber on naïve DCs in the other chamber. Green: effects of supernatant conditioned by NDV-infected DC subsequently digested with proteinase K in one transwell-chamber on naïve DCs in the other chamber. Blue: naïve DCs with unconditioned media in the other trans-well chamber. The data shown are representative of two replicate experiments using two different donors that showed similar results.
Paracrine effects of virus-infected human epithelial fibroblasts on naïve DCs. Fibroblasts were seeded in the lower compartment of the trans-well system and were infected, (green), or not infected, (blue), with NDV to see if they were capable of generating the same maturation surface markers pattern as AVDCs (red). Fluorescence minus one control (FL -1) is shown in black. The data shown are representative of three different experiments using three different donors that showed similar results.
Effect of extended trans-well culturing on naïve DCs after 18, 24 and 48 hours, (red, blue and green respectively). FL-1 control is shown in black. The data shown are representative of three different experiments using three different donors that showed similar results.
ACKNOWLEDGEMENTS
The following reagent was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CEF Control Peptide Pool from Dr. Josephine Cox and Dr. Jeffrey Currier.
Footnotes
This work was supported by grants from: The Damon Runyon Foundation, The Dana Foundation, The W.W. Smith Charitable Trust, The Bill and Melinda Gates Foundation; Leukemia & Lymphoma Society LLS 7040-03, and US Public Health Service grants K08 DA11880, U19 AI040035, R01 DK57998, R 37 CA33084, and R01 CA18029. MW and JK were supported by a research fellowship of the Deutsche Krebshilfe, Germany and TK by a research fellowship from the Deutsche Forschungsgemeinschaft, Germany.
Reference List
- 1.Weiss RA, McMichael AJ. Social and environmental risk factors in the emergence of infectious diseases. Nat.Med. 2004;10:S70–S76. doi: 10.1038/nm1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox AL, Mosbruger T, Lauer GM, Pardoll D, Thomas DL, Ray SC. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology. 2005;42:104–112. doi: 10.1002/hep.20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lechner F, Gruener NH, Urbani S, Uggeri J, Santantonio T, Kammer AR, Cerny A, Phillips R, Ferrari C, Pape GR, Klenerman P. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur.J.Immunol. 2000;30:2479–2487. doi: 10.1002/1521-4141(200009)30:9<2479::AID-IMMU2479>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 4.Cucchiarini M, Kammer AR, Grabscheid B, Diepolder HM, Gerlach TJ, Gruner N, Santantonio T, Reichen J, Pape GR, Cerny A. Vigorous peripheral blood cytotoxic T cell response during the acute phase of hepatitis C virus infection. Cell Immunol. 2000;203:111–123. doi: 10.1006/cimm.2000.1683. [DOI] [PubMed] [Google Scholar]
- 5.Rehermann B, Chang KM, McHutchison JG, Kokka R, Houghton M, Chisari FV. Quantitative analysis of the peripheral blood cytotoxic T lymphocyte response in patients with chronic hepatitis C virus infection. J.Clin.Invest. 1996;98:1432–1440. doi: 10.1172/JCI118931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, McKinney D, Sette A, Hughes AL, Walker CM. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 2001;15:883–895. doi: 10.1016/s1074-7613(01)00245-x. [DOI] [PubMed] [Google Scholar]
- 7.Cox AL, Mosbruger T, Mao Q, Liu Z, Wang XH, Yang HC, Sidney J, Sette A, Pardoll D, Thomas DL, Ray SC. Cellular immune selection with hepatitis C virus persistence in humans. J Exp.Med. 2005;201:1741–1752. doi: 10.1084/jem.20050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Timm J, Lauer GM, Kavanagh DG, Sheridan I, Kim AY, Lucas M, Pillay T, Ouchi K, Reyor LL, Zur Wiesch JS, Gandhi RT, Chung RT, Bhardwaj N, Klenerman P, Walker BD, Allen TM. CD8 epitope escape and reversion in acute HCV infection. J.Exp.Med. 2004;200:1593–1604. doi: 10.1084/jem.20041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimura Y, Gushima T, Rawale S, Kaumaya P, Walker CM. Escape mutations alter proteasome processing of major histocompatibility complex class I-restricted epitopes in persistent hepatitis C virus infection. J Virol. 2005;79:4870–4876. doi: 10.1128/JVI.79.8.4870-4876.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seifert U, Liermann H, Racanelli V, Halenius A, Wiese M, Wedemeyer H, Ruppert T, Rispeter K, Henklein P, Sijts A, Hengel H, Kloetzel PM, Rehermann B. Hepatitis C virus mutation affects proteasomal epitope processing. J.Clin.Invest. 2004;114:250–259. doi: 10.1172/JCI20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen TM, O'Connor DH, Jing P, Dzuris JL, Mothe BR, Vogel TU, Dunphy E, Liebl ME, Emerson C, Wilson N, Kunstman KJ, Wang X, Allison DB, Hughes AL, Desrosiers RC, Altman JD, Wolinsky SM, Sette A, Watkins DI. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 2000;407:386–390. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- 12.Allen TM, Altfeld M, Yu XG, O'Sullivan KM, Lichterfeld M, Le Gall S, John M, Mothe BR, Lee PK, Kalife ET, Cohen DE, Freedberg KA, Strick DA, Johnston MN, Sette A, Rosenberg ES, Mallal SA, Goulder PJ, Brander C, Walker BD. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J.Virol. 2004;78:7069–7078. doi: 10.1128/JVI.78.13.7069-7078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones NA, Wei X, Flower DR, Wong M, Michor F, Saag MS, Hahn BH, Nowak MA, Shaw GM, Borrow P. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J.Exp.Med. 2004;200:1243–1256. doi: 10.1084/jem.20040511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Draenert R, Le Gall S, Pfafferott KJ, Leslie AJ, Chetty P, Brander C, Holmes EC, Chang SC, Feeney ME, Addo MM, Ruiz L, Ramduth D, Jeena P, Altfeld M, Thomas S, Tang Y, Verrill CL, Dixon C, Prado JG, Kiepiela P, Martinez-Picado J, Walker BD, Goulder PJ. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp.Med. 2004;199:905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokomaku Y, Miura H, Tomiyama H, Kawana-Tachikawa A, Takiguchi M, Kojima A, Nagai Y, Iwamoto A, Matsuda Z, Ariyoshi K. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are major escape mechanisms from CTL immune pressure in human immunodeficiency virus type 1 infection. J Virol. 2004;78:1324–1332. doi: 10.1128/JVI.78.3.1324-1332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu.Rev.Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 17.Allen TM, Yu XG, Kalife ET, Reyor LL, Lichterfeld M, John M, Cheng M, Allgaier RL, Mui S, Frahm N, Alter G, Brown NV, Johnston MN, Rosenberg ES, Mallal SA, Brander C, Walker BD, Altfeld M. De novo generation of escape variant-specific CD8+ T-cell responses following cytotoxic T-lymphocyte escape in chronic human immunodeficiency virus type 1 infection. J Virol. 2005;79:12952–12960. doi: 10.1128/JVI.79.20.12952-12960.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klenerman P, Zinkernagel RM. Original antigenic sin impairs cytotoxic T lymphocyte responses to viruses bearing variant epitopes. Nature. 1998;394:482–485. doi: 10.1038/28860. [DOI] [PubMed] [Google Scholar]
- 19.Mongkolsapaya J, Dejnirattisai W, Xu XN, Vasanawathana S, Tangthawornchaikul N, Chairunsri A, Sawasdivorn S, Duangchinda T, Dong T, Rowland-Jones S, Yenchitsomanus PT, McMichael A, Malasit P, Screaton G. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat.Med. 2003;9:921–927. doi: 10.1038/nm887. [DOI] [PubMed] [Google Scholar]
- 20.Vidovic D, Matzinger P. Unresponsiveness to a foreign antigen can be caused by self-tolerance. Nature. 1988;336:222–225. doi: 10.1038/336222a0. [DOI] [PubMed] [Google Scholar]
- 21.Garfein RS, Doherty MC, Monterroso ER, Thomas DL, Nelson KE, Vlahov D. Prevalence and incidence of hepatitis C virus infection among young adult injection drug users. J.Acquir.Immune.Defic.Syndr.Hum.Retrovirol. 1998;18(Suppl 1):S11–S19. doi: 10.1097/00042560-199802001-00004. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Netski DM, Mao Q, Laeyendecker O, Ticehurst JR, Wang XH, Thomas DL, Ray SC. Accurate representation of the hepatitis C virus quasispecies in 5.2-kilobase amplicons. J Clin.Microbiol. 2004;42:4223–4229. doi: 10.1128/JCM.42.9.4223-4229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang YM, Ray SC, Laeyendecker O, Ticehurst JR, Thomas DL. Assessment of hepatitis C virus sequence complexity by electrophoretic mobilities of both single- and double-stranded DNAs. J.Clin.Microbiol. 1998;36:2982–2989. doi: 10.1128/jcm.36.10.2982-2989.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauer GM, Ouchi K, Chung RT, Nguyen TN, Day CL, Purkis DR, Reiser M, Kim AY, Lucas M, Klenerman P, Walker BD. Comprehensive analysis of CD8(+)-T-cell responses against hepatitis C virus reveals multiple unpredicted specificities. J.Virol. 2002;76:6104–6113. doi: 10.1128/JVI.76.12.6104-6113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Currier JR, Kuta EG, Turk E, Earhart LB, Loomis-Price L, Janetzki S, Ferrari G, Birx DL, Cox JH. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J.Immunol.Methods. 2002;260:157–172. doi: 10.1016/s0022-1759(01)00535-x. [DOI] [PubMed] [Google Scholar]
- 26.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol.Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 27.Jung T, Schauer U, Heusser C, Neumann C, Rieger C. Detection of intracellular cytokines by flow cytometry. J Immunol.Methods. 1993;159:197–207. doi: 10.1016/0022-1759(93)90158-4. [DOI] [PubMed] [Google Scholar]
- 28.Sidney J, Southwood S, Oseroff C, Del Guercio JF, Sette A, Grey H. Measurement of MHC/Peptide Interactions by Gel Filtration. In: Bierer B, Coligan JE, Margulies DH, Shevach EM, Strober W, Kruisbeek A, editors. Current Protocols in Immunology. John Wiley & Sons, Inc: 1998. pp. 18.3.1–18.3.19.. [Google Scholar]
- 29.Ho WY, Nguyen HN, Wolfl M, Kuball J, Greenberg PD. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naive repertoire. J Immunol.Methods. 2006;310:40–52. doi: 10.1016/j.jim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 30.Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, Nemazee D, Ponomarenko JV, Sathiamurthy M, Schoenberger S, Stewart S, Surko P, Way S, Wilson S, Sette A. The immune epitope database and analysis resource: from vision to blueprint. PLoS.Biol. 2005;3:pe91. doi: 10.1371/journal.pbio.0030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeger SL, Liang KY, Albert PS. Models for longitudinal data: a generalized estimating equation approach. Biometrics. 1988;44:1049–1060. [PubMed] [Google Scholar]
- 32.Chang KM, Rehermann B, McHutchison JG, Pasquinelli C, Southwood S, Sette A, Chisari FV. Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J.Clin.Invest. 1997;100:2376–2385. doi: 10.1172/JCI119778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopez-Labrador FX, He XS, Berenguer M, Cheung RC, Gonzalez-Candelas F, Wright TL, Greenberg HB. Genetic variability of hepatitis C virus non-structural protein 3 and virus-specific CD8+ response in patients with chronic hepatitis C. J Med Virol. 2004;72:575–585. doi: 10.1002/jmv.20036. [DOI] [PubMed] [Google Scholar]
- 34.Garboczi DN, Biddison WE. Shapes of MHC restriction. Immunity. 1999;10:1–7. doi: 10.1016/s1074-7613(00)80001-1. [DOI] [PubMed] [Google Scholar]
- 35.La Gruta NL, Doherty PC, Turner SJ. A correlation between function and selected measures of T cell avidity in influenza virus-specific CD8+ T cell responses. Eur.J Immunol. 2006;36:2951–2959. doi: 10.1002/eji.200636390. [DOI] [PubMed] [Google Scholar]
- 36.Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, Greenberg PD. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110:201–210. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pittet MJ, Valmori D, Dunbar PR, Speiser DE, Lienard D, Lejeune F, Fleischhauer K, Cerundolo V, Cerottini JC, Romero P. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J.Exp.Med. 1999;190:705–715. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Driessche A, Gao L, Stauss HJ, Ponsaerts P, Van Bockstaele DR, Berneman ZN, Van Tendeloo VF. Antigen-specific cellular immunotherapy of leukemia. Leukemia. 2005;19:1863–1871. doi: 10.1038/sj.leu.2403930. [DOI] [PubMed] [Google Scholar]
- 39.Uhlin M, Masucci M, Levitsky V. Is the activity of partially agonistic MHC:peptide ligands dependent on the quality of immunological help? Scand.J Immunol. 2006;64:581–587. doi: 10.1111/j.1365-3083.2006.01850.x. [DOI] [PubMed] [Google Scholar]
- 40.Quaratino S, Thorpe CJ, Travers PJ, Londei M. Similar antigenic surfaces, rather than sequence homology, dictate T-cell epitope molecular mimicry. Proc.Natl.Acad.Sci.U.S A. 1995;92:10398–10402. doi: 10.1073/pnas.92.22.10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, Depierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006 doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 43.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of proteinase K in the generation of AVDCs. Three surface markers were studied, CD86, MHC-I and MHC-II. Red: effects of supernatant conditioned by NDV-infected DCs in one trans-well-chamber on naïve DCs in the other chamber. Green: effects of supernatant conditioned by NDV-infected DC subsequently digested with proteinase K in one transwell-chamber on naïve DCs in the other chamber. Blue: naïve DCs with unconditioned media in the other trans-well chamber. The data shown are representative of two replicate experiments using two different donors that showed similar results.
Paracrine effects of virus-infected human epithelial fibroblasts on naïve DCs. Fibroblasts were seeded in the lower compartment of the trans-well system and were infected, (green), or not infected, (blue), with NDV to see if they were capable of generating the same maturation surface markers pattern as AVDCs (red). Fluorescence minus one control (FL -1) is shown in black. The data shown are representative of three different experiments using three different donors that showed similar results.
Effect of extended trans-well culturing on naïve DCs after 18, 24 and 48 hours, (red, blue and green respectively). FL-1 control is shown in black. The data shown are representative of three different experiments using three different donors that showed similar results.