

Abstract
Background
The neurobiological mechanisms by which only a minority of stress-exposed individuals develop psychiatric diseases remain largely unknown. Recent evidence suggests that dopaminergic neurons of the ventral tegmental area (VTA) play a key role in the manifestation of stress vulnerability.
Methods
Using a social defeat paradigm, we segregated susceptible mice (socially avoidant) from unsusceptible mice (socially interactive) and examined VTA punches for changes in neurotrophic signaling. Employing a series of viral vectors, we sought to causally implicate these neurotrophic changes in the development of avoidance behavior.
Results
Susceptibility to social defeat was associated with a significant reduction in levels of active/phosphorylated AKT (thymoma viral proto-oncogene) within the VTA, whereas chronic antidepressant treatment (in mice and humans) increased active AKT levels. This defeat-induced reduction in AKT activation in susceptible mice was both necessary and sufficient to recapitulate depressive behaviors associated with susceptibility. Pharmacologic reductions in AKT activity also significantly raised the firing frequency of VTA dopamine neurons, an important electrophysiologic hallmark of the susceptible phenotype.
Conclusions
These studies highlight a crucial role for decreases in VTA AKT signaling as a key mediator of the maladaptive cellular and behavioral response to chronic stress.
Keywords: Depression, mesolimbic dopamine, PTSD, resilience, social defeat, vulnerability
Stressful life events are causally related to major depression (1,2) and may also lead to persistent conditions such as posttraumatic stress disorder (PTSD) (3). Preclinical and postmortem studies aimed at elucidating the underlying neurobiology of these syndromes have mostly focused on describing the neurochemical correlates of the “depressed state” (4,5). However, it is often unknown whether these neuroadaptations represent 1) “pro-depressant” pathological mediators of maladaptive behavior or 2) “pro-resilient” homeostatic mechanisms that serve to promote active coping in times of adversity (6). A detailed understanding of these neuroadaptations is vital for the development of fundamentally novel therapeutic agents for stress-related disorders.
Among neural substrates involved in emotional processing, we have focused on the mesolimbic dopamine circuit, which is composed of dopaminergic neurons of the ventral tegmental area (VTA) and their projections to the nucleus accumbens (NAc), a circuit best known for its role in encoding the incentive-motivational valence of drug and natural rewards (7–9). This pathway has also been implicated in the pathophysiology of major depression, which includes symptoms of anhedonia (an inability to experience naturally rewarding and exciting stimuli) and a high incidence of comorbid substance abuse (10–13). Using a social defeat model adapted to explore individual differences, our group has recently identified this reward circuit as an important neural substrate in the manifestation of stress vulnerability (14,15). Mice that are vulnerable to social defeat display depression-like phenotypic features such as anhedonia, social avoidance, and weight loss, symptoms that are mediated via enhanced VTA dopamine neuron excitability and the consequent increased activity-dependent release of brain-derived neurotrophic factor (BDNF) in the NAc. Preventing the increase in VTA firing, NAc BDNF, or downstream BDNF signaling in the NAc was sufficient to reduce vulnerability, that is, these manipulations promoted a resilient phenotype. Because neuroadaptations within the VTA serve as the origin for this form of vulnerability (15) and given that neurotrophic signaling pathways in this region are potent regulators of the behavioral and cellular responses to stress and drugs of abuse (16–22), the purpose of this study was to explore stress-induced changes in neurotrophic signaling within the VTA. Through a combination of behavioral, molecular, and electrophysiologic techniques, we demonstrate that decreased AKT (thymoma viral proto-oncogene) activity in this brain region is an important mediator of stress vulnerability.
Methods and Materials
Animals and Drugs
For all experiments, animals were 1) male, 2) fed ad libitum, 3) allowed a 1-week habituation period before experimental manipulation, and 4) housed at 23°–25°C on a 12-hour light/dark cycle (lights on at 7 AM). Sprague-Dawley rats (350–375 g; Charles River, Wilmington, Massachusetts), 9 week-old male c57BL/6 mice (Jackson Labs, Bar Harbor, Maine), and CD1 retired breeders (Charles River) were used for this study. Rats (two per cage), c57BL/6 (four per cage), and CD1 (one per cage) mice were housed in clear polypropylene boxes containing wood shavings. Chronic fluoxetine was administered via subcutaneous pellets (Innovative Research, Novi, Michigan) implanted in the dorsal interscapular region under brief isoflurane anesthesia (Henry Schein, Melville, New York). Pellets were designed to administer 20 mg/kg/day of fluoxetine (or placebo) over a 20-day interval. All experiments were conducted in accordance with guidelines of the University of Texas Southwestern and Florida State University Animal Care and Use Committees.
Stress
Social defeat was performed as described previously (15). Resident CD1 mice with consistent attack latencies (≤3 0 sec on three consecutive screening tests) were housed in cages fitted with perforated Plexiglas separators (Ace Acrylics, Irving, Texas), which allow sensory contact without permitting physical contact. For 10 min a day for 10 days, mice were introduced into the aggressor's half of the cage and physically interacted and were then separated for the remainder of the day. Defeated mice were exposed to a new resident and cage every day and displayed classical murine signs of subordination stress (submissive posturing, vocalizations, etc.) during agonistic encounters. Instances of severe fighting were immediately interrupted, and topical Neosporin (Henry Schein) was applied to minor wounds. Control mice were handled daily and housed opposite another c57BL/6. Immediately after the 1st (in the case of single defeat) or 10th defeat, control and defeated mice were singly housed in preparation for behavioral testing the following day (Day 11). To test for pro-vulnerability effects of viral manipulation, naïve mice were exposed to a submaximal defeat protocol: three 5-min-long defeat episodes interspersed by 15-min rest periods on a single day (15). Social avoidance was tested the following day. Social isolation stress was applied by singly housing c57BL/6 mice for a period of 10 weeks (between 10 and 20 weeks of age); control mice for this experiment were housed in groups of 4.
Viral Vectors and Surgery
Herpes simplex viral (HSV) vectors encoding green fluorescent protein (GFP), wildtype insulin receptor substrate-2 (IRS2wt), IRS2dn (dn, dominant negative), AKTca (ca, constitutively active), or AKTdn have been previously used and validated (20,23,24). DNA constructs were ligated into the HSV plasmid (HSV-PrPUC) and virus was packaged via cotransfection with the helper 5dl1.2 (25). For stereotaxic delivery of the virus, mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). We employed established mouse VTA coordinates (anteroposterior: −3.2 mm, lateral: +1.0 mm, dorsoventral: −4.6 mm, 7° angle) (15), and bilaterally infused .5 μL over 5 min. Rats were anesthetized with a ketamine (90 mg/kg) and xylazine (10 mg/kg). We used established rat VTA coordinates (anteroposterior: −5.3 mm, lateral: +2.2 mm, dorsoventral: −7.6 mm, 10° angle) (17) and unilaterally infused 2 μL of virus over 10 min. The VTA placements in rats ranged from −5 to −5.5 mm posterior to the bregma, and injection sites were confirmed in all animals by standard histologic methods. All behavioral experiments were performed on Day 3 after viral surgery, when maximal transgene expression is observed (26).
Behavior
A two-trial social interaction test was used to assay avoidance behaviors toward an aversive social cue as described previously (14). In the first 2.5-min trial (“target absent”), the experimental c57BL/6 mouse was allowed to explore freely a square-shaped open-field arena (44 × 44 cm) possessing a wire-mesh cage (10 × 6 cm) apposed to one side (Figure 1A). During the second 2.5-min trial (“target present”), the mouse was reintroduced into this arena now containing an unfamiliar CD1 retired breeder mouse within the cage. Video-tracking software was employed to measure the time spent in the “interaction zone” (14 × 26 cm). Interaction ratios (IRs) were calculated as the ratio of the interaction zone durations in the target-present condition over the target-absent condition (expressed as a percentage), allowing a distinction of susceptible (IR < 100) and unsusceptible mice (IR ≥ 100) (15).
Figure 1.
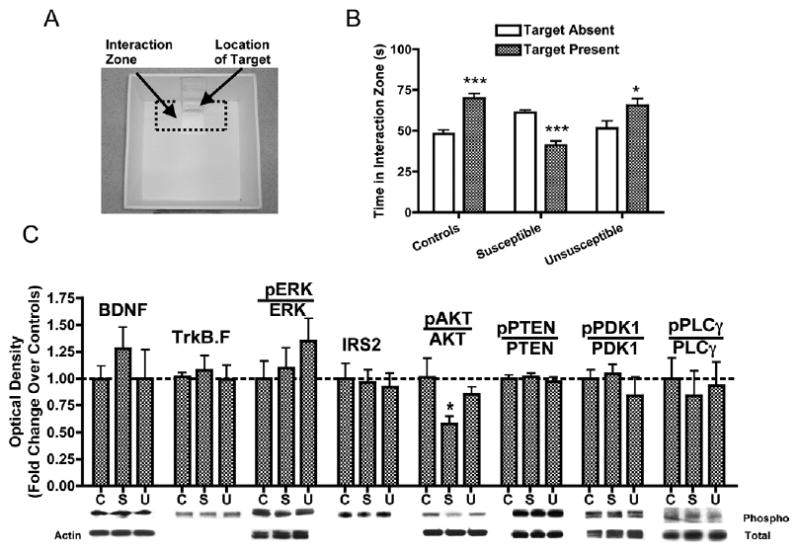
Chronic social defeat decreases phospho-AKT levels in the ventral tegmental area (VTA). (A) The photograph depicts the location of the “interaction zone” (dotted lines) within an open-field arena used for mouse social interaction testing. An unfamiliar CD1 mouse (“target”) is placed inside the wire mesh cage during the second trial. (B) Social interaction times for nondefeated control mice (t54 = 5.82, p< .0001, n = 28), susceptible (interaction ratio [IR] < 100, t52 = 6.52, p < .0001, n = 27), and Unsusceptible mice (IR ≥ 100, t32 = 2.30, p < .05, n = 17) mice (see Methods and Materials). (C) The VTA punches from c57BL/6 mice 1 day after interaction testing showed that chronic social defeat causes a approximately 40% reduction in phospho-AKT levels [F(2,42) = 3.41, p < .05, n = 11–18), without changes in the total level of AKT or in the total or phosphorylated levels of several other signaling molecules. Data are shown as mean + SEM, with * and *** indicating significant (p < .05, p < .01, or p < .001, respectively) comparisons from respective controls. C, controls; BDNF, brain-derived neurotrophic factor; ERK, extracellular-signal regulated kinase; IRS2, insulin-receptor substrate 2; PTEN, phosphatase and tensin homolog; PDK1, phospho-inositol dependent kinase 1; PLCγ, phospholipase Cγ; S, susceptible; TrkB.F, full-length tropomyosin-related kinase B; U, unsusceptible.
Sucrose preference was assayed using published protocols (17). Briefly, rats were habituated to a 1% sucrose solution. On pretest day, rats were singly housed and given access to a two-bottle choice for 30 min; the position of the bottles was balanced between the groups. Sucrose preference was calculated by dividing the volume of sucrose solution consumed by their total fluid intake. Rats with a preference ≥ 60% were randomly assigned to viral conditions such that average pretest scores were similar, and 3 days following stereotaxic surgeries, sucrose preference “posttest” scores were obtained. A 2-day forced-swim test protocol was employed to obtain immobility measures (18). Three days after surgery, rats were placed in plastic cylinders (30 × 45 cm) filled to 30 cm depth with 25°C tap water and were forced to swim for 15 min. Rats were retested the next day under identical conditions for 5 min, and all sessions were videotaped and scored by a blinded observer (27). For mice, locomotor activity was measured using a standard locomotor beam break apparatus (15) designed to detect ambulations within a novel cage. Rats were assayed on an automated open field (63 × 63 × 26 cm) apparatus.
Immunoblotting
Tissue punches of VTA (1.25 mm diameter) and NAc (1 mm diameter) from c57BL/6 mice were sonicated in a standard lysis buffer (15) and then centrifuged at 14,000 rpm for 15 min. Samples (20 μg; estimated through the Bradford assay) were treated with β-mercaptoethanol and subsequently electrophoresed on precast 4%–20% gradient gels (Biorad, Hercules, California). Proteins were transferred to polyvinylidene difluoride (PVDF), washed in 1× Tris-buffered saline with .1% Tween-20 (TBS-T), and blocked in milk dissolved in TBS-T (5% weight/volume) for 1 hour at 25°C. Incubations with the appropriate primary antibody solution were performed overnight at 4°C. After further washes, membranes were incubated with peroxi-dase-labeled goat antirabbit immunoglobulin G (IgG) or horse antimouse IgG (1:40,000; Vector Labs, Burlingame, California). Bands were visualized with SuperSignal West Dura substrate (Pierce Biotechnology, Rockford, Illinois) and quantified with Scion Image (National Institutes of Health).
Human Postmortem Study
Human VTA specimens were obtained from the Dallas Brain Collection and processed as previously described (15,28). Only depressed subjects with a recent history of chronic antidepressants were included; depressed subjects without antidepressant use were not available. Dissected VTA was placed immediately in a 1:1 mixture of dry ice and isopentane (volume/volume). Frozen tissue was then pulverized on dry ice and stored at −80°C. For immunoblotting, frozen pulverized tissue was sonicated in a lysis buffer (phosphate-buffered saline, protease and phosphatase inhibitor cocktails; Sigma, St. Louis, Missouri), centrifuged at 14,000 rpm for 15 min, and the protein concentration of the supernatant was estimated by the Bradford assay (between 1 and 4 μg/μL). Total BDNF levels in this fraction were estimated using a BDNF Emax ImmunoAssay kit (Promega, Madison, Wisconsin) with strict adherence to manufacturer protocols (15,29).
Electrophysiology
Brains removed from anesthetized mice were placed in sucrose-artificial cerebrospinal fluid (aCSF). Slices containing VTA were transferred into a recording chamber (aCSF flow ∼2.5 mL/min, 34°C). Single-unit extracellular potentials were recorded with glass microelectrodes filled with 2.0 M NaCl. The VTA dopamine neurons were identified by location and well-established electrophysiologic criteria: regular spontaneous firing, action potential (AP) with triphasic waveforms (positive, negative, positive) (15,30,31), and AP width (from start to trough) > 1.1 milliseconds. For whole-cell voltage-clamp recording experiments, cells were visualized with an upright microscope using infrared differential interference contrast illumination, and recordings were made under continuous single-electrode voltage clamp mode. Putative dopamine neurons in the VTA were identified in whole-cell recordings by the presence of a large hyperpolarization-activated current (Ih) (32,33), which was evoked by holding cells at −60 mV and stepping to −120 mV in 10-mV increments. Kynurenic acid (1 mmol/L) was added to the aCSF to block N-methyl-D-aspartate and alpha-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid/kainate receptor-mediated excitatory postsynaptic responses. Spontaneous inhibitory postsynaptic currents (sIPSCs) were obtained at the holding potential −60 mV and confirmed to be γ-aminobutyric acid (GABA)-mediated in each recording by adding 2 μmol/L SR95531 (a selective GABAA receptor antagonist) or 100 μmol/L picrotoxin (a chloride channel blocker).
Statistical Analysis
Statistical significance was measured using an unpaired two-tailed Student t test (for comparing two groups) and one-way analyses of variance (ANOVAs) followed by a Dunnett's post hoc test (for comparing three or more groups). For locomotor testing, we used a repeated-measure ANOVA. Main effects were considered significant at a p < .05. Data are expressed as the mean + SEM, with *, **, and *** representing significant differences from controls at p < .05, p < .01, and p < .0001 respectively.
See Supplement 1 for additional methodologic details.
Results
Chronic Social Defeat Stress Decreases AKT Phosphorylation in the VTA
Social defeat stress (15) was accomplished by introducing a c57BL/6 mouse into a cage territorialized by an aggressive CD1 retired breeder mouse. Twenty-four hours after the last defeat episode, control and defeated mice were tested on a two-trial social interaction test (Figure 1A). Whereas nondefeated control and unsusceptible mice displayed a significant increase in interaction times (Figure 1B), susceptible mice (∼60% of all defeated mice, IRs < 100) displayed a strong avoidance phenotype (15). Twenty-four hours after social avoidance testing, VTA punches were obtained from c57BL6 mice for immunoblotting studies in which we examined a panel of antibodies against molecules involved in BDNF signaling (Figure 1C). Although social defeat did not produce significant changes in the levels of BDNF or TrkB.F (full length tropomyosin-related kinase B), VTAs from susceptible mice displayed a significant reduction in AKT phosphorylation (Ser473, a site associated with catalytic activation) (34), with no changes observed in total levels of AKT nor in the phosphorylated or total levels of several other signaling proteins. This decrease in AKT activation was not observed in unsusceptible mice, after a single defeat or after a prolonged period of social isolation, suggesting that this change is unique to chronic and active forms of stress (Figure 2A). In contrast, chronic fluoxetine treatment significantly increased phospho-AKT levels (Figure 2A), and this effect was also observed in postmortem VTA samples from depressed humans chronically treated with antidepressants (Figure 2B, Supplement 2). In these samples, levels of total BDNF protein were unchanged (Figure 2C).
Figure 2.
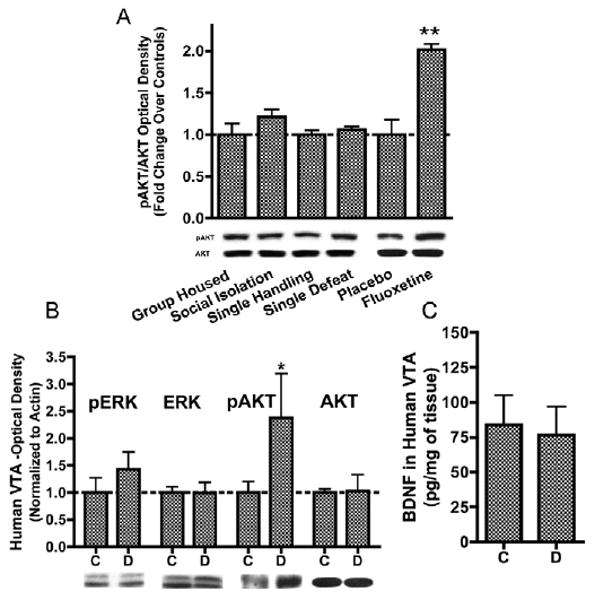
Antidepressant treatments produce increases in AKT activation in the ventral tegmental area (VTA). (A) Whereas prolonged social isolation (10 weeks, t12 = 1.37, p > .1) ora single episode of defeat (t12 = .94, p> .1) did not alter levels of phospho-AKT in the VTA, chronic fluoxetine administration significantly increased phospho-AKT levels (t7 = 3.77,p < .05, n = 4). (B) Postmortem VTA samples from depressed humans treated chronically with antidepressants (Supplement 2) also displayed a significant increase in phospho-AKT levels (t15 = 2.16, p < .05, n = 6-12/group) without altering ERK activation. (C) Results of a BDNF immunoassay experiment demonstrating no change in levels of total BDNF protein in human depressed VTA (t16 = .22, p > .5). Data are shown as mean + SEM, with * and ** indicating significant (p < .05 or p < .01. respectively) comparisons from respective control VTAs. BDNF, brain-derived neurotrophic factor; C, controls; D, depressed; ERK, extracellular-signal regulated kinase.
Modulating AKT Activity in the VTA Regulates Vulnerability to Social Defeat
To causally implicate decreased AKT phosphorylation as a mediator of defeat-induced social avoidance, we tested the ability of HSV-AKTca and HSV-AKTdn, infused into the VTA (Figure 3A), to alter behavior in the social defeat model. Because activated IRS2 binds to PI3K (phosphatidylinositol-3-kinase) and functions upstream of AKT (35), we also examined the effects of wildtype IRS2 (HSV-IRS2wt) and a dominant negative mutant of IRS2 (HSV-IRS2dn), which have previously been shown to increase and decrease, respectively, AKT phosphorylation (20). This approach enabled us experimentally to manipulate levels of AKT activity without affecting the total amount of the enzyme and hence better mimic the effects of chronic stress. In the first experiment, we injected HSV-GFP, HSV-IRS2dn, or HSV-AKTdn into the VTA of a group of age- and weight-matched naïve c57BL/6 mice (n = 6–8). Three days later, these groups were exposed to a submaximal defeat paradigm (Figure 3B schematic), consisting of three short defeats interspersed over 1 hour (15). On the fifth day, GFP-expressing mice displayed levels of social interaction comparable to nondefeated control mice (Figure 3B). In contrast, IRS2dn- and AKTdn-expressing mice did not significantly increase their interaction rates in this way, indicating an increased vulnerability. To obtain the converse type of information, we obtained three groups of susceptible mice matched for Day 11 interaction times (Figure 3C schematic). On Day 12, we stereotaxically infused HSV-GFP, HSV-IRS2wt, or HSV-AKTca into the VTA of these three groups, respectively, and measured social interaction 3 days later. Whereas GFP-expressing mice demonstrated the expected reduction in interaction times in response to a CD1 social cue, IRS2wt- and AKTca-expressing mice did not display social avoidance (Figure 3C), showing that restoring AKT function in the VTA ameliorates the effects of chronic social defeat.
Figure 3.
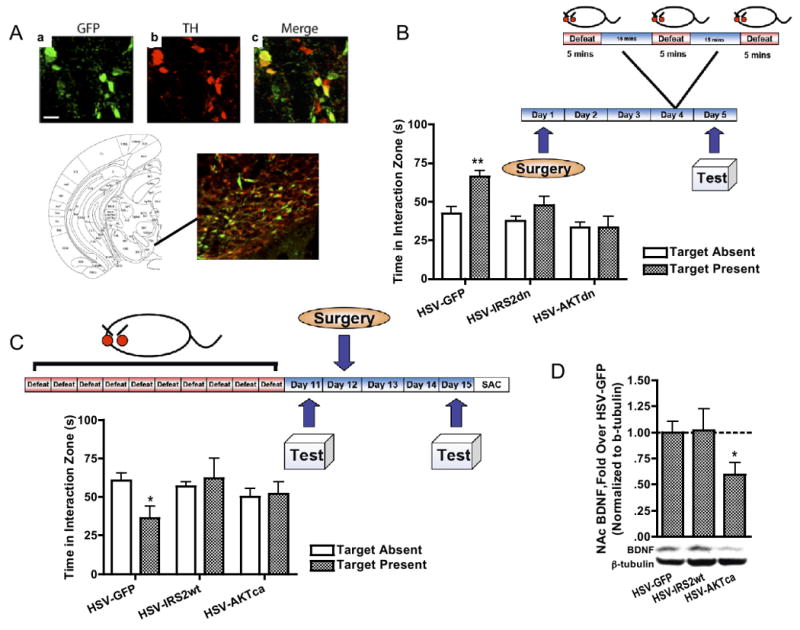
Decreased ventral tegmental area (VTA) AKT activity mediates social avoidance following chronic social defeat stress. (A) Validation of viral-mediated gene transfer in VTA (scale bar = 20 μm), showing cells expressing virally encoded GFP (a, Cy2), tyrosine hydroxylase (TH)-positive cells (b, Cy3), and merged confocal image of A and B showing double labeling (C) in region of VTA as shown in schematic (50). (B) Naïve mice infused with HSV-GFP and subjected to submaximal social defeat showed high levels of interaction (t10 = 3.83, p< .01), whereas IRS2dn or AKTdn overexpression in the VTA decreased interaction levels (p > .1 in both cases). (C) In the converse experiment, we examined the effects of viral manipulation on three matched groups of susceptible mice [30.2 ± 7.8 seconds, 36.5 ± 5.4 seconds, and 35.0 ± 6.2 seconds, F(2,28) = .23, p>.5, n = 7−11). Whereas a clear avoidance phenotype was observed after an intra-VTA injection of HSV-GFP (t19 = 2.65, p < .05), HSV-IRS2wt, or HSV-AKTca alleviated this form of avoidance (p > .5). (D) One day after the avoidance test in C, NAc BDNF protein levels in HSV-AKTca-infected mice are significantly lower than those in control animals (t19 = 2.52, p < .05). Data are shown as mean + SEM, with * and ** indicating significant (p < .05 or p < .01, respectively) comparisons from respective control animals. BDNF, brain-derived neurotrophic factor; GFP, green fluorescent protein; HSV, herpes simplex virus; TH, tyrosine hydroxylase.
Interestingly, when NAc samples from these groups were analyzed for levels of BDNF, HSV-AKTca infused mice displayed significantly lower levels of BDNF (Figure 3D), suggesting that the stress-resistant properties of AKT restoration are associated with blockade of stress-induced increases in BDNF signaling to the NAc, which we have previously shown is important for the Susceptible phenotype (14,15). As a control, separate groups of naïve c57BL/6 mice were infused with these viral vectors and examined for changes in locomotor activity. Modulating IRS2 or AKT function in this way had no effect on overall locomotor activity (Figure 4A), the motivation to socially interact with a CD1 mouse (Figure 4B), or the accumulation of BDNF protein within the NAc (Figure 4C). Collectively, these data show that a defeat-induced decrease in AKT function is both necessary and sufficient for the display of social avoidance, in a manner that cannot be explained simply by alterations in locomotor activity or sociability.
Figure 4.
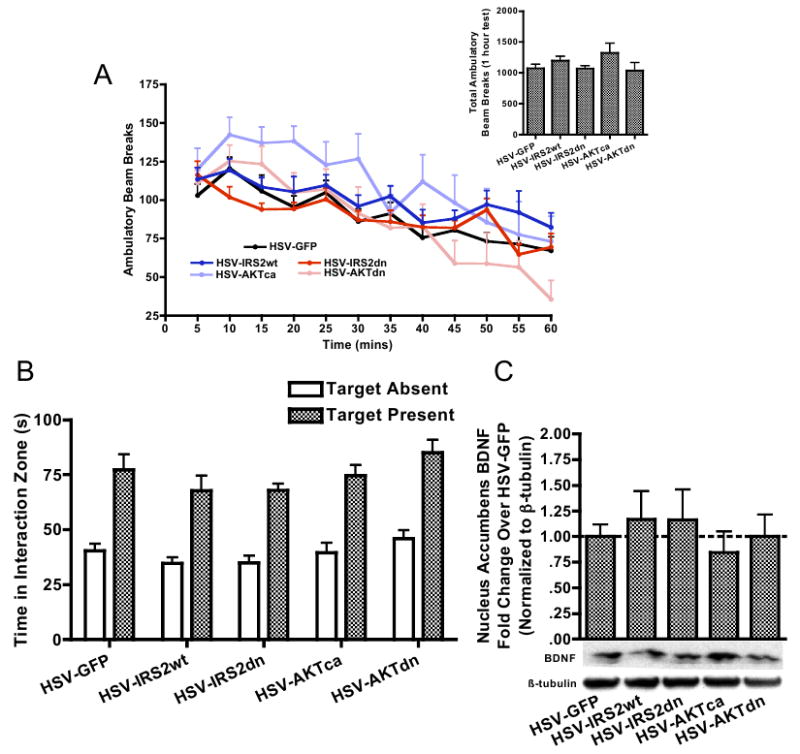
(A) Ventral tegmental area (VTA) overexpression of GFP, IRS2, or AKT in nonstressed mice does not significantly alter ambulatory locomotor hyperactivity or habituation over a 1-hour beam break locomotor test [virus main effect, F(4,43) = 1.44, p > .2]. Overall locomotor activity was also comparable across viral groups (inset). (B) VTA overexpression of GFP, IRS2, or AKT in nonstressed mice does not alter baseline social interaction rates [virus × target interaction, F(4,43) = .98, p > .3], as well as (C) levels of BDNF in the nucleus accumbens measured 3 days after viral overexpression [F(4,33) = .32, p > .5]. AKT, thymoma viral proto-oncogene; BDNF, brain-derived neurotrophic factor; GFP, green fluorescent protein; HSV, herpes simplex virus.
VTA AKT Activity Is a Regulator of Depression-Related Behaviors in Rats
To explore further a possible role for AKT activity in the VTA in regulating responses to stressful stimuli, and to extend our observations to a second species, we carried out a series of experiments in rats. In the forced-swim test (FST), rats placed in a cylinder of water initially struggle but soon display immobile posturing, which is taken to be an analog of the behavioral despair and helplessness often observed in stress-related disorders (36,37) and is indeed reversed by antidepressant treatments (38). Three days following intra-VTA stereotaxic infusions (FST Day 2), we found that the latency to (and duration of) immobility was dependent on viral treatment (Figure 5A and 5B): IRS2dn- and AKTdn-expressing rats displayed significantly greater immobility durations, a pro-depression-like effect. During this test, viral groups were not different on counts of swimming but displayed significant differences on counts of climbing and immobility (Figure 5C). These effects were unrelated to locomotor activity, which was unchanged by viral treatment (Figure 5D).
Figure 5.
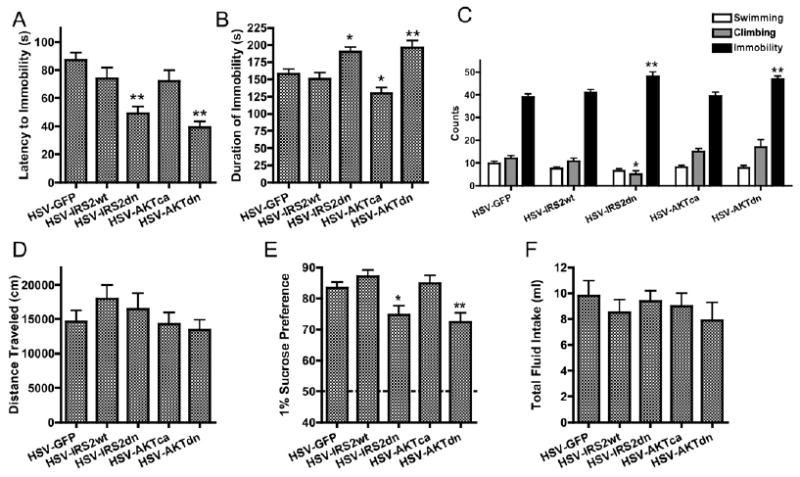
RAT: Decreases in insulin receptor substrate-2 (IRS2) and AKT signaling ventral tegmental area (VTA) enhance depression-related behaviors. (A) VTA-specific reductions in IRS2 or AKT function significantly decreased the latency to immobility on Day 2 of the rat forced-swim test [F(4,98) = 9.99, p<.0001,n = 13–28], as well as (B) significantly enhanced immobility durations [F(4,98) = 9.93,p<.0001]. (C) On this day, although viral groups were no different on counts of swimming [F(4,90) = 1.78, p > .1], IRS2dn overexpressing rats displayed significantly lower climbing counts [F(4,90) = 5.57, p < .0001], and both IRS2dn and AKTdn overexpressing ratsdisplayed significantly enhanced total immobility counts [F(4,90) = 6.84, p < .0001, n = 13–28]. (D) VTA overexpression of GFP, IRS2, or AKT in rats does not significantly change the distance moved in an open field [F(4,95) = 1.01, p > .3, n = 16–23). (E) In this experiment, we tested the effects of these manipulations on five groups of rats matched for equal “pretest” sucrose preference scores [F(4,92) = .33, p> .5, n = 13–28]. Three days postsurgery, IRS2dn and AKTdn overexpression produced significantly lower sucrose preference scores [F(4,92) = 6.00, p < .001]. (F) The total fluid consumed during the sucrose preference test was not changed by viral manipulation [F(4,92) = .47, p > .5). Data are shown as mean + SEM, with * and ** indicating significant (p < .05 orp < .01, respectively) post hoc comparisons from respective control rats. AKT, thymoma viral proto-oncogene; GFP, green fluorescent protein; HSV, herpes simplex virus.
Similar results were obtained on measures of sucrose preference, which is reduced following various forms of chronic stress (39,40). When tested 3 days after viral infusions, sucrose preference varied as a function of viral treatment (Figure 5E): IRS2dn- and AKTdn-expressing rats displayed significantly lower preference scores, without clear differences in fluid intake (Figure 5F).
PI3K Inhibition Increases the Spontaneous Firing Frequency of VTA Dopamine Neurons
Long-lasting increases in the excitability of VTA dopamine neurons, but not of nondopaminergic neurons, mediate defeat-induced increases in NAc BDNF levels as well as several behaviors associated with susceptibility (15). However, VTA-intrinsic mechanisms that underlie this electrophysiologic adaptation are unknown. Because AKTca overexpression in socially defeated mice was able to reduce significantly BDNF protein levels in the NAc (Figure 3D), we sought to explore how modulating AKT function would alter the spontaneous firing frequency of VTA dopamine neurons. LY294002 is a reversible PI3K inhibitor capable of blocking AKT phosphorylation (41) and 100 μmol/L LY294002 blocks the expression of long-term potentiation in the hippocampal CA1 region (42). Extracellular recordings of VTA dopamine neurons showed that 100 μmol/L LY294002 caused a significant increase in the pacemaker frequency of these neurons (Figure 6A), and this effect was reversed by washout. LY294002's effects on VTA excitability were blocked in the presence of 2 μmol/L SR95531 (GABAA antagonist). These findings indicate that inhibition of AKT is sufficient to increase VTA neuronal excitability through GABAA-dependent mechanisms. One of AKT's known targets is the β2 subunit of the GABAA receptor: AKT phosphorylates Ser410 of the β2 subunit, which increases membrane insertion and GABAergic transmission (43). Through whole-cell recording techniques, we observed that LY294002 bath application caused a significant reduction in both the amplitude and frequency of spontaneous GABA-mediated inhibitory postsynaptic currents (IPSCs) recorded in dopaminergic neurons (Figure 6B). These data strongly implicate reduced AKT activity and subsequent reductions in GABAergic transmission as a candidate mechanism for the heightened firing rates of VTA dopamine neurons and BDNF release observed in socially defeated mice (Figure 7).
Figure 6.

Decreased AKT signaling increases the excitability of ventral tegmental area (VTA) dopamine neurons. (A) Extracellular recordings from VTA dopamine neurons reveal that 100 μmol/L LY294002, an inhibitor of PI3K that reduces thymoma viral proto-oncogene (AKT) phosphorylation and activity (51), significantly increases spontaneous pacemaker firing frequency (t10 = 2.0, p < .05, n = 5–6, 3–4 mice/group, paired t test), which is reversed by washout (representative traces shown on right). SR95531, 2 μmol/L, (a γ-aminobutyric acid [GABA]A antagonist) also increases VTA firing frequency and blocks further LY294002 mediated increases in VTA excitability (n = 7/group). (B) In whole-cell voltage-clamp recording experiments, 100 μmol/L LY294002 also produced significant decrements in the amplitude (t6 = 3.09, p < .05) and frequency (t6 = 3.16, p < .05, n = 7 neurons/group, 3–4 mice/group, paired t tests) of spontaneous GABA-mediated inhibitory postsynaptic currents (IPSCs) onto VTA dopamine neurons (representative IPSC traces shown above). Data are shown as mean + SEM, with * and ** indicating significant (p < .05 or p < .01) paired t test comparisons against the control condition.
Figure 7.
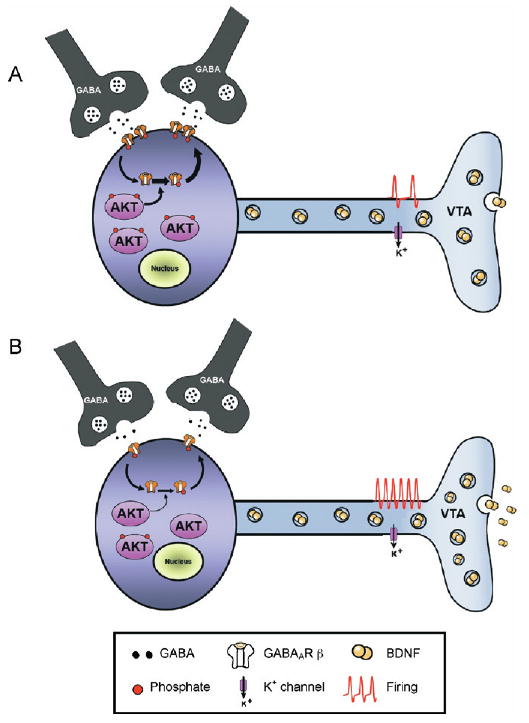
Working model of the role of thymoma viral proto-oncogene (AKT) in the ventral tegmental area (VTA) after chronic stress. (A) At baseline, VTA dopamine neurons receive tonic inhibition from surrounding GABAergic interneurons as well as projection neurons. High basal levels of AKT activation support the membrane insertion of GABAA receptors (GABAAR) via the AKT-medi-ated phosphorylation of GABAAR β2 subunits. (B) Following chronic exposure to social defeat stress, reduced AKT tone leads to reduced GABAergic transmission, through a combination of reduced GABAAR membrane expression and reduced GABA release. This leads to enhanced excitability and subsequent increased rates of activity-dependent BDNF release in nucleus accumbens. BDNF, brain-derived neurotrophic factor; GABA, γ-amino butyric acid.
Discussion
The most significant nongenetic risk factors for depression and PTSD include 1) adverse childhood experiences, 2) neuroticism, and 3) female gender (5,44,45). These data identify at-risk populations, but they offer little insight into neurobiological and molecular mechanisms underlying vulnerability. By coupling a chronic stress paradigm to a quantitative measure of social withdrawal, we showed that vulnerability to defeat-induced social avoidance was mediated through enhanced firing of VTA dopamine neurons and a consequent increase in the activity-dependent release of BDNF from the VTA to the NAc (15). Although the site of BDNF's pro-vulnerability effects appears to be the NAc, it is clear that primary neuroadaptations within the VTA that modulate dopamine neuron excitability serve as an origin for the manifestation of vulnerability. This study illustrates how one such adaptation in stress-susceptible mice, decreased AKT phosphorylation, is both necessary and sufficient to recapitulate the behavioral and electrophysiologic signatures of vulnerability to social defeat stress.
Postmortem studies of cortical regions obtained from depressed humans (46,47) have also described reductions in AKT function. In our study, we observed a significant increase in AKT phosphorylation in depressed VTA postmortem samples, without a change in total AKT levels. This effect was recapitulated in mice chronically treated with the selective serotonin reuptake inhibitor (SSRI), fluoxetine, which suggests that the changes we observed in postmortem VTA are likely a consequence of chronic antide-pressant exposure. Given our small sample size and our lack of access to cases of nonmedicated depression, further studies are warranted to dissect the contribution of depression-related disease processes versus antidepressant-induced changes in the VTA.
It remains unclear whether reduced AKT function represents a preexisting risk factor or the result of an interaction between social defeat stress and other, as yet undefined mechanisms. In particular, the upstream mechanisms underlying the stress-induced downregulation in AKT signaling in susceptible mice are currently unknown, as well as those mechanisms that allostati-cally maintain AKT activity within normal levels in VTAs from unsusceptible mice. Our immunoblotting studies did not reveal changes in the activity of BDNF, IRS2, PDK1, or PTEN, all of which are upstream nodes capable of altering AKT activity. Indeed, there was a trend for increased BDNF levels, which is consistent with the marked pro-depressant effect of BDNF within the mesolimbic dopamine circuit (14,15,18). BDNF and its receptor are only one among a constellation of growth factor-receptor tyrosine kinase systems linked to IRS-PI3K-AKT signaling, and further studies are required to determine whether the defeat-induced downregulation of AKT signaling in the VTA is secondary to changes in any of these other systems. As just one example, psychological stress is thought to play a key role in the development of metabolic syndrome (48), a constellation of symptoms that includes abdominal obesity, insulin resistance, and hypertension. Insulin receptors are expressed on VTA dopamine neurons (49), and decreased AKT signaling within the VTA (and other brain regions) may be a central correlate of systemwide insulin resistance brought about by exposure to chronic stress.
Through viral-mediated gene transfer, we showed that decreased AKT activation within the VTA is necessary and sufficient to confer vulnerability to defeat-induced social avoidance. In rats, whereas HSV-IRS2dn and HSV-AKTdn significantly enhanced depression-related behaviors, overexpression of IRS2wt or AKTca had no measurable effects. These viruses did promote a resilient phenotype when overexpressed in the background of abnormally low levels of AKT phosphorylation, that is, following social defeat. In addition, the overexpression of AKTca significantly reduced stress-induced increases in NAc BDNF protein levels (Figure 3). This effect was not observed following IRS2 overexpression; we speculate that this unique property of AKTca may be related to the enhanced ability of a constitutively active form of AKT (which lies more “downstream” in the PI3K cascade) to alter VTA firing rates.
Collectively, these data suggest that, at baseline, the activity of this pathway may be sufficiently high in the VTA to maximally drive downstream effects. Thus small decrements in the activity of this cascade may have significant cellular (and behavioral) ramifications, such as in our observation that 100-μM LY294002, a PI3K inhibitor that is known to reduce AKT phosphorylation, significantly increased the pacemaker frequency of VTA dopamine neurons. We have previously identified heightened rates of VTA firing as a crucial mediator of the vulnerability to social defeat stress (15). Thus the robust pro-depressant effects of HSV-IRS2dn and HSV-AKTdn may be mediated through their ability to potentiate VTA dopamine neuron excitability. AKT has a diverse array of known substrates (34), including the β2 subunit of the GABAA receptor: phosphorylation of Ser410 of the GABAaβ2 by AKT has been shown to increase the insertion of GABAA receptors into the plasma membrane, thereby enhancing GABA-mediated inhibition (43). Whole-cell recording experiments allowed us to observe a decrease in the amplitude of spontaneous GABAA-receptor-mediated IPSCs, indicative of reductions in membrane GABAAR expression. We also observed a reduction in the frequency of GABAergic IPSCs, suggesting that presynaptic effects on basal GABAergic transmission cannot be ruled out. Indeed, reductions in AKT activation in both interneu-rons and dopaminergic neurons may synergistically increase VTA excitability through overall reductions in GABAergic neurotransmission (Figure 7). Further experiments to examine the functional consequences of PI3K inhibition on VTA GABAergic interneurons are clearly warranted.
In conclusion, we show that the stress-induced downregulation of AKT activity in the VTA serves a crucial role in regulating the vulnerability to develop depressive behaviors in both mouse and rat models. Our study further substantiates the important role of neuroplasticity within the mesolimbic dopamine circuit in the control of emotional homeostasis and resilience (4,14,15). A clearer understanding of the detailed intracellular mechanisms downstream of AKT changes and how they may regulate the physiology and morphology of VTA dopamine neurons could provide novel insights into the development of therapeutic agents against depression and other stress-related disorders.
Supplementary Material
Acknowledgments
We thank Bryan Potts and Kelly Lewis-Amezcua for outstanding technical assistance. This study would not have been possible without the generosity and foresight of the families of our brain tissue donors. This work was supported by grants from the National Institute on Drug Abuse (to CAB and EJN), the National Institute of Mental Health (to EJN), a research alliance with AstraZeneca, and Young Investigator Awards from the National Alliance for Research in Schizophrenia and Depression (to CAB, MHH and SJR).
Footnotes
All the authors reported no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online.
Contributor Information
Vaishnav Krishnan, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas.
Ming-Hu Han, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas.
Michelle Mazei-Robison, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas.
Sergio D. Iñiguez, Department of Psychology and Program in Neuroscience, Florida State University, Tallahassee
Jessica L. Ables, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas
Vincent Vialou, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas.
Olivier Berton, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas.
Subroto Ghose, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas.
Herbert E. Covington, III, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas.
Matthew D. Wiley, Department of Psychology and Program in Neuroscience, Florida State University, Tallahassee
Ross P. Henderson, Department of Psychology and Program in Neuroscience, Florida State University, Tallahassee
Rachael L. Neve, Harvard Medical School, McLean Hospital, Belmont, Massachusetts
Amelia J. Eisch, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas
Carol A. Tamminga, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas
Scott J. Russo, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas
Carlos A. Bolaños, Department of Psychology and Program in Neuroscience, Florida State University, Tallahassee
Eric J. Nestler, Departments of Psychiatry and Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas
References
- 1.Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- 2.Charney DS, Manji HK. Life stress, genes, and depression: Multiple pathways lead to increased risk and new opportunities for intervention. Sci STKE. 2004 March 16;:re5. doi: 10.1126/stke.2252004re5. [DOI] [PubMed] [Google Scholar]
- 3.Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- 4.Charney DS. Psychobiological mechanisms of resilience and vulnerability: Implications for successful adaptation to extreme stress. Am J Psychiatry. 2004;161:195–216. doi: 10.1176/appi.ajp.161.2.195. [DOI] [PubMed] [Google Scholar]
- 5.Yehuda R, Ledoux J. Response variation following trauma: A translational neuroscience approach to understanding PTSD. Neuron. 2007;56:19–32. doi: 10.1016/j.neuron.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Yehuda R, Flory JD. Differentiating biological correlates of risk, PTSD, and resilience following trauma exposure. J Traum Stress. 2007;20:435–447. doi: 10.1002/jts.20260. [DOI] [PubMed] [Google Scholar]
- 7.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: The role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 8.Kelley AE, Berridge KC. The neuroscience of natural rewards: Relevance to addictive drugs. J Neurosci. 2002;22:3306–3311. doi: 10.1523/JNEUROSCI.22-09-03306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wise RA. Dopamine, learning and motivation. Nature Rev. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- 10.Dunlop BW, Nemeroff CB. The role of dopamine in the patho-physiology of depression. Arch Gen Psychiatry. 2007;64:327–337. doi: 10.1001/archpsyc.64.3.327. [DOI] [PubMed] [Google Scholar]
- 11.Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 12.Brady KT, Sinha R. Co-occurring mental and substance use disorders: The neurobiological effects of chronic stress. Am J Psychiatry. 2005;162:1483–1493. doi: 10.1176/appi.ajp.162.8.1483. [DOI] [PubMed] [Google Scholar]
- 13.Rush AJ. The varied clinical presentations of major depressive disorder. J Clin Psychiatry. 2007;68(suppl 8):4–10. [PubMed] [Google Scholar]
- 14.Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 15.Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 16.Bolanos CA, Nestler EJ. Neurotrophic mechanisms in drug addiction. Neuromolecular Med. 2004;5:69–83. doi: 10.1385/NMM:5:1:069. [DOI] [PubMed] [Google Scholar]
- 17.Bolanos CA, Perrotti LI, Edwards S, Eisch AJ, Barrot M, Olson VG, et al. Phospholipase Cgamma in distinct regions of the ventral tegmental area differentially modulates mood-related behaviors. J Neurosci. 2003;23:7569–7576. doi: 10.1523/JNEUROSCI.23-20-07569.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisch AJ, Bolanos CA, de Wit J, Simonak RD, Pudiak CM, Barrot M, et al. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: A role in depression. Biol Psychiatry. 2003;54:994–1005. doi: 10.1016/j.biopsych.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russo SJ, Bolanos CA, Theobald DE, Decarolis NA, Renthal W, Kumar A, et al. IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nature Neurosci. 2007;10:93–99. doi: 10.1038/nn1812. [DOI] [PubMed] [Google Scholar]
- 21.Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: Implications for incubation of cocaine craving. J Neurosci. 2003;23:742–747. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pierce RC, Bari AA. The role of neurotrophic factors in psycho-stimulant-induced behavioral and neuronal plasticity. Rev Neurosci. 2001;12:95–110. doi: 10.1515/revneuro.2001.12.2.95. [DOI] [PubMed] [Google Scholar]
- 23.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 24.Papapetropoulos A, Fulton D, Mahboubi K, Kalb RG, O'Connor DS, Li F, et al. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/sur-vivin pathway. J Biol Chem. 2000;275:9102–9105. doi: 10.1074/jbc.275.13.9102. [DOI] [PubMed] [Google Scholar]
- 25.Neve RL, Howe JR, Hong S, Kalb RG. Introduction of the glutamate receptor subunit 1 into motor neurons in vitro and in vivo using a recombinant herpes simplex virus. Neuroscience. 1997;79:435–447. doi: 10.1016/s0306-4522(96)00645-8. [DOI] [PubMed] [Google Scholar]
- 26.Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, et al. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Nat Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cryan JF, Page ME, Lucki I. Differential behavioral effects of the antidepressants reboxetine, fluoxetine, and moclobemide in a modified forced swim test following chronic treatment. Psychopharmacology. 2005;182:335–344. doi: 10.1007/s00213-005-0093-5. [DOI] [PubMed] [Google Scholar]
- 28.Stan AD, Ghose S, Gao XM, Roberts RC, Lewis-Amezcua K, Hatanpaa KJ, et al. Human postmortem tissue: what quality markers matter? Brain Res. 2006;1123:1–11. doi: 10.1016/j.brainres.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- 31.Werkman TR, Olijslagers JE, Perlstein B, Jansen AH, McCreary AC, Kruse CG, et al. Quetiapine increases the firing rate of rat substantia nigra and ventral tegmental area dopamine neurons in vitro. EurJ Pharmacol. 2004;506:47–53. doi: 10.1016/j.ejphar.2004.10.053. [DOI] [PubMed] [Google Scholar]
- 32.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 33.Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A. Cortico-tropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron. 2003;39:401–407. doi: 10.1016/s0896-6273(03)00461-6. [DOI] [PubMed] [Google Scholar]
- 34.Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamada M, Ohnishi H, Sano S, Nakatani A, Ikeuchi T, Hatanaka H. Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J Biol Chemistry. 1997;272:30334–30339. doi: 10.1074/jbc.272.48.30334. [DOI] [PubMed] [Google Scholar]
- 36.Lucki I, Dalvi A, Mayorga AJ. Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psycho-pharmacol. 2001;155:315–322. doi: 10.1007/s002130100694. [DOI] [PubMed] [Google Scholar]
- 37.Cryan JF, Markou A, Lucki I. Assessing antidepressant activity in rodents: Recent developments and future needs. Trend Pharmacol Sci. 2002;23:238–245. doi: 10.1016/s0165-6147(02)02017-5. [DOI] [PubMed] [Google Scholar]
- 38.Berton O, Nestler EJ. New approaches to antidepressant drug discovery: Beyond monoamines. Nature Rev. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- 39.Papp M, Moryl E, Willner P. Pharmacological validation of the chronic mild stress model of depression. Eur J Pharmacol. 1996;296:129–136. doi: 10.1016/0014-2999(95)00697-4. [DOI] [PubMed] [Google Scholar]
- 40.Willner P. Chronic mild stress (CMS) revisited: Consistency and behavioural-neurobiological concordance in the effects of CMS. Neuro-psychobiology. 2005;52:90–110. doi: 10.1159/000087097. [DOI] [PubMed] [Google Scholar]
- 41.Kuzman JA, Gerdes AM, Kobayashi S, Liang Q. Thyroid hormone activates Akt and prevents serum starvation-induced cell death in neonatal rat cardiomyocytes. Journal of molecular and cellular cardiology. 2005;39:841–844. doi: 10.1016/j.yjmcc.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 42.Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, et al. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, et al. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- 44.Fava M, Kendler KS. Major depressive disorder. Neuron. 2000;28:335–341. doi: 10.1016/s0896-6273(00)00112-4. [DOI] [PubMed] [Google Scholar]
- 45.Yehuda R. Post-traumatic stress disorder. New Engl J Med. 2002;346:108–114. doi: 10.1056/NEJMra012941. [DOI] [PubMed] [Google Scholar]
- 46.Hsiung SC, Adlersberg M, Arango V, Mann JJ, Tamir H, Liu KP. Attenuated 5-HT1A receptor signaling in brains of suicide victims: Involvement of adenylyl cyclase, phosphatidylinositol 3-kinase, Akt and mitogen-activated protein kinase. J Neurochem. 2003;87:182–194. doi: 10.1046/j.1471-4159.2003.01987.x. [DOI] [PubMed] [Google Scholar]
- 47.Karege F, Perroud N, Burkhardt S, Schwald M, Ballmann E, La Harpe R, et al. Alteration in kinase activity but not in protein levels of protein kinase B and glycogen synthase kinase-3beta in ventral prefrontal cortex of depressed suicide victims. Biol Psychiatry. 2007;61:240–245. doi: 10.1016/j.biopsych.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 48.Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30:1–10. doi: 10.1016/j.psyneuen.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG. Expression of receptors for insulin and leptin in the ventral tegmental area/sub-stantia nigra (VTA/SN) of the rat. Brain Res. 2003;964:107–115. doi: 10.1016/s0006-8993(02)04087-8. [DOI] [PubMed] [Google Scholar]
- 50.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press; 2001. [Google Scholar]
- 51.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-ben-zopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.