

Abstract
The role of extracellular matrix (ECM) in neurological development, function and degeneration has evolved from a simplistic physical adhesion to a system of intricate cellular signaling. While most cells require ECM adhesion to survive, it is now clear that differentiated function is intimately dependent upon cellular interaction with the ECM. Therefore, it is not surprising that the ECM is increasingly found to be involved in the enigmatic process of neurodegeneration. Descriptive studies of human neurodegenerative disorders and experimental studies of animal models of neurodegeneration have begun to define potential mechanisms of ECM disruption that can lead to synaptic and neuronal loss.
Keywords: ECM, neurodegeneration, CNS
BRAIN EXTRACELLULAR MATRIX (ECM) STRUCTURE, MODULATION AND FUNCTION
The structure of the brain ECM
Compared to systemic ECM, the adult brain ECM is unusual in at least two respects. First, unlike other organs, the brain exhibits limited ultrastructurally well‐defined stromal space. Despite elegant descriptions by Camillo Golgi and Santiago Ramon y Cajal at the turn of the previous century, prior to the 1970s, it has been generally accepted that brain tissue consists predominantly of closely apposed neurons and glia, leaving little room for significant amounts of ECM. Several newer technologies have been used to gradually revise this theory to accommodate the presence of a variety of ECM molecules filling significant amounts of ECM space (93). Second, common ECM components of systemic organs (eg, fibronectin and collagen) are virtually absent from the adult brain ECM, while different types of proteoglycans are abundantly expressed in the adult brain and are localized to intercellular spaces between neurons and glia [For extensive review, see Yamaguchi (93), Dityatev and Schachner (31), and Bandtlow and Zimmermann (8).].
In retrospect, the neuronal cell surface feature called the perineuronal net (PNN), first described by Camillo Golgi and Santiago Ramon y Cajal in 1890, is consistent with the “new discoveries” of adult brain ECM. PNNs are reticular networks observed on the surface of neuronal cell bodies and proximal dendrites (Figure 1). As the name implies, the ECM materials deposited in the space between neurons and astrocytic processes ensheath the neuronal cell surface with a netlike structure (Figure 1). While histologically bland in appearance, PNNs vary in character and distribution, each with a potentially unique molecular structure surrounding a wide variety of neurons (31). This structure includes lecticans, hyaluronic acid (HA), tenascin‐C and tenascin‐R. It is thought that the HA–lecticans–tenascin complex deposited on neuronal surfaces may form a repulsive barrier against approaching axons and dendrites. The absence of synaptic contacts along neuronal surfaces covered by PNN supports this hypothesis. Although the physiological role of PNNs has not been fully elucidated, their postnatal appearance suggests a role in limiting the development of new synaptic contacts (93).
Figure 1.
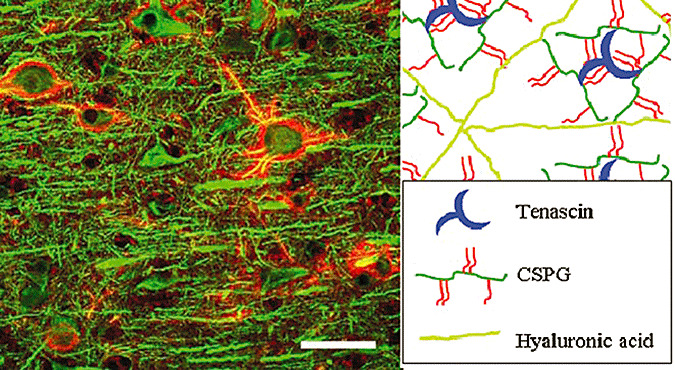
Perineuronal nets (PNNs). Macaque brain stained with microtubule‐associated protein 2 (MAP2) (green) and Wisteria floribunda agglutinin (WFA) (red) (A). Model of PNNs (B). Hypothetical PNN ternary complex of tenascin, chondroitin sulfate proteoglycans (CSPGs) and hyaluronic acid (HA).
PNNs are a structural defined part of the ECM composed of a variety of proteoglycans. The diversity of proteoglycans in the ECM is dependent on differential expression of genes encoding core proteins, alternative splicing and transcription–termination, as well as variations in the length and types of glycosaminoglycan (GAG) side chains. Built around a core protein, polymers of 20–200 disaccharide repeats are attached through serine residues. Classified on the basis of disaccharide composition, GAGs are grouped as chondroitin sulfate (CS)/dermatan sulfate (DS), heparan sulfate (HS) and keratan sulfate (KS). HA is a non‐sulfonated polymer of glucosamine and glucoronic acid, which exists as a protein‐free polysaccharide on cell surfaces and in the ECM (8). Chondroitin sulfate or HS side chains compose most central nervous system (CNS) proteoglycans; some of them are constituents of the ECM and others are bound to cell surfaces by a transmembrane domain or anchored with glycosyl phosphatidylinositol (GPI). Proteoglycans can either promote or inhibit neuritic growth and possibly synaptic remodeling. Both the protein core and GAG side chains contribute to promoting or inhibiting neurite growth (Table 1) (8).
Table 1.
Extracellular matrix (ECM) components. Abbreviations: PNN = perineuronal net; CSPG = chondroitin sulfate proteoglycan; GAG = glycosaminoglycan; CNS = central nervous system; GPI = glycosyl phosphatidylinositol; HSPG = heparan sulfate proteoglycan; FGF = fibroblast growth factor; VEGF = vascular endothelial growth factor; HB‐GAM = heparin‐binding growth‐associated molecule; aFGF = acidic FGF; bFGF = basic fibroblast growth factor; TGFβ = transforming growth factor β; LTP = long‐term potentiation; MMP = matrix metalloproteinase; TIMP = tissue inhibitors of metalloproteinase; tPA = tissue plasminogen activator; ADAMTS = A disintegrin and metalloproteinase with thrombospondin motifs; PDGF = platelet‐derived growth factor; IGF = Insulin growth factor; KGF = keratinocyte growth factor.
| Molecule | Description |
|---|---|
| GAGs | |
| Heparan sulfate (HS) | Polymer of disaccharide of N‐acetylglucosamine and glucoronic acid. HS moieties of cell surface proteoglycans modulate the biological responses to FGF. |
| Chondroitin sulfate (CS) | Polymer of disaccharide of N‐acetylgalactosamine and glucoronic acid. |
| Dermatan sulfate (DS) | Polymer of disaccharide of N‐acetylgalactosamine and iduronic acid. |
| Keratan sulfate (KS) | Polymer of disaccharide of N‐acetylgalactosamine and galactose. |
| Hyaluronic acid (HA) | Non‐sulfated polymer of N‐acetylglucosamine and glucoronic acid. As opposed to other GAGs, it is not attached to a core protein. Component of PNNs. The biological effects of HA are mediated through cell surface receptors including CD44 and receptor for HA‐mediated motility. |
| Proteoglycans | |
| Glypican | A family of six GPI‐anchored cell surface HSPGs that are widely expressed in the CNS. Bind laminin, thrombospondin, FGFs, VEGF and IGF. Associated with neurogenesis and neurite outgrowth. |
| Syndecan | A family of four membrane‐associated HSPGs. Bind tenascin‐C, fibronectin, laminin, HB‐GAM, bFGF and TGFβ. Associated with LTP and synaptic function and neurite outgrowth. |
| Phosphacan | A secreted CSPG. Bind tenascin‐R/C and HB‐GAM. Associated with neurogenesis, neuronal migration and neurite outgrowth. |
| Versican | Secreted CSPG. Binds to HA, tenascin‐R and fibronectin. Different alternative spliced variants have been identified. Associated with neuronal migration and neurite outgrowth. |
| Brevican | Secreted and GPI‐anchored CSPG. Binds to HA and tenascin‐R. Associated with neurite outgrowth and synaptic function. |
| Neurocan | A secreted CSPG. Binds to HA, tenascin‐R/C and bFGF. Associated with neurite outgrowth and synaptic function. |
| Other proteins | |
| Tenascin | ECM protein associated with PNNs. Binds to brevican, phosphacan, neurocan, HA, syndecan, glypican and integrins. Associated with neurite outgrowth and LTP and synaptic function. |
| Laminin | A secreted family of key members of the ECM. Binds predominantly to integrins. Associated with migration, survival and synaptic function. |
| ECM‐modulating enzymes | |
| MMP | Family of zinc‐dependent extracellular proteases. Associated with neurogenesis, survival and plasticity. |
| TIMP | Inhibitor of MMPs. Associated with neuronal survival and plasticity. |
| tPA | Serine protease that converts inactive plasminogen to active plasmin. Key initiator of MMPs. Associated with neurite outgrowth, neuronal migration, survival and synaptic function. |
| ADAMTS | Protease family with aggrecanase activity associated with the ECM through a thrombospondin motif. |
| Plasminogen activator inhibitor 1 | A serine proteinase inhibitor (serpin) plasminogen activator inhibitor. |
| Hyaluronidase | Family of enzymes that degrade HA. |
| Heparanase | An endo‐β‐D‐glucuronidase that catalyzes the hydrolytic cleavage of the β‐1,4‐glycosidic bond in HS. |
| Chondroitinase | Lyase that degrades CS. |
| ECM‐binding factors | |
| FGF | FGFs constitute a large family of polypeptides (eg, aFGF, bFGF, KGF) that are important in the control of cell growth and differentiation and play a key role in oncogenesis, developmental processes and neuronal development. |
| HB‐GAM | HB‐GAM (also designated as pleiotrophin) and midkine form a two‐member family of ECM proteins that bind tightly to sulfated carbohydrate structures such as HS. Binds to phosphacan and syndecan. Associated with neurite outgrowth, axon guidance and synaptogenesis. |
| PDGF | PDGF is one of the numerous growth factors that regulate cell growth and division. |
| VEGF | VEGF is an important signaling molecule involved in angiogenesis. |
Lecticans are large secreted proteoglycans that carry mainly CS side chains and include aggrecan, neurocan, brevican and versican. These proteoglycans are characterized by the presence of an HA‐binding domain. Two families of membrane‐bound heparan sulfate proteoglycans (HSPGs) abundantly expressed in the CNS are glypicans and syndecans. All glypicans are synthesized as precursor peptides with a signal peptide and a hydrophobic stretch at the C‐terminus, which is replaced in the mature polypeptide by a covalently linked GPI anchor. All four mammalian syndecans are transmembrane proteoglycans that carry predominantly HS side chains. Shedding of syndecan membrane‐bound ectodomains is key to their physiological function (8).
ECM capacity to bind growth factors via HS and CS modulates their interaction with the cell surface. In fact, the localization and biological activity of factors such as fibroblast growth factors (FGFs) depend strongly on the presence and composition of the ECM. Cells that express high‐affinity receptors but lack surface HS do not respond to these ligands (94). HSPG can stabilize FGF, protect it from proteolysis and serve as a co‐receptor influencing its interaction with cell surface high‐affinity receptors (Figure 2) 77, 91. Specific structural aspects, including sulfonation of HSPG, are required for the proper interaction of HSPG with FGF. Thus, the regulation of GAG biosynthesis and modification greatly influences the functions of HSPG. The differential binding characteristics of specific HS structures can potentiate or inhibit the biological activity of FGFs (8). Other growth factor families, such as vascular endothelial growth factor (VEGF) and platelet‐derived growth factor (PDGF), are known to bind to heparin/HS, which can modulate their biological activities 66, 72. The interaction between chondroitin sulfate proteoglycans (CSPGs) and growth factors has attracted less attention; however, recent evidence supports the ability of CS chains to bind growth factors and to modulate their cell signaling. Phosphacan binds basic fibroblast growth factor (bFGF) and potentiates its mitogenic activity almost as effectively as heparin (59). Oversulfated CS type E (CS‐E) was shown to bind midkine, heparin‐binding growth‐associated molecule (HB‐GAM), bFGF, heparin‐binding epidermal growth factor, FGF10, FGF16 and FGF18 (28). Appican, the proteoglycan form of amyloid precursor protein, contains oversulfated CS‐E and is capable of interacting with midkine and HB‐GAM; however, not much is known about its effect on their biological activities (88).
Figure 2.
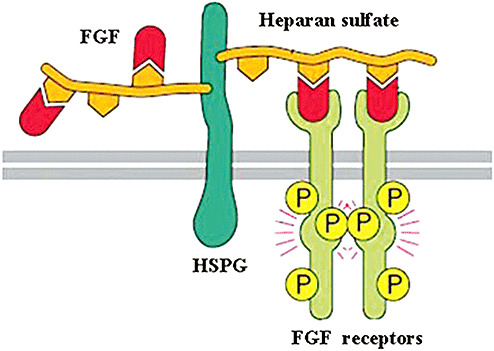
Interaction of heparan sulfate proteoglycans (HSPGs) and fibroblast growth factor (FGF) signaling.
Another class of soluble proteins with high affinity for HSPG is the secreted HB‐GAMs pleiotrophin and midkine. Pleiotrophin interacts with N‐syndecan and the CSPG phosphacan. N‐syndecan functions as receptor/co‐receptor in HB‐GAM‐induced neurite outgrowth in brain neurons and HS and HB‐GAM also cooperates in synaptic induction. These proteins play important roles in the regulation of mitogenesis, angiogenesis, and neurite and glial process outgrowth (73).
ECM modulators in the CNS
The two major systems that modify the adult brain matrix are the serine protease tissue plasminogen activators (tPAs) and matrix metalloproteinases (MMPs). The MMPs are synthesized as proenzymes with removal of a propeptide as a prerequisite for activation. They are synchronously expressed with tissue inhibitors of metalloproteinases (TIMPs), which form tight complexes with the enzyme. Activation of MMPs can occur through plasmin generation by tPAs, thus functionally linking the two proteolytic systems. The tPAs are also secreted in a precursor form as single‐chain polypeptides and in vitro are converted by plasmin to active double‐chain forms. Regulation of tPA activity in the nervous system can occur at multiple levels: transcription, translation, secretion and inhibition by anti‐proteases. Despite high levels of tPA mRNA in the hippocampal CA1 region, protein and enzymatic assays show no evidence of tPA, suggesting stringent translational control (39). Synaptic signaling can release this control and can lead to tPA secretion. Inhibition of tPA activity is under tight control by a variety of serpin family anti‐proteases (eg, protease‐nexin 1, neuroserpin and plasminogen activator inhibitor 1) (39). Some tPA effects are plasminogen independent (eg, cleavage of NR1 subunit of the NMDA receptor (NMDAR) enhancement of N‐methyl‐D‐aspartic acid (NMDA)‐mediated intracellular calcium levels or binding of the low‐density lipoprotein (LDL) receptor‐related protein) and promote upregulation of MMP‐9, degradation of the ECM resulting in synaptic plasticity or neuronal degeneration (55). The mechanisms of activation of tPAs and MMPs in vivo are still poorly understood. The detailed molecular biology and biochemistry of these enzyme systems have been extensively documented elsewhere 27, 34, 52, 63, 95, 96. For purposes of this review, these proteolytic and GAG‐degrading enzymes, such as heparanase and chondroitinase, are key components in modulating cellular interactions with ECM (eg, proteolytic cleavage of core proteins or degradation of GAG side chains). MMPs are generally secreted; however, they can be localized to cell surfaces by binding to cell adhesion molecules or cell surface proteoglycans and integrins or through transmembrane domains of membrane‐type MMPs. Another subgroup of metalloproteases is the A disintegrin and metalloproteinases (ADAMs) and A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS). ADAMs are membrane‐anchored enzymes that possess both proteolytic and adhesive activities. They are involved in shedding of cytokines, receptors and growth factors as well as adhesion to integrins. Tumor necrosis factor alpha (TNFα)‐converting enzyme or ADAM17 is the most characterized sheddase releasing TNFα from the cell surface (16). ADAMTSs are secreted enzymes that are involved in the cleavage of ECM proteoglycans and in collagen processing (71). MMP secretion has been shown to contribute to the migration ability of monocytes as well as astrocyte motility 56, 62, 67, 69, 74, 82. MMPs can also be secreted by neurons, oligodendrocytes, microglia and endothelial cells (52). The MMP inhibitors (TIMPs) are expressed in neurons and astrocytes in the adult CNS (25). tPA is expressed both in neurons and microglial cells and is highly expressed in regions involved in learning and memory. The activity of tPA in neural tissue is correlated with neurite outgrowth, regeneration and migration, suggesting that it might be involved in neuronal plasticity (58).
ECM and neuronal functionality
Several studies have shown that modulations of the neuronal matrix affect synapse morphology and function [eg, interfering with induction or maintenance of long‐term potentiation (LTP)] (Table 2) 15, 34, 35. Antibodies against neuronal cell adhesion molecules (NCAMs) or removal of the associated carbohydrate polysialic acid prevents LTP, whereas bFGF enhances LTP in the dentate gyrus. Stabilization of LTP has been suppressed in an early phase of LTP by recombinant HB‐GAM, which is known to bind to HSPGs in hippocampal slice cultures. Soluble syndecan‐3's potential to modulate cell matrix associated with synaptic plasticity has been demonstrated by its suppression of tetanus‐induced LTP after application to the CA1 area of hippocampal slice cultures. Synaptically localized syndecans may be involved in binding to certain growth factors, thereby potentiating their action on synaptic receptor kinases, leading to induction of cytoskeletal changes. The late temporal expression of syndecan‐2 within developing synapses suggests a role more closely associated with dendritic spine maturation rather than with early spine formation. This proposed role is supported by experimental evidence that syndecan‐2 overexpression in hippocampal neurons accelerates spine maturation without affecting the total number of synapses (8).
Table 2.
Neuronal functions modulated by extracellular matrix (ECM). Abbreviations: PNN = perineuronal net; MMP = matrix metalloproteinase; tPA = tissue plasminogen activator; LTP = long‐term potentiation; NCAM = neuronal cell adhesion molecule; HB‐GAM = heparin‐binding growth‐associated molecule; TIMP = tissue inhibitors of metalloproteinase; RPTP = Receptor protein tyrosine phosphatase beta.
| Neuronal function | ECM modulators | Reference |
|---|---|---|
| Neurogenesis | Glypican, phosphacan, MMPs | 8, 27 |
| Migration | Versican, receptor protein tyrosine phosphatase beta (RPTP‐β)/phosphacan | (8) |
| Neurite outgrowth and inhibition | Glypican‐1/3, N‐syndecan, neurocan, phosphacan, brevican, versican, HB‐GAM | 8, 27 |
| Survival | PNNs, laminin, TIMP‐1, MMPs, tPA | 1, 7, 17, 25, 46, 47, 52, 53, 54, 63, 68, 69, 79, 80, 92, 97 |
| LTP and synaptic function | NCAM, syndecans, MMPs, TIMPs, reelin, HB‐GAM, tenascin‐R, tenascin‐C, brevican, neurocan, laminin, tPA | 7, 8, 26, 52, 60, 61, 85, 88, 95 |
Several other ECM molecules have been identified as crucial modulators of the formation, maturation and plasticity of synapses [eg, pentraxin NP1, Narp (NP2), laminin, thrombospondin‐1 and −2, and reelin]. HSPGs directly interact with AMPA receptors to affect their activity and possibly to stimulate their aggregation. Injection of heparin or removal of HS by heparinase inhibits LTP, showing that LTP is dependent on endogenous HSs. HSPGs in association with polysialylated forms of NCAM regulate synaptogenesis and LTP‐induced formation of perforated synapses (32). Enzymatic removal of CS by chondroitinase has also shown reduction of LTP similar to mutant mice deficient in tenascin‐R 19, 76.
CHANGES IN THE ECM IN NEURODEGENERATIVE DISEASES AND IN ANIMAL MODELS
Brain ECM is organized not only to provide physical support but also to support homeostatic functions crucial to survival of terminally differentiated neurons that are believed to be incapable of regeneration. In addition to controlling a plethora of cell signaling molecules, the ECM can physically buffer ions and neurotransmitters associated with synaptic activity. Several studies reviewed below have shown changes in the ECM associated with neuropathological states, but few studies have demonstrated a mechanistic connection between ECM changes and neuronal death.
Ischemic stroke
In the penumbra of an infarct, gliosis delineates regions of apoptotic cell death. Within the region of glial scar, stroke induces the growth‐inhibitory CSPGs, such as aggrecan, phosphacan and versican, the semaphorin IIIa receptor neuropilin, and myelin‐associated glycoprotein. Stroke also induces growth‐promoting proteins in this region of glial scar, including GAP43, cytoskeleton‐associated protein 23, myristoylated alanine‐rich protein kinase C substrate and small proline repeat rich protein 1. Thus, the glial scar of a stroke is a narrow region that closely borders the infarct and contains increased expression of both growth‐promoting and growth‐inhibitory molecules. The peri‐infarct region that lies just outside of the glial scar has reduced levels of growth‐inhibitory molecules. This growth‐permissive zone corresponds to the region of post‐stroke axonal sprouting (21).
Al'Qteishat et al (2) performed a thorough immunohistochemical analysis of post‐mortem tissues from stroke patients and found enhanced deposition of HA around blood vessels and, paradoxically, intracellularly and in the nuclei of peri‐infarct neurons. Western blot and immunohistochemistry showed upregulation of HA synthases 1 and 2 and hyaluronidases 1 and 2 in inflammatory cells in stroke and peri‐infarct regions. Hyaluronidase 1 was upregulated in microvessels and intracellularly in neurons. The expression of HA‐binding protein CD44 and tumor necrosis factor‐stimulated gene 6 was mainly increased in infiltrating mononuclear cells from inflammatory regions. The results demonstrated a significant change in the enzymes responsible for HA synthesis and degradation together with upregulation of HA receptors. This may reflect tissue remodeling after stroke and may be partially responsible for increased angiogenesis and neuronal migration (2).
A commonly used animal model of human stroke is permanent middle cerebral artery occlusion. This model has demonstrated that, in addition to central neuronal loss, the peri‐infarct regions have shown loss of PNN marker Wisteria floribunda agglutinin (WFA) staining and, to a lesser degree, loss of CSPG core protein staining (41). Beginning at 24 h, osteopontin protein was expressed by endogenous microglia and by infiltrating macrophages at 48 h in the infarct. By day 5, osteopontin mRNA was restricted to the infarct region. The osteopontin receptor integrin αvβ3 was expressed in peri‐infarct astrocytes at 5 and 15 days. It has been hypothesized that the osteopontin is deposited in the matrix and serves as a barrier for the glial scar formation by astrocytes (33).
In summary, beyond the extensive destruction of ischemic tissue, in the penumbra of infarcts, abundant alterations occur in the ECM. Some of these alterations contribute to growth‐inhibiting glial scars, while others are associated with neuronal growth‐promoting regions. In addition to endogenous neuroglial cell modulation, infiltrating inflammatory cells take part in ECM remodeling.
Kainic acid (KA) excitotoxicity
KA‐induced seizures in rodents represent a well‐established animal model for human temporal lobe epilepsy (11). KA produces neuronal excitation resulting in seizures within hours following intraperitoneal injection. At 2–3 days after treatment, neurodegeneration of both necrotic and apoptotic character can be observed in the limbic system, in particular the hippocampus and amygdala, in addition to other brain regions (47). ECM changes that have been documented in this model include reduction in laminin and phosphacan, and increases in neurocan and ADAMTS‐cleaved brevican.
Laminin, an important constituent of the ECM, has been shown to play a key role in KA excitotoxicity (23). In wild‐type mice, 2 h after injection of KA, neurons had normal morphology, but laminin had almost completely disappeared in CA1 and CA3. Two days after injection, neurons in CA1, CA2 and CA3 had degenerated. These results indicate that laminin degradation precedes neuronal loss after KA treatment and occurs in exactly the same hippocampal regions that eventually experience neuronal degeneration. The CA3 region that showed more laminin expression was more resistant to excitotoxin‐induced neuronal cell death. This correlation between high laminin expression and neuronal resistance to excitotoxin further implicates laminin as an important factor in maintaining cellular viability. While loss of laminin attachment was deleterious in combination with kainate injection, it was not by itself significantly toxic. Infusion of anti‐laminin antibodies was toxic only in conjunction with KA injection (23).
KA‐induced convulsions caused prolonged changes in the CSPGs neurocan and phosphacan. Okamoto et al (68) showed increased neurocan staining that co‐localized with glial fibrillary‐associated protein staining. Phosphacan staining was reduced in areas of pyramidal cell loss in the hippocampus and around parvalbumin‐positive neurons. Interestingly, compared to other classes of neurons undergoing degeneration, the parvalbumin‐positive neurons were relatively conserved (68).
Mediators of ECM destruction in the KA model have been studied in mice deficient in tPA or plasminogen. Mice with either of these enzymes knocked out were resistant to neuronal destruction induced by excitotoxins. Because infusion of excess tPA into the wild‐type hippocampus does not kill neurons, excitotoxin treatment must cause additional cellular changes beyond ECM alterations to induce neurodegeneration.
Using the KA model, Yuan et al (98) assessed co‐localization of ADAMTS or its activity in regions of synaptic loss or neuronal death that could indicate a functional role for the protease in degeneration. They demonstrated that increased ADAMTS cleavage of brevican was found 24 h after KA administration and was sustained in several susceptible brain regions. An increase in the mRNA of ADAMTS‐1/4 in the seized animals was accompanied by elevation of the ADAMTS‐cleaved brevican (98).
In summary, many of the neurodegenerative changes associated with KA excitotoxicity are linked to ECM alterations. Degeneration of the ECM does not directly cause neurodegeneration but rather makes neurons susceptible to activation‐associated cell death. Therefore, supporting neuronal‐ECM interactions appears to be a reasonable approach to blocking neurodegeneration in this model.
Multiple sclerosis (MS)
MS is a demyelinating disease associated with inflammation, gliosis and variable axon loss. While most of the disease process is focused in the white matter, recently, gray matter lesions have been demonstrated (13). In a recent review, van Horssen summarized the ECM changes in MS (45). Classical MS lesions showed accumulation of laminin, HA and MMP‐19. The active MS lesions showed accumulation of laminin in the parenchyma, which was normally associated with the basement membrane of endothelial cells. This laminin accumulation was not seen in inactive MS lesions.
CD68‐positive macrophages in the vicinity of the lesion expressed high levels of transforming growth factor β1 (TGFβ1), which is known to modulate ECM production (43). MMP‐19 was upregulated in MS lesions and was expressed by microglia/macrophages (44). As opposed to tenascin‐R and tenascin‐C that accumulate in chronic MS lesions, there was a striking loss of both tenascin‐R and tenascin‐C in the edge of acute lesions where the macrophage density is greatest. This loss extended into adjacent, histologically normal white matter (40). In chronic demyelinated lesions, a high‐molecular‐weight HA synthesized by astrocytes accumulated. This was thought to inhibit remyelination as oligodendrocyte progenitor cells do not mature into myelin‐forming cells in demyelinating lesions where high‐molecular‐weight HA is present (6). The inflammatory response of MS is associated with modulation of the ECM that is incommensurate with regeneration. Manipulation of this response could be key to anatomical and functional recovery.
Human immunodeficiency virus dementia (HIVD)
Human immunodeficiency virus (HIV)‐1‐associated dementia (AIDS dementia complex) is marked by deficits in motor control, cognition and behavior (64). Achim and Wiley (1) reported that at autopsy, AIDS patients who had clinical dementia during terminal stages of their disease demonstrated human immunodeficiency virus encephalitis (HIVE). The constellation of neuropathological changes associated with HIVE includes activated and infected macrophages, astrocytosis and neurodegeneration (18). As neurons are not infected by HIV, the etiology of neurodegeneration must be indirect. Current leading theories suggest that neurodegeneration results from neurotoxic viral or inflammatory factors. The ability of activated monocytes and macrophages to penetrate the ECM and to migrate to target tissues is dependent on ECM‐degrading proteins like MMPs and heparanase. This degradation of ECM components could result in disintegration of the structure that physically and physiologically supports neurons, and thus could lead to degeneration.
Belichenko et alstudied the ECM in HIVE cases and found that each of the nine cases with HIVE was completely devoid of labeled ECM components, while only 44% of HIV‐infected non‐encephalitic cases had ECM loss (9). Although PNNs were lost in HIVE, there was no loss of parvalbumin‐, calbindin‐ and calretinin‐positive neurons that are frequently surrounded by these nets. ECM destruction was not confined to microglial nodules, suggesting that if macrophages were the source of released ECM‐degrading enzymes, these would need to diffuse throughout the brain parenchyma. Alternatively, macrophages could activate astrocytes to secrete enzymes responsible for the metabolism of the ECM. Staining for Vicia villosa agglutinin, a lectin that binds to N‐acetyl‐galactoseamine residues on CS or DS, was lost before the core protein staining, suggesting that the degradation of the GAG chains occurs before the actual degradation of the core protein (9). A more indirect indication of GAG chain degradation in lentiviral encephalitis is the presence of HA in the cerebrospinal fluid (CSF) (49).
Support of these findings of ECM degradation in AIDS comes from the simian immunodeficiency virus (SIV) encephalitis model in rhesus and pigtailed macaques. Disruption of brain ECM was present in SIV cases without encephalitis, but was more severe in cases with encephalitis (57). This suggests that ECM changes occur prior to parenchymal macrophage infiltration. ECM changes in SIV encephalitis (SIVE) occur concurrently with reduced staining for the pre‐ and postsynaptic marker synaptophysin and microtubule‐associated protein 2 (MAP2). One possible source of ECM degradation activity is the induced microglial/macrophage expression of ADAMTS‐1 and ADAMTS‐4 (57) and MMP‐2 and MMP‐7, as observed by their elevation in the CSF of patients with HIVD (24). Other sources of ECM‐degrading enzymes include astrocytes or lymphocytes as shown in the coronavirus‐induced encephalitis in mice, where increased mRNA levels of MMP‐3 and MMP‐12 as well as the inhibitor TIMP‐1 were found. In this model, MMP‐3 expression was restricted to astrocytes and TIMP‐1 expression was localized with CD4‐positive cells. However, when T‐cells were removed from this model using irradiation–immunosuppression, mice that had high viral load in the CNS showed increased mRNA levels of MMP‐3, MMP‐12 and TIMP‐1, suggesting that viral replication directly triggers upregulation of these proteins (100).
In summary, lentiviral infection is associated with neurodegeneration and disruption of CNS ECM. While SIV systemic infection may elicit brain ECM disruption, a more pronounced disruption is associated with SIVE. As observed in other viral encephalitides, this ECM disruption occurs independently of local lymphocyte response and appears tightly associated with macrophage activation and infiltration.
Alzheimer's disease (AD)
The pathological hallmark of AD is the deposition of β‐amyloid senile plaques (SPs), cerebral amyloid angiopathy and neurofibrillary tangles (NFTs), leading to synaptic loss. However, in recent years, evidence of changes in cell surface proteoglycans and/or ECM components has appeared as well. HSPGs such as glypican, syndecans 1–3 and agrin were found associated with SPs and NFTs. Glypican‐1 co‐localized in cerebral amyloid angiopathy (CAA) in AD and hereditary cerebral hemorrhage with amyloidosis of the Dutch type (90). Syndecan‐2 was frequently associated with vascular Aβ in AD but not with vascular Aβ deposits in hereditary cerebral hemorrhage with amyloidosis of the Dutch type (42).
Snow et al (84) demonstrated that HS staining was found in primitive plaques both in AD and in Down's syndrome, suggesting that their accumulation takes place during early stages of plaque development. In non‐affected areas, HSPG was associated with blood vessels within the brain parenchyma and was lost in affected areas, suggesting a change in basement membrane structure. Ultrastructural studies specifically localized the HSPGs to amyloid fibrils present in SPs and CAA. The close association between amyloid fibrils and HSPGs suggests that HSPGs may have an important role in amyloidosis 83, 84.
In addition to HSPGs, CSPGs and DS proteoglycans were also found to be associated with SPs. The basic pattern of CSPG immunoreactivity was similar in controls and in AD brains; however, CS and undersulfated chondroitin were found intracellularly with NFTs in dystrophic neurites and in the area around the plaque. Decorin was primarily localized to the periphery of the spherical plaques and to edges of amyloid fibril bundles within the plaque periphery as well as filaments within NFTs 29, 85.
Baig et al found loss of two‐thirds of the neurons with intact PNNs in AD as compared to controls; however, the density of parvalbumin‐positive neurons did not differ (7). Bruckner et al studied the spatiotemporal patterns of damaged neurons and PNNs and found that cortical neurons associated with PNNs were largely spared from neurofibrillary changes in AD even in severely damaged regions. Phosphorylated tau‐positive staining was not found in pyramidal or non‐pyramidal neurons surrounded by PNNs in six out of seven cases tested (17).
Aberrant ECM components are associated with the pathological hallmarks of AD, plaques and tangles. The pattern and abundance of HSPGs suggest an early association with neurodegeneration. It is an intriguing possibility that certain ECM structures may protect select neuronal elements from degeneration.
POSSIBLE MECHANISMS OF ECM‐RELATED NEURODEGENERATION
Proteolytic‐induced ECM changes
Induction of MMPs and their inhibitors is evident in neurodegenerative states
Degradation of the ECM associated with an imbalance of the MMP/TIMP ratio has been correlated with the invasive potential of brain tumor cells (70) and with the histopathogenesis of inflammatory‐related diseases like rheumatoid arthritis (97) or MS (5). In addition, abundance of evidence exists linking changes in MMPs and TIMPs to human neurodegenerative diseases and animal models.
Evidence for MMP induction
Upregulation of MMPs was found in a variety of neurodegenerative diseases. MMP‐2 and MMP‐7 were elevated in the CSF of patients with HIVD (24). MMP‐9 expression was significantly increased in both the frontal cortex and substantia nigra (SN) of progressive supranuclear palsy, whereas MMP‐1 levels were increased in the SN (54). MMP‐9 transcription and activation were induced after KA administration in the dentate gyrus (86). Inflammatory cells could produce or induce other cells to express MMPs. In vitro, HIV‐infected macrophages secreted MMP‐2 (99). MMP‐3 and MMP‐9 expressions were increased in astrocytes exposed to activated T‐lymphocytes (38).
Evidence for alterations of ADAMTS in neurodegenerative diseases is more limited
ADAMTSs lack transmembrane domains and contain carboxy thrombospondin motifs, which are thought to be responsible for binding to ECM GAGs. In the SIVE model, microglia/macrophages express ADAMTS‐1 and ADAMTS‐4 (57). In vitro, treatment of astrocyte culture with Aβ induced the expression of ADAMTS‐4 mRNA, which suggests that ECM degradation occurs in AD brain in regions of deposition (79).
Evidence for TIMP induction
MMP activity is under strict control by their inhibitors (TIMPs), with both molecules being co‐expressed. Therefore, induction of TIMPs can be paradoxically associated with increased MMP activity. TIMP‐1 was induced after KA administration in adult forebrain astrocytes surrounding experimental allergic encephalomyelitis (EAE) lesions 26, 75. TIMP‐1 knockout mice were found resistant to KA excitotoxicity and did not undergo the typical mossy fiber sprouting observed in wild‐type mice (46). Elevated TIMP‐1 has been observed in the CSF of people with Parkinson's disease, AD, Huntington's disease and amyotrophic lateral sclerosis, and elevated TIMP‐2 has been observed in the CSF of people with AD and Huntington's disease 53, 54.
Altered MMP activity induces changes in plasticity
Bilousova et al (12) showed that MMP‐7 disrupts dendritic spines in hippocampal cultures. These changes were accompanied by a dramatic redistribution of F‐actin. MMP‐7‐induced changes were associated with NMDAR activation as MK‐801 was able to block those effects. Thus, MMP‐7 could induce mild synaptic reorganization that is associated with excitotoxicity (12).
In summary, changes in proteolytic activity in several models of neurodegeneration can clearly influence ECM structure and function. MMP‐induced changes can induce changes in plasticity. Although TIMPs seem to be elevated in response to MMP activity, they also serve biological activities that are distinct from MMP inhibition. Therefore, it is not clear how the balance between MMPs and TIMPs is modulating ECM integrity and its exact role in neurodegeneration. The complexity of the sheer number of potential proteases and their inhibitors makes elucidating their mechanism of modulating ECM a daunting task. Added to the complex transcriptional and translational control of different neuroglial and immune cells, deciphering mechanisms of ECM modulation in various disease states remains an important challenge.
tPA activation is associated with laminin degradation, microglial activation and neurodegeneration
After exposure to kainate, neurons are massively depolarized, leading to a pathological influx of calcium. The depolarization results in two phases of tPA induction: release of presynthesized tPA and stimulation of tPA gene transcription. Increased tPA activates CNS plasminogen to plasmin, which can degrade laminin and other components of the ECM. tPA‐deficient mice exhibit less microglial activation in reaction to neuronal injury (87). In contrast, the microglial response of plasminogen‐deficient mice is comparable to that of wild‐type mice, suggesting a tPA‐mediated (plasminogen‐independent) pathway for activation of microglia. Infusion of inhibitors of tPA/plasmin proteolytic cascade into the hippocampus protects neurons against excitotoxic injury. Mice overexpressing neuroserpin, an inhibitor of tPA, show a decreased infarct size; however, infusion of tPA alone does not cause neuronal death.
Thus, tPA proteolytic activity plays a role in ECM remodeling but by itself is not sufficient to cause microglial activation and/or neuronal death, suggesting that other factors are involved. The generation of plasmin might be favorable or detrimental to the survival of neurons, depending on the context. In addition, plasmin has been shown to degrade amyloid aggregates, implying that the tPA–plasmin system might be involved in abnormal accumulation of amyloid plaques in the brains of patients with AD 55, 58.
Production of toxic proteolytic cleavage products
In addition to the above‐mentioned ECM changes mediated by MMPs and tPA, one study showed an additional mechanism that could contribute to neurodegeneration. HIV‐infected macrophages secreted MMP‐2, which metabolizes the chemokine stromal cell‐derived factor (SDF)‐1 to the cleaved form. This cleaved form is more toxic than the whole protein (99). Dose‐dependent neuronal death was observed in human cholinergic cell line incubated with increasing concentration of MMP‐2 but not with a mutant MMP‐2. Treatment with neutralizing antibodies against MMP‐2 or inhibitor of MMP‐2 reduced neuronal death. The cleaved SDF‐1 causes apoptosis and dose‐dependent neuronal death. Mice that were stereotactically injected with cleaved SDF‐1 showed more microglial reactivity and astrocytosis than animals injected with the whole SDF‐1 (99). Thus, in addition to ECM remodeling, protelolytic activity can induce protein gain of toxicity.
Induction of chemotaxis and neuroinflammtory response
Chemokines and their receptors play a key role in directing the migration of mononuclear cells into the CNS under normal physiological conditions and in pathological states. Plasmin or MMP‐mediated proteolysis of chemokines is one way by which these proteolytic enzymes can influence leukocyte trafficking. MMP proteolysis can activate or inhibit the biological functions of chemokines (89). Apoptotic cells trigger the recruitment of phagocytic cells through release of chemoattractants. It has been shown that apoptotic neurons release the active form of MMP‐3 that elicits microglial secretion of TNFα(48). The chemokine fractalkine is cleaved by MMPs after excitotoxic injury. Although inhibition of MMPs by batimastat inhibits fractalkine cleavage, it does not confer neuroprotection. Release of fractalkine from neurons plays a pivotal role in attracting microglia and other cells derived from the monocytic lineage (22). Mice genetically deficient for the chemokine monocyte chemotactic protein‐1 (MCP‐1) display decreased microglial recruitment and resist excitotoxic neurodegeneration. MCP‐1 undergoes proteolytic processing mediated by plasmin, enhancing the potency of MCP‐1 in in vitro migration assays. Infusion of the cleaved form of MCP‐1 into the CNS restores microglial recruitment and excitotoxicity in plasminogen‐deficient mice (81). In summary, plasmin‐ or MMP‐mediated proteolysis of chemokines can increase their potency in inducing microglial activation and trafficking of immune cells into the CNS.
Expression of regeneration‐inhibitory CSPGs
In the trauma model of unilateral knife lesion, increased staining of neurocan was observed. In vitro, neurocan was shown to inhibit neurite outgrowth and being deposited on the substratum around astrocytes but not on the cell surface. These findings support the role of astrocytes within the region of glial scar as a source of growth‐inhibitory CSPGs (4). Unsulfated chondroitin, chondroitin 4‐sulfate and chondroitin 6‐sulfate were found associated with SPs, NFTs and dystrophic neurites (29). These results suggest that CSPG could contribute or be responsible for the regression of neurites around SPs in AD. Abnormal CSPG accumulation could disrupt cell adhesion or growth factor utilization (29). Deposition of CSPG has been found at the periphery of SP cores. It has been hypothesized that neurotoxicity is not simply the consequence of β‐amyloid aggregation. Implantation of nitrocellulose impregnated with β‐amyloid in the cortex of neonatal rats caused reactive gliosis in addition to deposition of CSPGs. Secondary glial cell reaction to the insoluble molecule and CSPGs, along with other proteoglycans, may be an additional link in the chain of events leading to neuronal dystrophy and possibly neuronal death (20).
Thus, neurite growth‐inhibitory CSPGs are induced in neuropathological states like SPs or glial scar regions and, therefore, could play a role in inhibiting regeneration of damaged neurons.
Associated HSPG deposition and protein aggregation
The HSPGs agrin, glypican and syndecans 1–3 were found to be incorporated into SPs (90). HSPGs normally associated with basement membrane of blood vessels were lost in affected areas in AD but were present in congophilic angiopathy (CA) in AD. HSPGs were also demonstrated in “primitive plaques” (defined as those having little or no amyloid core), suggesting that their accumulation takes place during early stages of plaque development. HSPGs were localized to astrocytes and neurons in close proximity of SPs and CA, suggesting that both may be involved in the deposition of HSPGs at these sites and may, therefore, play a role in plaque development. Ultrastructural studies specifically localized the HSPGs to amyloid fibrils present in SPs and CA. This close association between amyloid fibrils and HSPGs suggests that HSPGs have an important role in amyloidosis (83). Other support for this idea comes from the work of Liu et al, who found that agrin co‐localizes with α‐synuclein in neuronal Lewy bodies in the SN of Parkinson's disease brain. Agrin was found to accelerate formation of protofibrils by α‐synuclein and to decrease the half‐time of fibril formation (51). In summary, HSPGs have been found to be associated with protein aggregation and deposition, and it has been hypothesized that changes in the metabolism of the ECM could contribute to amyloid neurodegenerative diseases.
Loss of protective PNN proteins
PNNs include lecticans, HA, tenascin‐C and tenascin‐R. Cortical neurons that were associated with PNNs in AD cases were largely spared from neurofibrillary changes. Also, PNNs were completely absent from regions in which the proteinase‐resistant prion protein of Creutzfeldt–Jakob disease (PrPCJD) was deposited, but parvalbumin‐positive cells themselves were lost only in those areas that were heavily infiltrated with PrPCJD. Destruction of the PNN thus appears to antecede the perineuronal deposition of PrPCJD. Loss of the PNN around parvalbumin‐positive neurons may lead to the retraction of synapses from their surface and finally to their death (10). Recent work by Franklin et al suggests loss of PNNs in murine prion disease soon after microglial activation that coincides with reduction in synaptic plasticity (36).
Angelov et al showed that downregulation of tenascin‐R after facial nerve axotomy is associated with loss of motoneuron protection and subsequent neurodegeneration. Tenascin‐R was found to be anti‐adhesive for activated microglia (3).
Mice deficient for tenascin‐R displayed alterations of the ECM. They showed less‐developed PNNs and WFA staining that does not extend well to the dendritic shaft and decreased axonal conduction velocities in the CNS (92).
In the rat trimethylin intoxication model, cell damage was seen in the pyramidal cell layer in regions CA1–4 and in the dentate gyrus. PNN immunoreactivity around CA2 pyramidal cells was reduced but independently of the presence of activated microglia. PNN‐associated neurons survived in the vicinity of damaged pyramidal cells. The results suggest that the type of the PNNs and the type of neurons could influence the fate of neurons during neurodegenerative diseases (80).
Treatment of Aβ1‐42 showed significant neurotoxicity on PNN‐free cortical neurons; however, it did not reveal neurotoxicity on PNN‐associated neurons. Aβ1‐42 was able to kill PNN‐associated neurons after treatment with chondroitinase. Treatment with glutamate killed both types of neurons. The results suggest that the neuroprotective actions of PNNs against Aβ are possibly caused by the inhibition of direct interaction of Aβ with neuronal membrane to attenuate the lipid peroxidation and reactive oxygen damage (60).
Neurons ensheathed by PNNs in the human cerebral cortex are less frequently affected by lipofuscin accumulation than neurons without PNNs, both in normal‐aged brain and AD. To some extent, clear lipofuscin deposits can also be detected in neurons with only delicate PNN CSPG immunoreactivity. Because lipofuscin is an intralysosomal pigment composed of cross‐linked proteins and lipids generated by iron‐catalyzed oxidative processes, the results suggest a neuroprotective function of PNNs against oxidative stress, and thus are potentially involved in neurodegeneration (61). Thus, PNNs are lost in neurodegenerative states, but neurons that preserve the PNNs seem to be protected from damage and/or loss of function.
ECM‐INDUCED INTRACELLULAR SIGNALING
Support for the importance of ECM‐induced intracellular signaling in maintaining differentiated epithelial functions comes from literature as diverse as oncology to inflammation. ECM–cell interaction can regulate gene expression through ECM‐response elements in different models. Interactions between ECM and mammary epithelial cells are critical for mammary gland homeostasis and apoptotic signaling; mammary epithelial cells organize into acinar three‐dimensional (3‐D) structures and secrete milk proteins in the presence of ECM and in response to lactogenic hormones. The laminin‐induced expression of β‐casein involves activation of ECM‐response elements in the promoter of the β‐casein gene while at the same time, laminin downregulates transforming growth factor β (TGFβ) through the ECM‐response element in the promoter region (65).
Interferon regulatory factor‐1 (IRF‐1) is a transcriptional regulator that promotes apoptosis during mammary gland involution and p53‐independent apoptosis as well as viral infections. It has been shown that reconstituted extracellular matrix (rECM) (growth factor depleted Matrigel™ (BD Biosciences, Franklin Lakes, NJ, USA)) promotes apoptosis by recruitment of STAT1 and cAMP response element‐binding protein to the γ‐activating sequence element of the IRF‐1 promoter and the subsequent induction of IRF‐1 and caspase‐1 and ‐3 activation (14). In addition, the cAMP response element‐binding protein regulates mammary epithelial cell proliferation and apoptosis through binding the LAMA3A promoter and transcriptional activation of laminin‐5 (30).
DISCUSSION
Neurodegenerative states can be divided into two groups based on the major ECM changes (Figure 3). Age‐related progressive neurodegenerative diseases, such as AD, are characterized by changes in the expression of proteoglycans and association with protein aggregation and amyloidosis. It is not known whether protein aggregation or deposition is a primary event leading to those secondary events of proteoglycan expression or whether the two processes are interrelated. Alternatively, changes in proteoglycans are the primary event that triggers protein aggregation. These changes are accompanied by a secondary microglial activation. The role and timing of microglial activation relative to Aβ deposition are controversial. The extent of microglial activation in Aβ deposition has been hypothesized to be limited to the transition from diffuse to mature plaques, when amounts of Aβ deposits and the degree of neuritic changes increase (78). As of now, no clear correlation between ECM breakdown and neuronal death is apparent. From the work of Bruckner et al, it seems that neurons surrounded by PNNs are spared from neurodegenerative changes in AD (17). Breakdown of the PNNs mostly around parvalbumin‐positive neurons without the actual loss of the neurons suggests a change in the neuronal environment that could affect their function or susceptibility to further insult.
Figure 3.
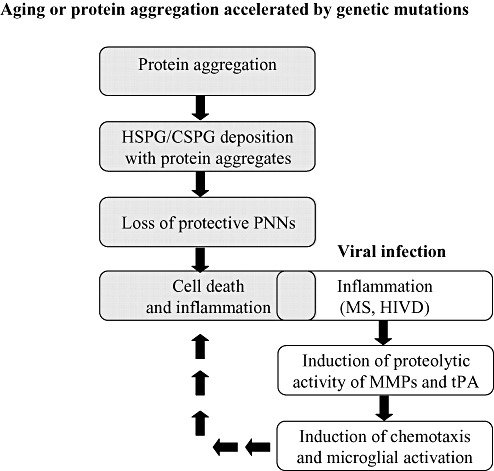
Possible mechanisms of extracellular matrix (ECM)‐related neurodegeneration. Aging or protein aggregation accelerated by genetic mutation can be associated with ECM alterations that would result to co‐deposition of ECM components [eg, heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate proteoglycans (CSPGs)]. Those ECM alterations can result in loss of protective perineuronal nets (PNNs) and increased susceptibility to cell death. Dying neurons can induce inflammation, degradation of ECM and induction of a more robust inflammatory response. Alternatively, inflammatory‐induced neurodegeneration can induce ECM degradation through proteolytic activity [eg, matrix metalloproteinase (MMPs) and tissue plasminogen activator (tPA)], induction of chemotaxis and microglial activation. The resulting trafficking of inflammatory cells and secretion of cytokines can induce neuronal death that would feed the vicious cycle. Abbreviations: MS = multiple sclerosis; HIVD = human immunodeficiency virus dementia.
MS, HIVD or KA excitotoxic insults are characterized by an increase in the expression of proteolytic enzymes that could damage ECM integrity and synaptic plasticity. The induction of MMPs and tPA activity can further enhance the inflammatory response by inducing chemotaxis and generation of products that increase microglial reactivity or augment ingress of blood monocytes. Apart from inducing ECM proteolytic activity, not much is known about how the changes in the ECM affect neurons and what are the signaling pathways and response elements involved in neurodegeneration. From other model systems, it is clear that cell–ECM interactions can regulate gene expression at the transcriptional level. Matrix‐stimulated transcription of differentiation‐specific genes has been demonstrated, for example, in hepatocytes and mammary epithelial cells.
Neurons grown on laminin exhibited increased resistance to glutamate‐induced apoptosis compared with neurons grown on polylysine. Treatment of cultures with an antibody against integrin β1 abolished the protective effect of laminin. As neurons maintained on laminin exhibited a sustained activation of the Akt signaling pathway, levels of the anti‐apoptotic protein Bcl‐2 were increased. This could be reversed by treatment with wortmannin (37). Rho GTPases are key transducers of integrin/ECM and growth factor signaling. Rac was found as a critical pro‐survival GTPase in cerebellar granule neurons. Clostridium difficile toxin B, a specific Rho family inhibitor, induced a selective caspase‐mediated degradation of Rac‐1. Both Tox B and dominant negative N17Rac‐1 elicited cerebellar granule neuron apoptosis, characterized by cytochrome C release and activation of caspase‐9 and 3. ToxB stimulated mitochondrial translocation and conformational activation of Bax, c‐Jun activation and induction of the BH3 protein Bim. These results indicate that Rac acts downstream of integrins and growth factors to promote neuronal survival by repressing c‐Jun/Bim‐mediated mitochondrial apoptosis (50).
In summary, neurodegeneration is accompanied by a myriad of ECM changes. Unfortunately, the complexity of potential factors and abundance of various models has not allowed resolution of whether ECM alterations precede or postdate neuronal dysfunction. The studies highlighted in this review support the importance of the ECM in maintaining the differentiated integrity of neuronal function and that disturbance of ECM in disease is causally related to neurodegeneration. Neurons surrounded by PNNs are less affected by these disease processes suggesting that laminin and integrin signaling are important for neuronal survival. From other model systems, it is apparent that cell–ECM interactions are important for the regulation of transcription factors that can promote apoptosis. Elucidation of the interaction between neurons and their surrounding ECM remains a daunting problem for neuropathology.
REFERENCES
- 1. Achim CL, Wiley CA (1996) Inflammation in AIDS and the role of the macrophage in brain pathology. Curr Opin Neurol 9:221–225. [DOI] [PubMed] [Google Scholar]
- 2. Al'Qteishat A, Gaffney J, Krupinski J, Rubio F, West D, Kumar S et al (2006) Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain 129:2158–2176. [DOI] [PubMed] [Google Scholar]
- 3. Angelov DN, Walther M, Streppel M, Guntinas‐Lichius O, Neiss WF, Probstmeier R et al (1998) Tenascin‐R is antiadhesive for activated microglia that induce downregulation of the protein after peripheral nerve injury: a new role in neuronal protection. J Neurosci 18:6218–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE et al (2000) Neurocan is upregulated in injured brain and in cytokine‐treated astrocytes. J Neurosci 20:2427–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avolio C, Ruggieri M, Giuliani F, Liuzzi GM, Leante R, Riccio P et al (2003) Serum MMP‐2 and MMP‐9 are elevated in different multiple sclerosis subtypes. J Neuroimmunol 136:46–53. [DOI] [PubMed] [Google Scholar]
- 6. Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J et al (2005) Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med 11:966–972. [DOI] [PubMed] [Google Scholar]
- 7. Baig S, Wilcock GK, Love S (2005) Loss of perineuronal net N‐acetylgalactosamine in Alzheimer's disease. Acta Neuropathol (Berl) 110:393–401. [DOI] [PubMed] [Google Scholar]
- 8. Bandtlow CE, Zimmermann DR (2000) Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol Rev 80:1267–1290. [DOI] [PubMed] [Google Scholar]
- 9. Belichenko PV, Miklossy J, Celio MR (1997) HIV‐I induced destruction of neocortical extracellular matrix components in AIDS victims. Neurobiol Dis 4:301–310. [DOI] [PubMed] [Google Scholar]
- 10. Belichenko PV, Miklossy J, Belser B, Budka H, Celio MR (1999) Early destruction of the extracellular matrix around parvalbumin‐immunoreactive interneurons in Creutzfeldt‐Jakob disease. Neurobiol Dis 6:269–279. [DOI] [PubMed] [Google Scholar]
- 11. Ben‐Ari Y (1985) Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14:375–403. [DOI] [PubMed] [Google Scholar]
- 12. Bilousova TV, Rusakov DA, Ethell DW, Ethell IM (2006) Matrix metalloproteinase‐7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J Neurochem 97:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bo L, Geurts JJ, Mork SJ, Van Der Valk P (2006) Grey matter pathology in multiple sclerosis. Acta Neurol Scand Suppl 183:48–50. [DOI] [PubMed] [Google Scholar]
- 14. Bowie ML, Troch MM, Delrow J, Dietze EC, Bean GR, Ibarra C et al (2007) Interferon regulatory factor‐1 regulates reconstituted extracellular matrix (rECM)‐mediated apoptosis in human mammary epithelial cells. Oncogene 26:2017–2026. [DOI] [PubMed] [Google Scholar]
- 15. Brakebusch C, Seidenbecher CI, Asztely F, Rauch U, Matthies H, Meyer H et al (2002) Brevican‐deficient mice display impaired hippocampal CA1 long‐term potentiation but show no obvious deficits in learning and memory. Mol Cell Biol 22:7417–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bridges LC, Bowditch RD (2005) ADAM‐integrin interactions: potential integrin regulated ectodomain shedding activity. Curr Pharm Des 11:837–847. [DOI] [PubMed] [Google Scholar]
- 17. Bruckner G, Hausen D, Hartig W, Drlicek M, Arendt T, Brauer K (1999) Cortical areas abundant in extracellular matrix chondroitin sulphate proteoglycans are less affected by cytoskeletal changes in Alzheimer's disease. Neuroscience 92:791–805. [DOI] [PubMed] [Google Scholar]
- 18. Budka H, Costanzi G, Cristina S, Lechi A, Parravicini C, Trabattoni R et al (1987) Brain pathology induced by infection with the human immunodeficiency virus (HIV). A histological, immunocytochemical, and electron microscopical study of 100 autopsy cases. Acta Neuropathol 75:185–198. [DOI] [PubMed] [Google Scholar]
- 19. Bukalo O, Schachner M, Dityatev A (2001) Modification of extracellular matrix by enzymatic removal of chondroitin sulfate and by lack of tenascin‐R differentially affects several forms of synaptic plasticity in the hippocampus. Neuroscience 104:359–369. [DOI] [PubMed] [Google Scholar]
- 20. Canning DR, McKeon RJ, DeWitt DA, Perry G, Wujek JR, Frederickson RC et al (1993) Beta‐amyloid of Alzheimer's disease induces reactive gliosis that inhibits axonal outgrowth. Exp Neurol 124:289–298. [DOI] [PubMed] [Google Scholar]
- 21. Carmichael ST (2006) Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann Neurol 59: 735–742. [DOI] [PubMed] [Google Scholar]
- 22. Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ (2000) Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci 20:RC87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen ZL, Strickland S (1997) Neuronal death in the hippocampus is promoted by plasmin‐catalyzed degradation of laminin. Cell 91:917–925. [DOI] [PubMed] [Google Scholar]
- 24. Conant K, McArthur JC, Griffin DE, Sjulson L, Wahl LM, Irani DN (1999) Cerebrospinal fluid levels of MMP‐2, 7, and 9 are elevated in association with human immunodeficiency virus dementia. Ann Neurol 46:391–398. [DOI] [PubMed] [Google Scholar]
- 25. Crocker SJ, Pagenstecher A, Campbell IL (2004) The TIMPs tango with MMPs and more in the central nervous system. J Neurosci Res 75:1–11. [DOI] [PubMed] [Google Scholar]
- 26. Crocker SJ, Whitmire JK, Frausto RF, Chertboonmuang P, Soloway PD, Whitton JL et al (2006) Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase‐1‐deficient mice. Am J Pathol 169:2104–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cuzner ML, Opdenakker G (1999) Plasminogen activators and matrix metalloproteases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol 94:1–14. [DOI] [PubMed] [Google Scholar]
- 28. Deepa SS, Umehara Y, Higashiyama S, Itoh N, Sugahara K (2002) Specific molecular interactions of oversulfated chondroitin sulfate E with various heparin‐binding growth factors. Implications as a physiological binding partner in the brain and other tissues. J Biol Chem 277:43707–43716. [DOI] [PubMed] [Google Scholar]
- 29. DeWitt DA, Silver J, Canning DR, Perry G (1993) Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer's disease. Exp Neurol 121:149–152. [DOI] [PubMed] [Google Scholar]
- 30. Dietze EC, Bowie ML, Mrozek K, Caldwell LE, Neal C, Marjoram RJ et al (2005) CREB‐binding protein regulates apoptosis and growth of HMECs grown in reconstituted ECM via laminin‐5. J Cell Sci 118:5005–5022. [DOI] [PubMed] [Google Scholar]
- 31. Dityatev A, Schachner M (2003) Extracellular matrix molecules and synaptic plasticity. Nat Rev Neurosci 4:456–468. [DOI] [PubMed] [Google Scholar]
- 32. Dityatev A, Schachner M (2006) The extracellular matrix and synapses. Cell Tissue Res 326:647–654. [DOI] [PubMed] [Google Scholar]
- 33. Ellison JA, Velier JJ, Spera P, Jonak ZL, Wang X, Barone FC et al (1998) Osteopontin and its integrin receptor alpha(v)beta3 are upregulated during formation of the glial scar after focal stroke. Stroke 29:1698–1706; discussion 707. [DOI] [PubMed] [Google Scholar]
- 34. Ethell IM, Ethell DW (2007) Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res 85:2813–2823. [DOI] [PubMed] [Google Scholar]
- 35. Evers MR, Salmen B, Bukalo O, Rollenhagen A, Bosl MR, Morellini F et al (2002) Impairment of L‐type Ca2+ channel‐dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin‐C. J Neurosci 22:7177–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Franklin SL, Love S, Greene JR, Betmouni S (2008) Loss of perineuronal net in ME7 prion disease. J Neuropathol Exp Neurol 67:189–199. [DOI] [PubMed] [Google Scholar]
- 37. Gary DS, Mattson MP (2001) Integrin signaling via the PI3‐kinase‐Akt pathway increases neuronal resistance to glutamate‐induced apoptosis. J Neurochem 76:1485–1496. [DOI] [PubMed] [Google Scholar]
- 38. Giraudon P, Szymocha R, Buart S, Bernard A, Cartier L, Belin MF et al (2000) T lymphocytes activated by persistent viral infection differentially modify the expression of metalloproteinases and their endogenous inhibitors, TIMPs, in human astrocytes: relevance to HTLV‐I‐induced neurological disease. J Immunol 164:2718–2727. [DOI] [PubMed] [Google Scholar]
- 39. Gualandris A, Jones TE, Strickland S, Tsirka SE (1996) Membrane depolarization induces calcium‐dependent secretion of tissue plasminogen activator. J Neurosci 16:2220–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gutowski NJ, Newcombe J, Cuzner ML. (1999) Tenascin‐R and C in multiple sclerosis lesions: relevance to extracellular matrix remodelling. Neuropathol Appl Neurobiol 25:207–214. [DOI] [PubMed] [Google Scholar]
- 41. Hobohm C, Gunther A, Grosche J, Rossner S, Schneider D, Bruckner G (2005) Decomposition and long‐lasting downregulation of extracellular matrix in perineuronal nets induced by focal cerebral ischemia in rats. J Neurosci Res 80:539–548. [DOI] [PubMed] [Google Scholar]
- 42. Van Horssen J, Otte‐Holler I, David G, Maat‐Schieman ML, Van Den Heuvel LP, Wesseling P et al (2001) Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer's disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol (Berl) 102: 604–614. [DOI] [PubMed] [Google Scholar]
- 43. Van Horssen J, Bo L, Dijkstra CD, De Vries HE (2006) Extensive extracellular matrix depositions in active multiple sclerosis lesions. Neurobiol Dis 24:484–491. [DOI] [PubMed] [Google Scholar]
- 44. Van Horssen J, Vos CM, Admiraal L, Van Haastert ES, Montagne L, Van Der Valk P et al (2006) Matrix metalloproteinase‐19 is highly expressed in active multiple sclerosis lesions. Neuropathol Appl Neurobiol 32:585–593. [DOI] [PubMed] [Google Scholar]
- 45. Van Horssen J, Dijkstra CD, De Vries HE (2007) The extracellular matrix in multiple sclerosis pathology. J Neurochem 103:1293–1301. [DOI] [PubMed] [Google Scholar]
- 46. Jourquin J, Tremblay E, Bernard A, Charton G, Chaillan FA, Marchetti E et al (2005) Tissue inhibitor of metalloproteinases‐1 (TIMP‐1) modulates neuronal death, axonal plasticity, and learning and memory. Eur J Neurosci 22:2569–2578. [DOI] [PubMed] [Google Scholar]
- 47. Kaminska B, Filipkowski RK, Biedermann IW, Konopka D, Nowicka D, Hetman M et al (1997) Kainate‐evoked modulation of gene expression in rat brain. Acta Biochim Pol 44:781–789. [PubMed] [Google Scholar]
- 48. Kim YS, Kim SS, Cho JJ, Choi DH, Hwang O, Shin DH et al (2005) Matrix metalloproteinase‐3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. J Neurosci 25:3701–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laurent UB, Laurent TC, Hellsing LK, Persson L, Hartman M, Lilja K (1996) Hyaluronan in human cerebrospinal fluid. Acta Neurol Scand 94:194–206. [DOI] [PubMed] [Google Scholar]
- 50. Le SS, Loucks FA, Udo H, Richardson‐Burns S, Phelps RA, Bouchard RJ et al (2005) Inhibition of Rac GTPase triggers a c‐Jun‐ and Bim‐dependent mitochondrial apoptotic cascade in cerebellar granule neurons. J Neurochem 94:1025–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu IH, Uversky VN, Munishkina LA, Fink AL, Halfter W, Cole GJ (2005) Agrin binds alpha‐synuclein and modulates alpha‐synuclein fibrillation. Glycobiology 15:1320–1331. [DOI] [PubMed] [Google Scholar]
- 52. Lo EH, Wang X, Cuzner ML (2002) Extracellular proteolysis in brain injury and inflammation: role for plasminogen activators and matrix metalloproteinases. J Neurosci Res 69:1–9. [DOI] [PubMed] [Google Scholar]
- 53. Lorenzl S, Albers DS, LeWitt PA, Chirichigno JW, Hilgenberg SL, Cudkowicz ME et al (2003) Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J Neurol Sci 207:71–76. [DOI] [PubMed] [Google Scholar]
- 54. Lorenzl S, Albers DS, Chirichigno JW, Augood SJ, Beal MF (2004) Elevated levels of matrix metalloproteinases‐9 and ‐1 and of tissue inhibitors of MMPs, TIMP‐1 and TIMP‐2 in postmortem brain tissue of progressive supranuclear palsy. J Neurol Sci 218:39–45. [DOI] [PubMed] [Google Scholar]
- 55. Madani R, Nef S, Vassalli JD (2003) Emotions are building up in the field of extracellular proteolysis. Trends Mol Med 9:183–185. [DOI] [PubMed] [Google Scholar]
- 56. Matias‐Roman S, Galvez BG, Genis L, Yanez‐Mo M, De La Rosa G, Sanchez‐Mateos P et al (2005) Membrane type 1‐matrix metalloproteinase is involved in migration of human monocytes and is regulated through their interaction with fibronectin or endothelium. Blood 105:3956–3964. [DOI] [PubMed] [Google Scholar]
- 57. Medina‐Flores R, Wang G, Bissel SJ, Murphey‐Corb M, Wiley CA (2004) Destruction of extracellular matrix proteoglycans is pervasive in simian retroviral neuroinfection. Neurobiol Dis 16:604–616. [DOI] [PubMed] [Google Scholar]
- 58. Melchor JP, Strickland S (2005) Tissue plasminogen activator in central nervous system physiology and pathology. Thromb Haemost 93:655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Milev P, Monnerie H, Popp S, Margolis RK, Margolis RU (1998) The core protein of the chondroitin sulfate proteoglycan phosphacan is a high‐affinity ligand of fibroblast growth factor‐2 and potentiates its mitogenic activity. J Biol Chem 273:21439–21442. [DOI] [PubMed] [Google Scholar]
- 60. Miyata S, Nishimura Y, Nakashima T (2007) Perineuronal nets protect against amyloid beta‐protein neurotoxicity in cultured cortical neurons. Brain Res 30:200–206. [DOI] [PubMed] [Google Scholar]
- 61. Morawski M, Bruckner MK, Riederer P, Bruckner G, Arendt T (2004) Perineuronal nets potentially protect against oxidative stress. Exp Neurol 188:309–315. [DOI] [PubMed] [Google Scholar]
- 62. Muir EM, Adcock KH, Morgenstern DA, Clayton R, Von Stillfried N, Rhodes K et al (2002) Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res Mol Brain Res 100:103–117. [DOI] [PubMed] [Google Scholar]
- 63. Nagase H, Woessner JF Jr (1999) Matrix metalloproteinases. J Biol Chem 274:21491–21494. [DOI] [PubMed] [Google Scholar]
- 64. Navia BA, Jordan BD, Price RW (1986) The AIDS dementia complex: I. Clinical features. Ann Neurol 19:517–524. [DOI] [PubMed] [Google Scholar]
- 65. Nelson CM, Bissell MJ (2006) Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol 22:287–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13:9–22. [PubMed] [Google Scholar]
- 67. Ogier C, Bernard A, Chollet AM, Led T, Hanessian S, Charton G et al (2006) Matrix metalloproteinase‐2 (MMP‐2) regulates astrocyte motility in connection with the actin cytoskeleton and integrins. Glia 54:272–284. [DOI] [PubMed] [Google Scholar]
- 68. Okamoto M, Sakiyama J, Mori S, Kurazono S, Usui S, Hasegawa M et al (2003) Kainic acid‐induced convulsions cause prolonged changes in the chondroitin sulfate proteoglycans neurocan and phosphacan in the limbic structures. Exp Neurol 184:179–195. [DOI] [PubMed] [Google Scholar]
- 69. Osman M, Tortorella M, Londei M, Quaratino S (2002) Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases define the migratory characteristics of human monocyte‐derived dendritic cells. Immunology 105:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pagenstecher A, Wussler EM, Opdenakker G, Volk B, Campbell IL (2001) Distinct expression patterns and levels of enzymatic activity of matrix metalloproteinases and their inhibitors in primary brain tumors. J Neuropathol Exp Neurol 60:598–612. [DOI] [PubMed] [Google Scholar]
- 71. Porter S, Clark IM, Kevorkian L, Edwards DR (2005) The ADAMTS metalloproteinases. Biochem J 386:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Raines EW, Ross R (1992) Compartmentalization of PDGF on extracellular binding sites dependent on exon‐6‐encoded sequences. J Cell Biol 116:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rauvala H, Peng HB (1997) HB‐GAM heparin‐binding growth‐associated molecule and heparin‐type glycans in the development and plasticity of neuron‐target contacts. Prog Neurobiol 52:127–144. [DOI] [PubMed] [Google Scholar]
- 74. Reijerkerk A, Kooij G, Van Der Pol SM, Khazen S, Dijkstra CD, De Vries HE (2006) Diapedesis of monocytes is associated with MMP‐mediated occludin disappearance in brain endothelial cells. FASEB J 20:2550–2552. [DOI] [PubMed] [Google Scholar]
- 75. Rivera S, Tremblay E, Timsit S, Canals O, Ben‐Ari Y, Khrestchatisky M (1997) Tissue inhibitor of metalloproteinases‐1 (TIMP‐1) is differentially induced in neurons and astrocytes after seizures: evidence for developmental, immediate early gene, and lesion response. J Neurosci 17:4223–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Saghatelyan AK, Dityatev A, Schmidt S, Schuster T, Bartsch U, Schachner M (2001) Reduced perisomatic inhibition, increased excitatory transmission, and impaired long‐term potentiation in mice deficient for the extracellular matrix glycoprotein tenascin‐R. Mol Cell Neurosci 17:226–240. [DOI] [PubMed] [Google Scholar]
- 77. Saksela O, Rifkin DB (1990) Release of basic fibroblast growth factor‐heparan sulfate complexes from endothelial cells by plasminogen activator‐mediated proteolytic activity. J Cell Biol 110:767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sasaki A, Yamaguchi H, Ogawa A, Sugihara S, Nakazato Y (1997) Microglial activation in early stages of amyloid beta protein deposition. Acta Neuropathol 94:316–322. [DOI] [PubMed] [Google Scholar]
- 79. Satoh K, Suzuki N, Yokota H (2000) ADAMTS‐4 (a disintegrin and metalloproteinase with thrombospondin motifs) is transcriptionally induced in beta‐amyloid treated rat astrocytes. Neurosci Lett 289:177–180. [DOI] [PubMed] [Google Scholar]
- 80. Schuppel K, Brauer K, Hartig W, Grosche J, Earley B, Leonard BE et al (2002) Perineuronal nets of extracellular matrix around hippocampal interneurons resist destruction by activated microglia in trimethyltin‐treated rats. Brain Res 958:448–453. [DOI] [PubMed] [Google Scholar]
- 81. Sheehan JJ, Zhou C, Gravanis I, Rogove AD, Wu YP, Bogenhagen DF et al (2007) Proteolytic activation of monocyte chemoattractant protein‐1 by plasmin underlies excitotoxic neurodegeneration in mice. J Neurosci 27:1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sithu SD, English WR, Olson P, Krubasik D, Baker AH, Murphy G et al (2007) Membrane‐type 1‐matrix metalloproteinase regulates intracellular adhesion molecule‐1 (ICAM‐1)‐mediated monocyte transmigration. J Biol Chem 282:25010–25019. [DOI] [PubMed] [Google Scholar]
- 83. Snow AD, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S et al (1988) The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer's disease. Am J Pathol 133:456–463. [PMC free article] [PubMed] [Google Scholar]
- 84. Snow AD, Mar H, Nochlin D, Sekiguchi RT, Kimata K, Koike Y et al (1990) Early accumulation of heparan sulfate in neurons and in the beta‐amyloid protein‐containing lesions of Alzheimer's disease and Down's syndrome. Am J Pathol 137:1253–1270. [PMC free article] [PubMed] [Google Scholar]
- 85. Snow AD, Mar H, Nochlin D, Kresse H, Wight TN (1992) Peripheral distribution of dermatan sulfate proteoglycans (decorin) in amyloid‐containing plaques and their presence in neurofibrillary tangles of Alzheimer's disease. J Histochem Cytochem 40:105–113. [DOI] [PubMed] [Google Scholar]
- 86. Szklarczyk A, Lapinska J, Rylski M, McKay RD, Kaczmarek L (2002) Matrix metalloproteinase‐9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J Neurosci 22:920–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tsirka SE, Gualandris A, Amaral DG, Strickland S (1995) Excitotoxin‐induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature 377:340–344. [DOI] [PubMed] [Google Scholar]
- 88. Umehara Y, Yamada S, Nishimura S, Shioi J, Robakis NK, Sugahara K (2004) Chondroitin sulfate of appican, the proteoglycan form of amyloid precursor protein, produced by C6 glioma cells interacts with heparin‐binding neuroregulatory factors. FEBS Lett 557:233–238. [DOI] [PubMed] [Google Scholar]
- 89. Van Lint P, Libert C (2007) Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol 82:1375–1381. [DOI] [PubMed] [Google Scholar]
- 90. Verbeek MM, Otte‐Holler I, Van Den Born J, Van Den Heuvel LP, David G, Wesseling P et al (1999) Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer's disease brain. Am J Pathol 155:2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Vlodavsky I, Miao HQ, Medalion B, Danagher P, Ron D (1996) Involvement of heparan sulfate and related molecules in sequestration and growth promoting activity of fibroblast growth factor. Cancer Metastasis Rev 15:177–186. [DOI] [PubMed] [Google Scholar]
- 92. Weber P, Bartsch U, Rasband MN, Czaniera R, Lang Y, Bluethmann H et al (1999) Mice deficient for tenascin‐R display alterations of the extracellular matrix and decreased axonal conduction velocities in the CNS. J Neurosci 19:4245–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yamaguchi Y (2000) Lecticans: organizers of the brain extracellular matrix. Cell Mol Life Sci 57:276–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM (1991) Cell surface, heparin‐like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 64:841–848. [DOI] [PubMed] [Google Scholar]
- 95. Yong VW (2005) Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 6:931–944. [DOI] [PubMed] [Google Scholar]
- 96. Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR (1998) Matrix metalloproteinases and diseases of the CNS. Trends Neurosci 21:75–80. [DOI] [PubMed] [Google Scholar]
- 97. Yoshihara Y, Nakamura H, Obata K, Yamada H, Hayakawa T, Fujikawa K et al (2000) Matrix metalloproteinases and tissue inhibitors of metalloproteinases in synovial fluids from patients with rheumatoid arthritis or osteoarthritis. Ann Rheum Dis 59:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yuan W, Matthews RT, Sandy JD, Gottschall PE (2002) Association between protease‐specific proteolytic cleavage of brevican and synaptic loss in the dentate gyrus of kainate‐treated rats. Neuroscience 114:1091–1101. [DOI] [PubMed] [Google Scholar]
- 99. Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J et al (2003) HIV‐induced metalloproteinase processing of the chemokine stromal cell derived factor‐1 causes neurodegeneration. Nat Neurosci 6:1064–1071. [DOI] [PubMed] [Google Scholar]
- 100. Zhou J, Marten NW, Bergmann CC, Macklin WB, Hinton DR, Stohlman SA (2005) Expression of matrix metalloproteinases and their tissue inhibitor during viral encephalitis. J Virol 79:4764–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]