

Abstract
Co‐deletion of chromosome arms 1p and 19q, characteristic of oligodendroglial tumors, was recently found to be mediated by t(1;19)(q10;p10). To evaluate the prevalence of 1p19q co‐deletion and t(1;19) in extraventricular neurocytomas (EVN), we studied tumors from 23 patients, including 13 females and 10 males (median age at diagnosis 34 years, range 2–76 years). Fluorescence in situ hybridization (FISH) studies were performed with probes targeting 1p36/1q25 and 19q13/19p13 to assess for 1p19q co‐deletion, as well as chromosome 1 α‐satellite and 19p12 to detect t(1;19)(q10;p10). FISH was successful in 21 (91%) cases and demonstrated 1p19q co‐deletion in five cases (24%) or isolated 1p loss in two cases (10%). Evidence for t(1;19) was found in four (of five) cases with 1p19q co‐deletion. Three tumors with 1p19q loss and t(1;19) demonstrated atypical histologic features, compared with one (of 17) tumors without 1p19q co‐deletion (P = 0.01, Fisher exact test). In addition, tumors with t(1;19) showed increased mitotic activity compared with tumors without t(1;19) (P = 0.045; Wilcoxon rank sum test). The four patients with t(1;19) developed tumor recurrence (n = 3), or expired (n = 2) 3.5 to 5.5 years after first resection. These results suggest that 1p19q loss and t(1;19) occur in a subset of EVN, and may be associated with aggressive histology in these tumors.
Keywords: 1p19q, FISH, neurocytoma, oligodendroglioma, t(1;19)
INTRODUCTION
Well‐differentiated neuronal neoplasms composed of small neurons superficially resembling oligodendrocytes (“neurocytes”) have a tendency to arise within the ventricular system, most often in the septum pellucidum nearby the foramen of Monro. The term “central neurocytoma” is reserved for these lesions. Morphologically similar neoplasms may occur in the cerebral hemispheres or in the spinal cord parenchyma and are therefore referred as “extraventricular neurocytomas” (EVN) (1).
Concurrent deletion of chromosome arms 1p and 19q, abnormalities frequently encountered in oligodendrogliomas, is associated with an improved response to treatment and/or a more favorable prognosis in these tumors 2, 3. Most recently, this cytogenetic alteration was reported to be mediated by the unbalanced translocation t(1;19)(q10;p10) 7, 9. Concordance between 1p19q co‐deletion and t(1;19) is approximately 90% (9).
Co‐deletion of 1p19q has been described in a minority of EVN 11, 14, but not in central neurocytomas 10, 14. This alteration, as well as the increased frequency of glial differentiation in EVN, suggests a possible biological relationship between EVN and oligodendroglioma. In addition, some oligodendrogliomas with focal neurocytic differentiation have demonstrated 1p19q deletion in both morphologic components (13). We hypothesized that both 1p19q co‐deletion and t(1;19)(q10;p10) may be present in a subset of EVN, unlike in their central counterparts, and that it may be of prognostic significance. Therefore, we sought to explore these alterations by interphase cytogenetics [fluorescence in situ hybridization (FISH)] in a series of these relatively uncommon neoplasms.
MATERIALS AND METHODS
We searched the pathology database of Mayo Clinic, Rochester, MN for cases of EVN. A total of 23 cases were identified. Overall, there was a slight female predominance (female to male ratio 13:10) and a median age at diagnosis of 34 years (range 2–76). Tumors available for special studies were obtained at first resection (n = 19) or recurrence (n = 4). Histologic slides were reviewed by at least two (of three) neuropathologists (C Giannini, FJ Rodriguez, BW Scheithauer) to confirm the morphological diagnosis of EVN according to the recent World Health Organization classification (5). Only cases with a diffuse neuronal phenotype were included (Figure 1). Oligodendrogliomas with focal neuronal differentiation, as previously described (13), were excluded, as well as tumors showing punctate/paranuclear synaptophysin immunoreactivity (21).
Figure 1.
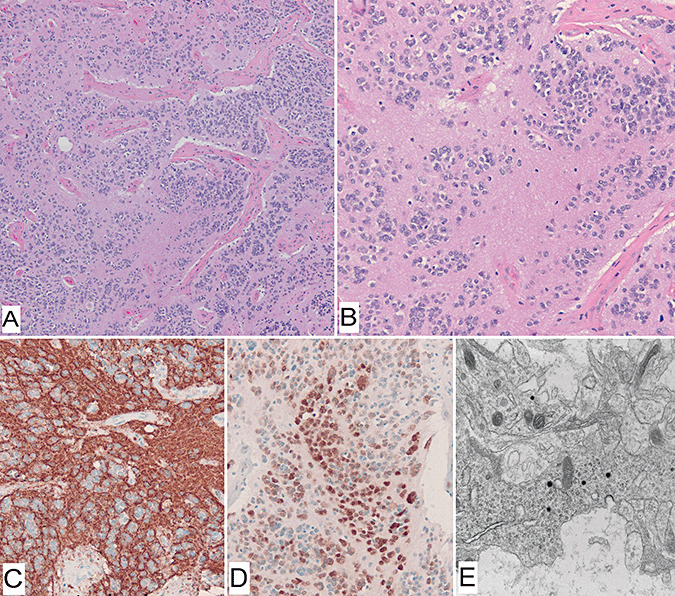
Histologic features of extraventricular neurocytoma (Case 8). Morphologic features of neuronal differentiation were evident on hematoxylin–eosin stain (A,B), including extensive neuropil formation and neurocytic cytology. Immunohistochemical stains for synaptophysin labels individual cells and neuropil (C). Neu‐N stain was positive in a nuclear fashion in all cases tested (D). Electron microscopy demonstrating dense core secretory granules (E).
Immunohistochemistry
Immunohistochemical studies were performed on 5 micron‐thick formalin‐fixed paraffin‐embedded tumor sections using antibodies directed against chromogranin (LK2H10; 1:500; Chemicon, Temecula, CA, USA), synaptophysin (clone SY38; 1:40; ICN, Costa Mesa, CA, USA), neurofilament protein (NF; clone 2F11, 1:75; Dako, Carpinteria, CA, USA), glial fibrillary acidic protein (GFAP; polyclonal; 1:4000; Dako), neuronal nuclei (Neu‐N; 1:10,000; Chemicon) and Ki67 [clone mindbomb homolog‐1 (MIB‐1); 1:300; Dako]. Quantitative evaluation of MIB‐1 labeling indices was performed using the Hamamatsu NanoZoomer Digital Pathology for scanning images and IHCScore software for computer assisted analyses (Bacus Laboratories, Inc, Chicago, IL, USA).
FISH
FISH studies were performed as previously described 8, 9. In brief, 5 micron‐thick formalin‐fixed paraffin‐embedded sections were baked for 2 h at 56°C and deparaffinized in Citrasolv (15 minutes × 2) followed by 100% ethanol (ETOH; 10 minutes). The slides were then immersed in 10 mM citric acid (pH 6.08) and was placed in a microwave on the high setting for 3 minutes, followed by pepsin digestion (4 mg pepsin/L 0.9% NaCl for 15 minutes in a 37°C water bath), and then dehydrated in ETOH at increasing concentrations. Dual‐color locus‐specific identifier probes targeting 1p36/1q25 and 19q13/19p13 (Vysis/Abbott Molecular Inc., Des Plaines, IL, USA) were used to assess 1p19q co‐deletion. To search for t(1;19)(q10;p10), a chromosome 1 α‐satellite probe (Spectrum Orange™, Vysis/Abbott Molecular Inc.) and a 19p12 locus‐specific probe [Spectrum Green™, bacterial artificial chromosome (BAC) contig, Vysis/Abbott Molecular Inc.] were used. All probe pairs were co‐denatured with the tissue sections and hybridized overnight at 37°C in separate slides. After hybridization the slides were washed on 2XSSC/0.1%NP‐40 for 2 minutes at 73°C, counterstained with 4′,6′‐diamidino‐2‐phenylindole dihydrochloride (DAPI), and then coverslipped. In a subset of cases, a three‐color FISH strategy involving a 1p36 BAC contig probe labeled with an aqua fluorochrome, added to the t(1;19) probe mixture, was performed in order to determine whether 1p loss and t(1;19) coexisted within the same cells. A total of 200 (for 1p19q) or 100 [for t(1;19)] non‐overlapping nuclei were scored by two independent observers and recorded as an average of both counts. Discordant cases were reviewed independently by a third observer. The threshold for 1q and 19p fusion was set to at least 60% of cells showing 1 α‐satellite (red) and 19p12 (green) signals within two‐signal widths of each other as previously described (9). Co‐deletion of 1p and 19q was defined as signal ratios of less than 0.8 for region of interest to control probe. Aneusomy was defined as loss or gain of the control probe in greater than 60% and 30% of cells, respectively.
Statistical analyses
Measurements of proliferative activity, including number of mitoses per 10 high‐power fields (HPF) and MIB‐1 labeling indices, were compared with the presence or absence of cytogenetic abnormalities using quantiles and the Wilcoxon rank sum test. The Fisher exact test was used to evaluate differences in proportions. All tests were two‐sided with any P values < 0.05 considered statistically significant. Statistical analyses were performed using JMP software (SAS Institute Inc., Cary, NC, USA).
RESULTS
FISH
FISH results are summarized in Table 1 and illustrated in 2, 3.
Table 1.
Fluorescence in situ hybridization studies in extraventricular neurocytomas. Abbreviation: NA = not applicable.
| Case | 1p/1q ratio | 19q/19p ratio | 1p | 19q | 1CEP‐19p12 % fusion | t(1;19) (>60% fused signals) | 1p19q distribution |
|---|---|---|---|---|---|---|---|
| 1 | 0.55 | 0.57 | Deletion | Deletion | 84 | Present | Same areas |
| 2 | 0.61 | 0.59 | Deletion | Deletion | 71 | Present | Same areas |
| 3 | 0.68 | 0.72 | Deletion | Deletion | 76 | Present | Same areas |
| 4 | 0.55 | 0.57 | Relative deletion* | Relative deletion | 75 | Present | Same areas |
| 5 | 0.66 | 0.66 | Relative deletion | Deletion | 24 | Absent | Different areas† |
| 6 | 0.69 | 0.93 | Deletion | Aneusomy | 38 | Absent | NA |
| 7 | 0.79 | 0.96 | Relative deletion | Normal | 41 | Absent | NA |
| 7 | 1.00 | 0.88 | Aneusomy | Aneusomy | 33 | Absent | NA |
| 9 | 1.01 | 0.96 | Aneusomy | Aneusomy | 24 | Absent | NA |
| 10 | 0.96 | 1.01 | Normal | Normal | 53 | Absent | NA |
| 11 | 0.88 | 1.06 | Aneusomy | Aneusomy | 15 | Absent | NA |
| 12 | 0.98 | 0.96 | Normal | Normal | 11 | Absent | NA |
| 13 | 1.00 | 0.98 | Normal | Normal | 11 | Absent | NA |
| 14 | 0.95 | 1.00 | Normal | Normal | 11 | Absent | NA |
| 15 | 0.96 | 0.99 | Aneusomy | Normal | 15 | Absent | NA |
| 16 | 0.98 | 0.98 | Normal | Normal | — | Failed | NA |
| 17 | 0.93 | 0.96 | Normal | Normal | — | Failed | NA |
| 18 | 0.95 | 1.01 | Normal | Aneusomy | — | Failed | NA |
| 19 | 0.98 | 1.02 | Aneusomy | Aneusomy | 33 | Absent | NA |
| 20 | 0.98 | 0.98 | Normal | Normal | — | Failed | NA |
| 21 | 1.03 | Failed | Normal | — | — | Failed | NA |
Relative deletion: region of interest/control probe count ratio <0.8 associated with aneusomy.
Different areas: 1p loss present in tumor areas with intact 19q and vice versa.
Figure 2.
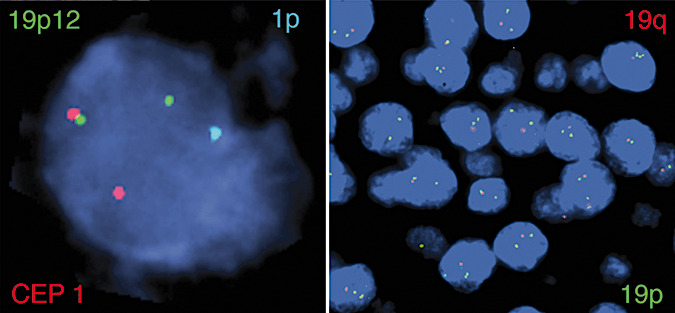
t(1;19) and 1p loss coexist in the same cells (Case 4). Three‐color fluorescence in situ hybridization strategy demonstrates a red/green fusion and a single aqua signal representing t(1;19) and 1p loss in the same cells, respectively (left). The same tumor also demonstrates a single red signal and two green signals in most cells reflecting 19q loss (right).
Figure 3.
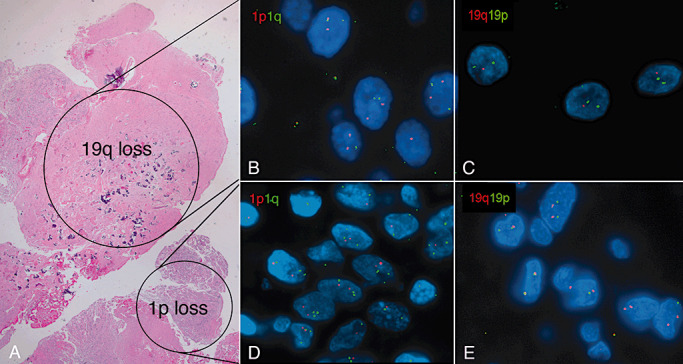
1p19q loss reflecting genetic heterogeneity (Case 1). The pattern of 1p19q loss in this case was unique, as it was limited to separate geographic areas (hematoxylin–eosin stain) (A). Dual‐color fluorescence in situ hybridization studies demonstrate a normal chromosome 1 (B) but 19q loss in most areas (C). Relative 1p loss with aneusomy (D) was a focal finding in an area showing no abnormalities in chromosome 19 (E). t(1;19) was absent (not shown).
FISH studies for 1p19q were successful in 21 (of 23) cases (91%) and demonstrated co‐deletion of 1p19q in five (of 21) cases (24%), isolated 1p loss in two (of 21) cases (10%), gains of chromosome 19 in six (of 21) cases (29%) and gains of chromosome 1 in five (of 21) cases (24%). FISH studies for t(1;19) were successful in 16 (of 23) cases (70%). As reflected by greater than 60% nuclei showing fused signals with the dual color chromosome 1 α‐satellite/19p12 probes, t(1;19) was present in four (of five) cases (80%) with 1p19q co‐deletion, and was absent in the two cases with isolated 1p loss and in the remainder of the cases tested. The three‐color probe set provided scorable signals in four cases and demonstrated: (i) concurrent 1p loss and t(1;19) in the same cells in two cases with 1p19q co‐deletion (Figure 2); and (ii) an intact 1p chromosome arm and no fusion in two cases with normal results by the standard probe set (not shown). The single case with 1p19q co‐deletion lacking t(1;19) demonstrated relative 1p loss in a focal region with intact 19q, and 19q loss in the majority of the tumor, 1p being preserved, consistent with genetic heterogeneity (Figure 3).
Clinicopathological characteristics
Important clinicopathologic features in cases with normal and abnormal 1p19q FISH results are summarized in Table 2. As previously described (1), the histology of EVN is variable. The majority of the tumors demonstrated morphologic evidence of neuronal differentiation including neurocytic cytology with speckled chromatin, rosette formation, delicate neuropil and ganglionic maturation (Figure 1). Partial microscopic infiltration, mainly at the interface with brain parenchyma, was present in six (of 21) cases (29%). Diffuse, non‐punctate synaptophysin immunoreactivity was evident in all cases (21 of 21), as well as chromogranin (n = 3) and Neu‐N (n = 4) in the subset of cases tested. Neuronal differentiation was further confirmed by electron microscopy in five cases in which primarily fixed tissue was available, and took the form of intracytoplasmic neurosecretory granules (Figure 1E). Rosenthal fibers (n = 3) and eosinophilic granular bodies (n = 4) were present only in tumors without t(1;19). Most tumors were low grade (n = 17) based upon low proliferation indices and absence of both necrosis and proliferative vascular changes. Four tumors showed atypical histologic features, including > 3 mitoses/10 HPF (n = 4), increased cellularity (n = 4), endothelial hypertrophy (n = 2) and necrosis (n = 1). No obvious morphologically distinct low grade precursor was noted in these tumors. MIB‐1 immunostaining was performed in 16 cases. The median labeling index was 1.5% (range 0.11–20). MIB‐1 labeling index was performed in only one (of four) case with t(1;19), but this tumor had the highest MIB‐1 labeling index (20%) of all the cases studied.
Table 2.
Clinicopathologic features of extraventricular neurocytomas (EVN) studied by fluorescence in situ hybridization.
| Characteristics n (%) | 1p19q co‐deletion and t(1;19) | |
|---|---|---|
| Present (n = 4) | Absent (n = 17) | |
| Age (years), (median, range) | 52 (39–67) | 29 (2–76) |
| Sex | ||
| Female | 4 (100) | 9 (53) |
| Male | 0 | 8 (47) |
| Location | ||
| Frontal lobe | 1 (25) | 7 (41) |
| Parietal lobe | 1 (25) | 3 (18) |
| Parieto‐occipital lobe | 1 (25) | 3 (18) |
| Temporal lobe | 1 (25) | 1 (5) |
| Brain not otherwise specified | 0 | 3 (18) |
| Histologic diagnosis | ||
| EVN without atypia | 1 (25) | 16 (94) |
| Atypical EVN | 3 (75) | 1 (6) |
| Histologic features | ||
| Mitoses/10 high‐power fields (median, range) | 3 (0–5) | 0 (0–3) |
| Necrosis | 1 (25) | 2 (12) |
| Endothelial hypertrophy | 2 (50) | 2 (12) |
| Rosettes | 2 (50) | 8 (47) |
| Granular bodies | 0 | 4 (24) |
| Rosenthal fibers | 0 | 3 (18) |
| Ganglionic maturation | 1 (25) | 11 (64) |
| Infiltration (partial) | 0 | 6 (35) |
| Surgery | ||
| Gross total resection | 1 (25) | 5 (29) |
| Subtotal resection | 2 (50) | 6 (35) |
| Unknown | 1 (25) | 6 (35) |
| Treatment | ||
| Radiation | 3 (75) | 3 (18) |
| Chemotherapy | 2 (50) | 1 (6) |
| Observation | 0 | 8 (47) |
| Unknown | 1 (25) | 5 (29) |
Of the four patients whose tumors showed 1p19q loss and t(1;19), three had a documented recurrence, and two expired 3.5 to 5.5 years after initial tumor resection, with a median follow‐up of 3.5 years (Table 3). They had received radiation therapy (n = 3) and/or chemotherapy including temozolamide (n = 2). The single patient with 1p19q deletion in different cell populations but without t(1;19) is stable without postoperative therapy, and showed no evidence of disease on a follow‐up MRI scan 10 months after surgery. Of the 17 patients, whose tumors did not show 1p19q co‐deletion/t(1;19), follow‐up was available in 71% and ranged from 1 to 11 years (median 4 years). Recurrences were documented in five patients: one of these patients, the single patient (of 17) with histologic atypia but without 1p19q co‐deletion and t(1;19), died of disease 11 years after multiple recurrences. Information about postoperative treatment was available in 11 (of 17) patients (65%) in this group, and consisted mostly of observation (n = 8; 73%), followed by radiation therapy (n = 3; 27%) and temozolamide chemotherapy (n = 1; 9%).
Table 3.
Clinicopathologic features of EVN with t(1;19) and 1p19q co‐deletion. Abbreviations: EVN = extraventricular neurocytomas; GTR = gross total resection; HPF = high‐powerfields; IHC = immunohistochemistry; ISH = in situ hybridization; MIB‐1 = mindbomb homolog‐1; NA = data not available; NED = no evidence of disease; STR = subtotal resection.
| Case | Age/sex | Location | Diagnosis | Histology | Mitoses/ 10 HPF | IHC | Surgery | Treatment | Follow‐up |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 51/F | Left parietal lobe | Atypical EVN | Calcification; palisading necrosis; apoptotic bodies; vascular hypertrophy. No infiltration. | 3 | Synaptophysin+ | STR | External beam radiation and Temodar® | NED 20 Mo postoperative |
| 2 | 52/F | Left parieto‐occipital lobe | Atypical EVN | Vascular hypertrophy. No infiltration. | 5 | Synaptophysin+ Chromogranin+ (IHC and ISH) Neu‐N + (focal) MIB‐1 labeling index × 20% | GTR | Craniospinal radiation (54 Gy to tumor bed) and multiple chemotherapies with recurrences including temozolamide most recently | Expired 5.5 years after resection, after at least three recurrences |
| 3 | 67/F | Right frontal lobe | Atypical EVN | Rosettes, ganglionic maturation, increased apoptosis. No infiltration. | 4 | Synaptophysin+ Neurofilament+ | STR | Radiation: 50 Gy | Recurrence; expired 3.5 years after resection |
| 4 | 39/F | Temporal lobe | Well‐differentiated EVN | Rosettes. No infiltration. | 0 | Synaptophysin+ | NA | NA | Recurrence |
Statistical analysis
When comparing the groups with 1p19q loss and t(1;19) (n = 4) with the rest (n = 17), the group with 1p19q loss/t(1;19) demonstrated histologic atypia in three of four cases vs. one of 17 (P = 0.012, Fisher exact test), increased mitotic activity (P = 0.045, Wilcoxon rank sum test; Figure 4), and increased median age (52 vs. 29 years; P = 0.02, Wilcoxon rank sum test). There were no statistically significant differences with other clinicopathologic features including sex, necrosis, endothelial proliferation, rosettes, eosinophilic granular bodies, rosenthal fibers, ganglionic maturation and infiltration, neither in proliferation between tumors with aneusomy of chromosomes 1 or 19 and those without it (P > 0.05). These findings should be interpreted with caution given the small number (n = 4) of cases with 1p19q loss and t(1;19).
Figure 4.
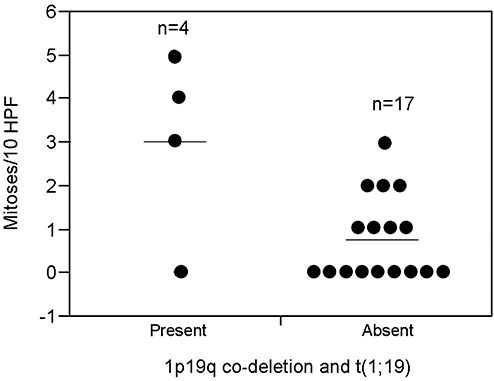
Fluorescence in situ hybridization studies identify two distinct subgroups in EVN. Scatterplot demonstrates increased mitoses per 10 high‐power fields in extraventricular neurocytomas (EVN) with 1p19 co‐deletion and t(1;19) as compared with EVN without 1p19q loss and t(1;19) (P = 0.045, Wilcoxon rank sum test).
DISCUSSION
In recent years there has been increased recognition of well‐differentiated neurocytic neoplasms that involve the cerebral hemispheres. The recent 2007 WHO classification recognized EVN to distinguish it from its intraventricular/central counterpart (5). Compared with the latter, EVN is morphologically diverse, and shows a higher frequency of ganglion cell formation and focal GFAP expression. This tumor may be difficult to distinguish from infiltrating gliomas, specifically oligodendrogliomas. However, EVN frequently demonstrates some patently neuronal features, including rosette and neuropil formation, in addition to immunoexpression of multiple markers of neuronal differentiation, particularly synaptophysin, and in a diffuse manner (1).
Given the rarity of EVN, its molecular genetic anomalies, as compared with diffuse gliomas, have not been well characterized. The presence of 1p19q co‐deletion has been reported in a small number of cases. For example Perry et al (14) reported this abnormality in two (of 12) EVN (17%), one additional case demonstrating isolated 1p loss. In addition, Mrak et al. reported co‐deletion of 1p19q by FISH and loss‐of‐heterozygosity studies in one case of atypical EVN with an infiltrative pattern of growth (11). Our study indicates that 1p19q co‐deletion occurs in 24% of EVN and, as in oligodendroglial tumors, is mediated in most cases by t(1;19).
In contrast to EVN, 1p19q co‐deletion is not a feature of central neurocytoma 10, 14. Although loss of heterozygosity of 1p was reported in six (of nine) (67%), and in 19q in five (of nine) cases (56%) in one study (20), the majority of the informative markers for both chromosome arms were retained, which is not usually the case with classic 1p19q co‐deletion. We did not find 1p19q co‐deletion in 17 central neurocytomas examined by FISH (10). Similar results were reported by others (14). Rather, gains of chromosome 7 have been found in roughly one‐third of central neurocytomas (18) but in the absence of EGFR amplification (20).
The main finding of our study is that 1p19q co‐deletion, usually with associated t(1;19)(q10;p10), is seen in roughly one‐fourth of EVN. This molecular abnormality has been associated only with diffuse gliomas, in particular oligodendrogliomas. For many years it has been observed that within the spectrum of diffuse gliomas, oligodendrogliomas are associated with an overall better prognosis and/or respond more favorably to treatment. This outcome has been most closely associated with 1p19q co‐deletion 2, 3. Although the group with 1p19q co‐deletion and t(1;19) was small and therefore the statistical analyses should be interpreted with caution, our findings suggest that worrisome histologic features occur in patients with EVN, associated with 1p19q loss/t(1;19) (three of four of our tumors), and raises important conceptual issues. One is that this subset of tumors may be more closely related to diffuse oligodendrogliomas which, although more indolent in behavior than infiltrating astrocytomas, nonetheless show a significant frequency of high‐grade transformation and a fatal outcome. In contrast, the presence of Rosenthal fibers and eosinophilic granular bodies in some of our cases without t(1;19), as well as lower proliferative activity, suggests that tumors without t(1;19) may have a different genetic etiology. Such tumors may be more closely related to tumors curable by gross total resection, and even have a more favorable prognosis than diffusely infiltrating tumors, including oligodendrogliomas.
Therefore, even though 1p19q loss and t(1;19) confer a better prognosis when present in diffuse gliomas, these tumors still have a significant risk for recurrence, although the same is not true for WHO grade I—more circumscribed gliomas, for instance pilocytic astrocytomas—which have a negligible risk for malignant transformation and may be cured by a gross total resection despite lacking 1p19q loss. The significance of these observations awaits further studies. However, it is possible that the presence or absence of t(1;19), more so than 1p19q co‐deletion, separates EVN into two biologic groups. These groups may be of prognostic significance and prompt interesting speculation regarding their pathogeneses. Prior investigators have raised the possibility of oligodendrogliomas arising from a glioneuronal progenitor based on the presence of focal neurocytic differentiation in four oligodendrogliomas, and 1p19q loss in glial and neuronal components of three (of four) tumors (13). One possible scenario, taking into account the cancer stem cell hypothesis, is t(1;19) occurring early in a pluripotential stem cell capable of multilineage differentiation. Subsequent genetic alterations in glial and neuronal progenitors may then lead to oligodendroglial tumors and atypical EVN, respectively, the latter more closely related to diffuse gliomas than EVN lacking t(1;19). Recent studies of gene expression in oligodendrogliomas with 1p loss as well as 1p19q co‐deletion have identified a proneural gene expression profile, which partially supports these speculations and narrows the gap between oligodendroglial morphology and neuronal differentiation 4, 12.
The significance of histologic atypia has been explored in central neurocytoma, “atypical neurocytoma” being defined as showing a MIB‐1 labeling index greater than 2%–3% 15, 17, a finding associated with decreased recurrence free and overall survival. In EVN the presence of increased MIB‐1 labeling indices, mitotic activity (≥3/10 HPF), geographic necrosis and/or vascular proliferation may also be of prognostic significance (1).
One important caveat is that focal neuronal differentiation is a well‐recognized occurrence in oligodendroglioma. One prior report described four oligodendrogliomas that focally had areas of neurocytic differentiation including rosette formation (13). Others have reported immunoreactivity for neuronal markers in oligodendroglioma, including NR‐1 and embryonic neural cell adhesion molecule (23), as well as synaptophysin and unphosphorylated neurofilament protein (22). Synaptophysin may be positive in a minority of oligodendroglial tumors but, when present, has been said to be extracellular and focal (6), or located in areas of neurocytic differentiation (13). A more recent study found immunoreactivity for synaptophysin in 100% of oligodendrogliomas using more sensitive immunohistochemical methods but usually in a peculiar paranuclear pattern (21). The presence of neurocytic differentiation in the form of rosettes is not limited to oligodendrogliomas, but has been described in infiltrating astrocytomas (19) and in ependymomas (16).
A weakness of our study is, unfortunately, the relatively small number of cases, the lack of frozen material for additional studies and limited follow‐up. These are not unexpected given the rarity of EVN. Increased recognition of this tumor and optimization of molecular techniques that work in formalin‐fixed, paraffin‐embedded tissue should provide further insight into the pathogenesis of EVN specifically and the larger, more clinically relevant group of diffuse gliomas in general.
In conclusion, we have demonstrated the presence of both 1p19q loss and t(1;19) in a subset of pathologically characterized EVN. The presence of both these abnormalities may define a subgroup of tumors with histologic atypia and less favorable behavior.
ACKNOWLEDGMENTS
This work was supported in part by NIH Training Grant T32 NS07494‐04 (FJ Rodriguez).
The authors also thank the Cytogenetic and the Tissue and Cell Molecular Analysis Shared Resources of the Mayo Clinic for excellent technical assistance, as well as Sarah Jenkins from the Division of Biostatistics, for helpful suggestions with the manuscript.
REFERENCES
- 1. Brat DJ, Scheithauer BW, Eberhart CG, Burger PC (2001) Extraventricular neurocytomas: pathologic features and clinical outcome. Am J Surg Pathol 25:1252–1260. [DOI] [PubMed] [Google Scholar]
- 2. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR et al (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479. [DOI] [PubMed] [Google Scholar]
- 3. Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, Brachman D et al (2006) Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 24:2707–2714. [DOI] [PubMed] [Google Scholar]
- 4. Ducray F, Idbaih A, Reynies A, Bieche I, Thillet J, Mokhtari K et al (2008) Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile. Mol Cancer 7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Figarella‐Branger D, Soylemezoglu F, Burger P (2007) Central neurocytoma and extraventricular neurocytoma. In: Who Classification of Tumours of the Central Nervous System, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 106–109. International Agency For Research On Cancer: Lyon, France. [Google Scholar]
- 6. Fujisawa H, Marukawa K, Hasegawa M, Tohma Y, Hayashi Y, Uchiyama N et al (2002) Genetic differences between neurocytoma and dysembryoplastic neuroepithelial tumor and oligodendroglial tumors. J Neurosurg 97:1350–1355. [DOI] [PubMed] [Google Scholar]
- 7. Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD, Murphy KM (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 65:988–994. [DOI] [PubMed] [Google Scholar]
- 8. Jenkins RB, Curran W, Scott CB, Cairncross G (2001) Pilot evaluation of 1p and 19q deletions in anaplastic oligodendrogliomas collected by a national cooperative cancer treatment group. Am J Clin Oncol 24:506–508. [DOI] [PubMed] [Google Scholar]
- 9. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861. [DOI] [PubMed] [Google Scholar]
- 10. Leenstra JL, Rodriguez FJ, Frechette CM, Giannini C, Stafford SL, Pollock BE et al (2007) Central neurocytoma: management recommendations based on a 35‐year experience. Int J Radiat Oncol Biol Phys 67:1145–1154. [DOI] [PubMed] [Google Scholar]
- 11. Mrak RE, Yasargil MG, Mohapatra G, Earel J Jr., Louis, DN (2004) Atypical extraventricular neurocytoma with oligodendroglioma‐like spread and an unusual pattern of chromosome 1p and 19q loss. Hum Pathol 35:1156–1159. [DOI] [PubMed] [Google Scholar]
- 12. Mukasa A, Ueki K, Ge X, Ishikawa S, Ide T, Fujimaki T et al (2004) Selective expression of a subset of neuronal genes in oligodendroglioma with chromosome 1p loss. Brain Pathol 14:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM (2002) Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropathol Exp Neurol 61:947–955. [DOI] [PubMed] [Google Scholar]
- 14. Perry A, Fuller CE, Banerjee R, Brat DJ, Scheithauer BW (2003) Ancillary FISH analysis for 1p and 19q status: preliminary observations in 287 gliomas and oligodendroglioma mimics. Front Biosci 8:a1–9. [DOI] [PubMed] [Google Scholar]
- 15. Rades D, Schild SE, Fehlauer F (2004) Prognostic value of the MIB‐1 labeling index for central neurocytomas. Neurology 62:987–989. [DOI] [PubMed] [Google Scholar]
- 16. Rodriguez FJ, Scheithauer BW, Robbins PD, Burger PC, Hessler RB, Perry A et al (2007) Ependymomas with neuronal differentiation: a morphologic and immunohistochemical spectrum. Acta Neuropathol 113:313–324. [DOI] [PubMed] [Google Scholar]
- 17. Soylemezoglu F, Scheithauer BW, Esteve J, Kleihues P (1997) Atypical central neurocytoma. J Neuropathol Exp Neurol 56:551–556. [DOI] [PubMed] [Google Scholar]
- 18. Taruscio D, Danesi R, Montaldi A, Cerasoli S, Cenacchi G, Giangaspero F (1997) Nonrandom gain of chromosome 7 in central neurocytoma: a chromosomal analysis and fluorescence in situ hybridization study. Virchows Arch 430:47–51. [DOI] [PubMed] [Google Scholar]
- 19. Teo JG, Gultekin SH, Bilsky M, Gutin P, Rosenblum MK (1999) A distinctive glioneuronal tumor of the adult cerebrum with neuropil‐like (including “rosetted”) islands: report of 4 cases. Am J Surg Pathol 23:502–510. [DOI] [PubMed] [Google Scholar]
- 20. Tong CY, Ng HK, Pang JC, Hu J, Hui AB, Poon WS (2000) Central neurocytomas are genetically distinct from oligodendrogliomas and neuroblastomas. Histopathology 37:160–165. [DOI] [PubMed] [Google Scholar]
- 21. Vyberg M, Ulhoi BP, Teglbjaerg PS (2007) Neuronal features of oligodendrogliomas—an ultrastructural and immunohistochemical study. Histopathology 50:887–896. [DOI] [PubMed] [Google Scholar]
- 22. Wharton SB, Chan KK, Hamilton FA, Anderson JR (1998) Expression of neuronal markers in oligodendrogliomas: an immunohistochemical study. Neuropathol Appl Neurobiol 24:302–308. [DOI] [PubMed] [Google Scholar]
- 23. Wolf HK, Buslei R, Blumcke I, Wiestler OD, Pietsch T (1997) Neural antigens in oligodendrogliomas and dysembryoplastic neuroepithelial tumors. Acta Neuropathol 94:436–443. [DOI] [PubMed] [Google Scholar]