

Abstract
Rationale
Plakophilin-2 (PKP2) is an essential component of the cardiac desmosome. Recent data show it interacts with other molecules of the intercalated disc. Separate studies show preferential localization of the voltage-gated sodium channel (NaV1.5) to this region.
Objective
to establish the association of PKP2 with sodium channels and its role on action potential propagation.
Methods and Results
Biochemical, patch clamp and optical mapping experiments demonstrate that PKP2 associates with NaV1.5, and that knockdown of PKP2 expression alters the properties of the sodium current, and the velocity of action potential propagation in cultured cardiomyocytes.
Conclusions
These results emphasize the importance of intermolecular interactions between proteins relevant to mechanical junctions, and those involved in electrical synchrony. Possible relevance to the pathogenesis of arrhythmogenic right ventricular cardiomyopathy is discussed.
Keywords: Plakophilin-2, Intercalated disc, ARVC, Cardiac desmosomes
INTRODUCTION
A high resolution image of the site of end-end contact between cardiomyocytes reveals an electron-dense organization called “the intercalated disc.” Its classical definition involves three structures: desmosomes and adherens junctions, providing mechanical coupling, and gap junctions, allowing electrical/metabolic synchronization between cells. Recent studies show that other molecules, not directly involved in intercellular coupling, also reside preferentially at the intercalated disc. Among them is NaV1.5, the major α subunit of the cardiac sodium channel.1 Here, we ask whether NaV1.5 and the desmosomal protein plakophilin-2 (PKP2) co-exist in the same molecular complex, and whether loss of PKP2 expression affects a) the amplitude and kinetics of the sodium current, and b) action potential propagation in a monolayer of cardiomyocytes. Our data demonstrate a functional cross-talk between a protein defined in the context of intercellular junctions (PKP2), and another one that is fundamental to the electrical behavior of the single myocyte.
MATERIALS AND METHODS
Specifics can be found in online supplement.
RESULTS AND DISCUSSION
Initial experiments aimed at whether NaV1.5 and PKP2 are present in the same molecular complex. A recombinant protein formed by GST concatenated to the head domain of PKP2 (GST-PKP2-H) was bound to glutathione beads, and presented to an adult heart lysate. The precipitate was immunoblotted for NaV1.5 (Figure 1A). GST, bound to beads but not PKP2-concatenated, was used as control (first two lanes). PKP2-H pulled-down NaV1.5 from heart lysate, suggesting a physical interaction, direct or indirect, between the two proteins. Interaction between NaV1.5 and native PKP2 was tested by co-immunoprecipitation. NaV1.5 was precipitated by antibody-coated beads (Figure 1B, top). Western blots demonstrated PKP2 in the precipitate (bottom). Immunofluorescence studies in freshly dissociated myocytes showed co-localization of NaV1.5 with PKP2 at the site originally occupied by the intercalated disc (Figure 1C and 1E). This subcellular distribution shifted with time in culture. After six days (timeframe required for silencing experiments; see below), the density of immunoreactive signal at the cell ends decreased, coincident with the appearance of punctate immunoreactive spots on the lateral membranes, better resolved for PKP2 staining (1D and 1F). NaV1.5 staining was less sharply defined, making it difficult to establish the extent of co-localization, though a coincidence of signals could be found (Figure 1F). Protein re-localization did not affect the kinetics of INa (online Figure I). Next, we assessed the properties of INa as a function of PKP2 expression.
Figure 1.
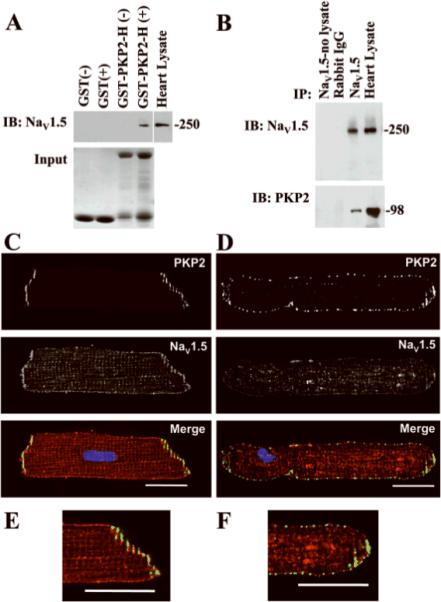
A: Head domain of PKP2 (PKP2-H; amino acids 1-to-335) concatenated to GST pulled down NaV1.5 from adult heart lysate. Top: western blot for NaV1.5. Bottom: Coomassie-blue staining of parallel gel from same samples demonstrating abundance of GST(first two lanes) or GST-PKP2-H (lanes 3 and 4). Symbols “+” and “−“, exposure, or not, of heart lysate to GST proteins. B: Co-immunoprecipitation of PKP2 with NaV1.5. Immunoblots (IB) for NaV1.5 (top) and PKP2 (bottom) from samples exposed to protein A/G beads coated with rabbit IgG (negative control), or NaV1.5 antibody. Sample not exposed to heart lysate used as control. Heart lysate (1:12 dilution, top panel; 1:6 dilution, bottom panel) ran as positive control. Numbers on right, molecular weights (kDa). C and E: Immunolocalization of NaV1.5 (red) and PKP2 (green) in isolated adult cardiocytes within 24 hours after dissociation. D and F: Day-6 after dissociation. Calibration bars: 20μm.
PKP2 level was decreased (<80% from control) by use of shRNA. In a first group, a mixture of 4 separate oligonucleotides, designed for selective PKP2 mRNA knockdown (KD) was introduced into myocytes using a transfection reagent (Dharmafect-1). Alternative non-targeted siRNA was used as control (ϕKD; details online). For every experiment, PKP2 silencing was confirmed by immunoblot (online Figure II). Sodium currents were recorded by voltage clamp (methods online). Figure 2A shows an example of currents obtained from cells either untreated (UNT) or treated with an oligonucleotide mixture. Composite data are displayed in Figures 2B–D. Treatment with control ϕKD constructs did not affect current parameters. However, a decrease in peak current density (2B), a shift in voltage dependence of steady-state inactivation (2C), and a prolongation of time-dependence of recovery from inactivation (2D) were observed in PKP2-silenced cells.
Figure 2.
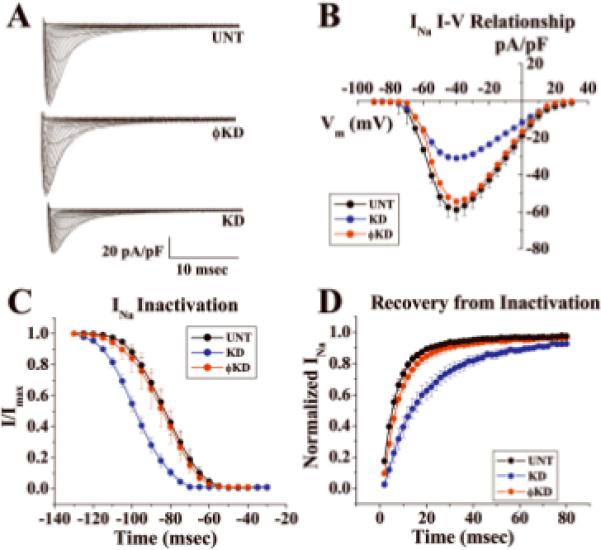
Voltage clamp data from adult cardiomyocytes after PKP2 knockdown (KD). Cells untreated (UNT) or treated with non-silencing constructs (ϕKD) used as control. A: Currents elicited by increasing voltage steps from holding potential −120 mV. B: Mean amplitude of peak inward current density as function of membrane potential during same voltage steps as in A. N=6, 6 and 6 for UNT, ϕKD and KD, respectively. C: Voltage dependence of steady-state inactivation. Cells held at Vm from −130 to −40 mV followed by 30ms test pulse to −40mV. Voltage corresponding to 50% inactivation (V1/2): −81.51±2.62, −99.84±2.07 and −85.01±3.54 For UNT, KD and ϕKD (Mean+/−SD; n=6, 6 and 6; p values: 0.100 UNTvsϕKD; .009 UNTvsKD; 0.015 ϕKDvsKD). D: Time course of recovery from inactivation. Amplitude of current elicited by S2 relative to amplitude elicited by S1 as function of S1-S2 interval. Holding potential:−120mV. Pulse potential:−40mV; curves fitted by exponential functions. Time constants: UNT:Tau1:5.03msec, Tau2:20.9msec; KD:Tau1:11.51msec, Tau2:43.7msec; ϕKD:Tau1:6.5msec; Tau2:37.5msec. N=6 (UNT), 6 (KD), 6 (ϕKD).
To reduce the possibility of off-target effects, additional experiments were conducted where a single silencing construct, of different sequence than those in the KD mixture and yet selective for PKP2, was transferred into myocytes via adenoviral infection (shRNA). Results were compared with those obtained from cells untreated (UNT), or treated with a non-silencing construct (ϕshRNA2). These results (online Figures III and IV), consistently demonstrated that loss of PKP2 expression associated with reduced peak current density, negatively shifted voltage-dependence of inactivation, and prolonged recovery from inactivation of sodium currents. Overall, we show that loss of PKP2 expression affects sodium current properties, regardless of the experimental procedure to achieve PKP2 knockdown.
Previously, we demonstrated that loss of PKP2 expression in neonatal rat ventricular myocytes (NRVMs) causes Cx43 remodeling,2 and an ~60% decrease in cell-cell dye coupling.2 We predicted that, combined with the change in sodium current function reported here, loss of PKP2 expression would significantly affect propagation properties in cardiomyocytes. Optical mapping experiments in monolayers of NRVMs revealed that loss of PKP2 expression caused slowing of action potential propagation, rate-dependent activation failure and arrhythmic behavior. Results are shown in Figure 3. Cells were treated with adenovirus containing either the ϕshRNA or the PKP2-shRNA constructs. We chose this method of silencing for consistency with our previous studies.2 Cells were paced at constant frequency from a stimulating electrode in the center of the dish. Only quiescent preparations were utilized for the study. Care was taken to minimize the presence of fibroblasts. Average conduction velocity in control monolayers paced at 1 Hz was 24.55 cm/s (n=9). Examples of isochrone maps from preparations treated with either ϕshRNA or PKP2-shRNA are shown in 3A and 3B, respectively. A Western blot demonstrating loss of PKP2 is shown in online Figure V. Loss of PKP2 expression resulted in significant decrease in conduction velocity, shown by the crowding of isochrone lines (Figure 3B). A plot of average conduction velocity as a function of pacing frequency under control conditions (black) or after treatment with either non-silencing (ϕshRNA; red) or silencing PKP2-shRNA (blue), is shown in 3C. Consistent with the increase in sodium current amplitude in single ventricular myocytes after adenoviral treatment (Online Figure IV), we observed an increase in average conduction velocity in monolayers treated with ϕshRNA virus. In contrast, we observed a large decrease in conduction velocity in preparations treated with PKP2-shRNA. In addition, a 1:1 stimulus:response capture at pacing frequencies higher than 8 Hz was possible in a fraction of preparations either untreated, or treated with ϕshRNA, whereas we failed to obtain 1:1 capture in all PKP2 knockdown monolayers. Instead, we observed non-paced sustained reentrant activity within the preparation (see phase map in Figure 3D). This is the first report demonstrating a link between loss of PKP2 expression, impaired cardiac propagation, and loss of electrical synchrony.
Figure 3.
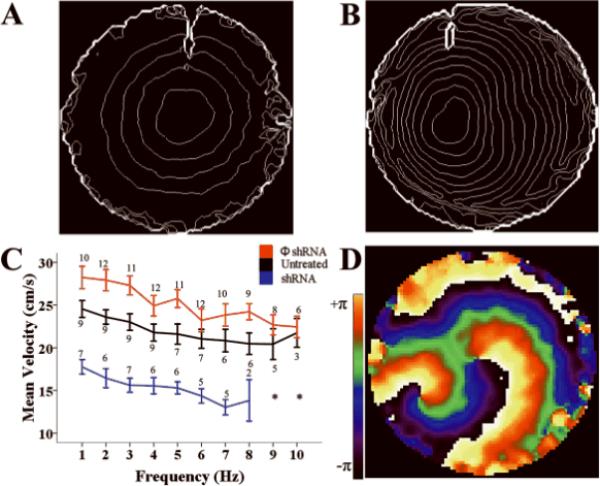
Optical mapping of propagation in monolayers of NRVMs. A–B: Isochrone maps (lines at 10 ms intervals) from monolayers treated with non-silencing construct (A) or silenced for PKP2 (B). C: Conduction velocity as function of pacing frequency. Stimulus frequency initiated at 1 or 2 Hz and progressed in increasing steps (1–2 Hz) to 6 Hz, and in 1 Hz thereafter, up to 10 Hz. “n” values by each data point. Decreasing n values after 6 Hz resulted from failure to capture. Asterisk notes failure to capture in all preparations. In range 1–7 Hz, p<0.001 for all shRNA vs ϕshRNA. p<0.005 for all shRNA vs UNT except at 4 and 6 Hz (p=0.007 and 0.02, respectively). For UNT vs ϕshRNA p<0.05 at 2, 3 and 5 Hz; p=0.03 at 8Hz. otherwise, p=NS. D: Phase map of spontaneous reentrant activity in an shRNA-treated preparation.
The relative contributions of decreased junctional conductance versus sodium current on the observed changes in conduction velocity remain undefined. Cable equations predict a decrease in conduction velocity for an increase in axial resistivity.3 Yet, studies in genetically-modified animals indicate that a 50% decrease in Cx43 content, and a concurrent decrease in electrical coupling, are not enough to significantly decrease conduction velocity, perhaps due to the relatively large contribution of myoplasmic resistivity to the total internal resistance (ref4 and others within). Likely, the observed decrease in conduction velocity resulting from loss of PKP2 expression is consequent to both, decreased electrical coupling2 and decreased sodium current, though dissecting the relative contributions will require further experimentation. In addition, our current results do not discard a possible effect of PKP2 silencing on membrane potential; membrane depolarization could be another factor leading to slow conduction velocity in these preparations. Future experiments will be necessary to address this possibility. Our data suggest a link between three components of the intercalated disc: desmosomes, gap junctions, and the voltage gated sodium channel NaV1.5 complex. Crosstalk between PKP2 and Cx43, and co-precipitation of Cx43 and NaV1.5 have been demonstrated.2,5 Yet, this is the first evidence of association between PKP2 expression and function of the sodium channel complex, and the first demonstration that molecular integrity of mechanical junctions can be relevant to ion channel function. We further demonstrate that loss of PKP2 expression leads to slow conduction velocity and propensity for frequency-dependent arrhythmias. The mechanisms by which this triad (desmosomes, gap junctions, sodium channels) is linked remain unknown. We emphasize that, because of the time required for PKP2 silencing, patch-clamp experiments were carried out within a time frame that overlaps with intercalated disc remodeling.6 As such, the PKP2 –NaV1.5 interaction did not occur within the confines of the intercalated disc. Yet, it was only after PKP2 knockdown that the change in INa properties became apparent. The latter indicates that during intercalated disc remodeling, PKP2 retains a physical interaction, direct or indirect, with NaV1.5, which carries a functional effect. We speculate that PKP2 may be important in the association of NaV1.5 with its non-covalently linked□ subunit(s), and/or the cytoskeletal adaptor protein, ankyrin-G; alternatively (or in addition) a direct PKP2-Nav1.5 association with impact on the biophysical properties of the channel cannot be discarded. Furthermore, whether these interactions occur in the heart in vivo, and/or in the confines of the intercalated disc of an intact heart, remains to be demonstrated. Of relevance, mutations in PKP2 are linked to arrhythmogenic right ventricular cardiomyopathy (ARVC), an inherited disease associated with ventricular arrhythmias and sudden death in the young. The mechanisms by which mutations in mechanical junction proteins affect electrical synchrony remain undefined. Whether the results presented here, in an in vitro system, bear relevance to the pathogenesis of ARVC is a tantalizing hypothesis for future investigation.
Supplementary Material
Acknowledgments
Source of Funding: NIH-GM057691-12, NIH-HL039707, NIH-HL087226 (MD), and NIH MH 059980 (LLI).
Glossary
Non-standard Abbreviations and Acronyms
- (PKP2)
Plakophilin 2
- (NaV1.5)
Sodium alpha subunit 1.5
- (GST)
Gluthathione Sepharose Transferase
- (GST-PKP2-H)
GST concatenated to the head domain of PKP2, amino acids 1–355
- (KD)
PKP2 Knockdown
- (ϕKD)
Non-targeted Knockdown
- (UNT)
Untreated
- (NRVMs)
Neonatal Rat Ventricular Myocytes
Footnotes
Disclosures: None
REFERENCES
- 1.Roden DM, Balser JR, George AL, Jr, Anderson ME. Cardiac ion channels. Annu Rev Physiol. 2002;64:431–475. doi: 10.1146/annurev.physiol.64.083101.145105. [DOI] [PubMed] [Google Scholar]
- 2.Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 2007;101:703–711. doi: 10.1161/CIRCRESAHA.107.154252. [DOI] [PubMed] [Google Scholar]
- 3.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 4.Beauchamp P, Choby C, Desplantez T, de Peyer K, Green K, Yamada KA, Weingart R, Saffitz JE, Kléber AG. Electrical propagation in synthetic ventricular myocyte strands from germline connexin43 knockout mice. Circ Res. 2004;95:170–178. doi: 10.1161/01.RES.0000134923.05174.2f. [DOI] [PubMed] [Google Scholar]
- 5.Malhotra JD, Thyagarajan V, Chen C, Isom LL. Tyrosine-phosphorylated and nonphosphorylated sodium channel beta1 subunits are differentially localized in cardiac myocytes. J Biol Chem. 2004;279:40748–40754. doi: 10.1074/jbc.M407243200. [DOI] [PubMed] [Google Scholar]
- 6.Kostin S, Hein S, Bauer EP, Schaper J. Spatiotemporal development and distribution of intercellular junctions in adult rat cardiomyocytes in culture. Circ Res. 1999;85:154–167. doi: 10.1161/01.res.85.2.154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.