

Abstract
Altered redox homeostasis involved in the control of cancer cell survival and proliferative signaling represents a chemical vulnerability that can be targeted by prooxidant redox intervention. Here, we demonstrate that the redox dye 2,6-dichlorophenolindophenol (DCPIP) may serve as a prooxidant chemotherapeutic targeting human melanoma cells in vitro and in vivo. DCPIP-apoptogenicity observed in the human melanoma cell lines A375 and G361 was inversely correlated with NAD(P)H:quinone oxidoreductase (NQO1) expression levels. In A375 cells displaying low NQO1 activity, DCPIP induced apoptosis with procaspase-3 and PARP cleavage, whereas G361 cells expressing high levels of enzymatically active NQO1 were resistant to DCPIP-cytotoxicity. Genetic (siRNA) or pharmacological (dicoumarol) antagonism of NQO1 strongly sensitized G361 cells to DCPIP apoptogenic activity. DCPIP-cytotoxicity was associated with the induction of oxidative stress and rapid depletion of glutathione in A375 and NQO1-modulated G361 cells. Expression array analysis revealed a DCPIP-induced stress response in A375 cells with massive up-regulation of genes encoding Hsp70B’ (HSPA6), Hsp70 (HSPA1A), heme oxygenase-1 (HMOX1), and early growth response protein 1 (EGR1) further confirmed by immunodetection. Systemic administration of DCPIP displayed significant antimelanoma activity in the A375 murine xenograft model. These findings suggest feasibility of targeting tumors that display low NQO1 enzymatic activity using DCPIP.
Keywords: melanoma; oxidative stress; NQO1; 2,6-dichlorophenolindophenol; Hsp70B’; xenograft
1. Introduction
Recent research indicates that redox dysregulation in melanoma and other cancer cells represents a chemical vulnerability that can be targeted by small molecule redox chemotherapeutics with prooxidant activity. According to this hypothesis, prooxidant pharmacological agents substantially increase cellular reactive oxygen species (ROS) and thereby induce deviations from redox homeostasis that do not reduce viability of untransformed cells, but cannot be tolerated by malignant cells already under high constitutive oxidative stress [1–5]. A variety of small molecule prooxidants containing redox active pharmacophores, such as organic endoperoxides [6, 7], Michael acceptor-and quinone-based electrophiles [8–11], and redox cyclers [12–14], have shown chemotherapeutic efficacy in cell culture-based systems and animal xenograft models [3, 4]. In addition, anticancer activity of investigational prooxidant chemotherapeutics, including artemisinin [7, 15], disulfiram [16], and elesclomol [17], is currently evaluated in advanced clinical trials [3, 5, 18].
Differential redox regulation between untransformed primary melanocytes and melanoma cells is known to alter proliferative and survival signaling through ROS-inhibition of the tumor suppressor phosphatase PTEN and ROS-activation of NFκB transcriptional activity, contributing to the notorious chemoresistance of metastatic melanoma and potentially providing a therapeutic opportunity for pharmacological redox intervention [5, 19–21]. Led by the emerging chemotherapeutic benefit provided by experimental prooxidants, we aimed at the identification of promising redox pharmacophores with acceptable systemic toxicity profile that would target melanoma cells by induction of oxidative stress and focused our interest on the established redox dye 2,6-dichlorophenolindophenol {2,6-dichloro-4-[(p-hydroxyphenyl)imino]-2,5-cyclohexadien-1-one sodium salt, DCPIP; CAS# 620-45-1}. This membrane-permeable dihalogenated 1,4-benzoquinoneimine-type oxidant (E’o (DCPIP) = + 0.22 V; log p (octanol/water) = 0.13) is widely used as a specific standard substrate for the colorimetric determination of cellular NAD(P)H:quinone oxidoreductase (NQO1; EC 1.6.99.2, also referred to as DT-diaphorase) enzymatic activity, thereby reductively converted into the hydroquinone-leucoform [23, 24]; DCPIP also serves as an oxidizing reactant (Tillman’s reagent) for analytical redox titration of the reducing agent L-ascorbic acid (Fig. 1) [25]. As a redox probe DCPIP can serve as an electron acceptor for electron carriers of the mitochondrial respiratory chain and as a substrate (Hill reagent) for chloroplast-mediated photoreduction [26–28]. Importantly, other simple quinone systems with related redox chemistry including menadione [E’o (menadione) = + 0.14 V] have shown efficacy as prooxidant experimental cancer chemotherapeutics [8], and antimelanoma activity of N-acetyl-para-aminophenol (acetaminophen) tested in ongoing clinical studies has recently been attributed to glutathione depletion by the tyrosinase-derived o-quinone-metabolite [29, 30].
Figure 1. The dihalogenated benzoquinoneimine redox dye 2,6-dichlorophenolindophenol (DCPIP).
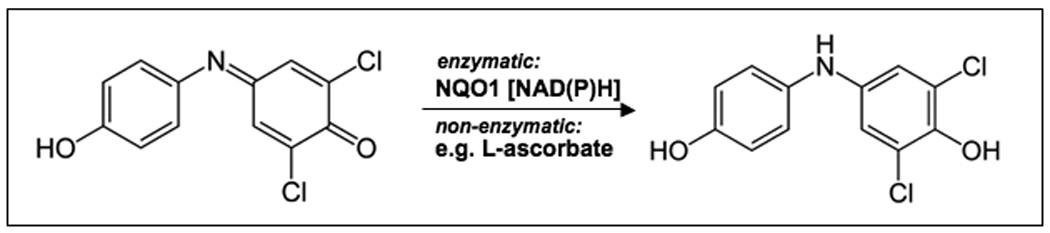
Reduction of the blue dye DCPIP (λmax = 600 nm) with formation of the colorless p-aminophenol hydroquinone leuco form occurs through enzymatic transformation by NAD(P)H quinone oxidoreductase (NQO1) or nonenzymatic reaction with reducing agents such as ascorbic acid.
Based on prooxidant redox reactivity and drug-like properties of DCPIP that include chemical stability, systemic deliverability, membrane permeability, and low systemic toxicity established earlier in mice (LD50 = 180 mg/kg; intravenous administration [31]), we tested feasibility of using this 1,4-benzoquinoneimine-derivative as an experimental redox chemotherapeutic targeting human melanoma cells in vitro and in vivo. Here we demonstrate that (I) DCPIP induces apoptosis in cultured melanoma cells associated with upregulation of cellular oxidative stress and oxidative stress-induced gene expression, that (II) DCPIP cytotoxicity in human melanoma cell lines is inversely correlated with NQO1 expression levels, and that (III) systemic administration of DCPIP displays significant antimelanoma activity in an animal model of the disease.
2. Material and methods
2.1.Materials
All chemicals were purchased from Sigma Chemical Co (St. Louis, MO, USA). The cell-permeable pancaspase inhibitor Z-VAD-(OMe)-fmk was from Calbiochem-Novabiochem (San Diego, CA, USA).
2.2.General cell culture
G-361 Human metastatic melanoma cells from ATCC (Manassas, VA, USA) were cultured in McCoy’s 5a medium containing 10% bovine calf serum (BCS). A375 human metastatic melanoma cells (ATCC) were cultured in RPMI medium containing 10% BCS and 2 mM L-glutamine. Cells were maintained at 37 °C in 5% CO2, 95% air in a humidified incubator.
2.3.Human melanoma SCID mouse xenograft model
A375 human melanoma cells were grown in HyQ RPMI-1640 media supplemented with 10% fetal bovine serum, and o maintained in 5% CO2–95% air humidified atmosphere at 37°C. Subconfluent cells were harvested by using 0.25% trypsin-EDTA. Cells (>90% viability) were resuspended at the concentration of 10 × 106 cells /100µl of sterile saline. A SCID mouse colony was developed at the University of Arizona using original SCID (C.B-17/IcrACCSCID) obtained from Taconic (Germantown, NY, USA). The mice were housed in microisolator cages (Allentown Caging Equipment Company, Allentown, NJ, USA) and maintained under specific pathogen-free conditions. Every month mice were screened by ELISA serology for mycoplasma, mouse hepatitis virus, pinworms, and Sendai virus and tested negative. SCID mice 6–8 weeks of age were bled (<200 µl) by retroorbital puncture in order to screen for the presence of mouse IgG using an ELISA. Only mice with IgG levels ≤ 20 ug/ml were used for the experiments. The mice ate NIH-31 irradiated pellets (Tekland Premier, Madison, WI, USA) and received autoclaved water. Animal facilities are approved by the Association for the Assessment and Accreditation of Laboratory Animal Care International and in accordance with United States Department of Agriculture, Department of Health and Human Services, and NIH regulations. A375 cell injections (10 × 106 cells) were given subcutaneously on the mouse's lower right flank on day 0, and after tumors became established (~65 mm3 ) mice were pair-matched into the treatment groups. The following day, treatment with PBS or PBS only was initiated. The chemotherapeutic test agent DCPIP (1mg/ml PBS) was prepared and administered by intraperitoneal injection in less than 1 hour. DCPIP (low dose group: 4 mg/kg/d, 100 µl, q.d., n=12; high dose group: 16 mg/kg/d, 200 µl, b.i.d., n=11) was given on days 1–7 and 10–12 post the day of pair-matching, whereas control animals received carrier only (PBS, 200 µl, b.i.d., n=12). Subcutaneous tumors were measured twice weekly for tumor volume estimation (mm3) in accordance with the formula (a2 × b)/2 where a is the smallest diameter and b is the largest diameter. The mice were sacrificed individually by CO2 when the tumors reached a volume of 2000 mm3. Tumor growth curves were obtained by determining average tumor volumes until day 30 after cell injection, and data points were analyzed using the two-sided Student’s t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001). All procedures were completed in accordance with the University of Arizona Institutional Animal Care and Use Committee (IACUC) protocol (# 07–029, approved May 24, 2007).
2.4. Cell proliferation assay
Cells were seeded at 10,000 cells/dish on 35-mm dishes. After 24 h, cells were treated with test compound. Cell number at the time of compound addition and 72 h later were determined using a Z2 Analyzer (Beckman Coulter, Inc., Fullerton, CA, USA). Proliferation was compared with cells that received mock treatment. The same methodology was used to establish IC50 values (drug concentration that induces 50% inhibition of proliferation of treated cells within 72 h exposure ± SD, n = 3) of anti-proliferative potency.
2.5. Apoptosis analysis
Viability and induction of cell death (early and late apoptosis/necrosis) were examined by annexin-V-FITC/propidium iodide (PI) dual staining of cells followed by flow cytometric analysis as published previously [32]. Cells (100,000) were seeded on 35 mm dishes and received drug treatment 24 hours later. Cells were harvested at various time points after treatment and cell staining was performed using an apoptosis detection kit according to the manufacturer’s specifications (APO-AF, Sigma, St. Louis, MO, USA).
2.6. Caspase-3 activation assay
Treatment-induced caspase-3 activation was examined in A375 melanoma cells using a cleaved/activated caspase-3 (asp 175) antibody (Alexa Fluor 488 conjugate, Cell Signaling, Danvers, MA, USA) followed by flow cytometric analysis as published recently [14]. Briefly, cells were harvested 24h after treatment, resuspended in PBS and fixed in 1% formaldehyde. Cells were then permabilized using 90% methanol and resuspended in incubation buffer (PBS, 0.5% BSA). After rinsing by centrifugation, cells were resuspended in incubation buffer (90 µl) and cleaved caspase-3 antibody (10 µl) was added. After incubation (40 min) followed by rinsing and centrifugation in incubation buffer, cells were resuspended in PBS and analyzed by flow cytometry.
2.7. PARP immunoblot analysis
Cells were treated with 40 µM DCPIP for 24 hours and lysed with RIPA buffer (100 µl, 50mM Tris-HCl, pH 7.4, 150mM NaCl, 1mM EDTA, 25% deoxycholic acid, and 1% NP-40). After sample separation (30 µg protein) by SDS-PAGE (4–15% gradient gel, Bio-Rad, Hercules, CA, USA), semidry transfer onto a nitrocellulose membrane (Optitran, Whatman, Bedford, MA, USA) was performed, followed by incubation in blocking buffer [PBST (0.1% Tween 20), 5% nonfat dry milk] for 1 hour at 25°C. Membranes were washed three times with PBST and incubated overnight at 4°C with a monoclonal rabbit anti-PARP antibody diluted 1:1000 (46D11, Cell Signaling, Danvers, MA, USA) in incubation buffer (PBST, 5% BSA). Membranes were washed three times with PBST followed by incubation for 1 hour at 25°C with HRP-linked anti-rabbit IgG antibody (Cell Signaling) diluted 1:2000 in blocking buffer. Visualization occurred by enhanced chemiluminescence.
2.8.Heme oxygenase-1 (HO-1) immunoblot analysis
One day before treatment, 2×106 cells were seeded in T-75 flasks. Cell growth medium was replaced 24 h after seeding, followed by addition of test compounds 60 min after medium change. Cells were incubated for 24 h (37 °C, 5% CO2), then washed with PBS, lysed in 1x SDS-PAGE sample buffer (200 µl, 0.375 M Tris HCl pH 6.8, 50% glycerol, 10% SDS, 5% β-mercaptoethanol, 0.25% bromophenol blue), and heated for 3 min at 95 oC. Samples (10 µL, containing approximately 45 µg total protein as determined by the BCA assay) were separated by 15% SDS-PAGE followed by immediate transfer to nitrocellulose membranes (Optitran, Whatman, Piscataway, NJ, USA). The membrane was blocked with 5% milk in 0.1% PBST for 1 h. Rabbit anti-HO-1 polyclonal antibody (Stressgen Bioreagents, Ann Arbor, MI) was used 1:5000 in 5% milk-PBST overnight at 4˚ C. The membrane was washed three times for 10 min in 0.1% PBST before adding HRP-conjugated goat anti-rabbit antibody (Jackson Immunological Research, West Grove, PA, USA) at 1:10000 dilution followed by visualization using enhanced chemiluminescence detection reagents. Equal protein loading was examined by α-actin-detection using a mouse anti-actin monoclonal antibody (Sigma) at 1:1500 dilution.
2.9.NQO1 immunoblot analysis
Cell extraction, separation by 12% SDS–PAGE, and Western analysis were performed as specified for HO-1. Immunoblot analysis was performed using a mouse anti-NQO1 monoclonal primary antibody (Abcam, Cambridge, MA, USA; 1:1000 dilution) and an HRP-conjugated goat anti-mouse secondary antibody (Jackson Immunological Research; 1:10,000 dilution).
2.10. Early Growth Response-1 (EGR-1) immunoblot analysis
Cell extraction, separation by 12% SDS–PAGE, and Western analysis were performed as specified for HO-1. Rabbit anti-EGR1 monoclonal primary antibody (44D5, Cell Signaling; 1:2000 dilution) and HRP-conjugated anti-rabbit IgG secondary antibody (Cell Signaling; 1:1500 dilution) were used.
2.11. Hsp70B’ ELISA
The enzyme-linked immunosorbent assay for Hsp70B’ was performed in 96 well format on cell lysates extracted from treated cells following kit instructions (Assay Designs, Inc., Ann Arbor, MI, USA). Briefly, 1×106 cells were seeded per T-75 flask one day before treatment. Cell growth medium was replaced 24 h after seeding, followed by addition of test compound 60 min after medium change. Cells (3×106 per group) were incubated for 24 h (37°C, 5% CO2) and then harvested, washed with PBS, and lysed in 1x extraction reagent. After protein quantification using the BCA assay, samples were diluted to a range within the Hsp70B’ standard curve and processed according to the manufacturer’s instructions. Absorbance (450 nm) was determined on a microtiter plate reader (Versamax, Molecular Devices, Sunnyvale, CA, USA). Data represent the average of three independent experiments.
2.10. Measurement of NQO1-specific activity
Determination of NQO1 specific activity was performed according to a published standard procedure [14]. In brief, cells (2 × 106) were harvested by trysinization and resuspended in ice-cold TE (20 mM Tris-HCl with 2 mM EDTA, pH 7.4). Cells were disrupted in three cycles of freeze/thawing using liquid nitrogen and a 37°C waterbath, followed by centrifugation (12,000 g, 5 min). Protein concentration in the supernatant was determined using the BCA assay (Pierce, Rockford, IL, USA). For determination of NQO1 specific activity the reaction mixture (1 mL final volume) contained: 25 mM Tris-HCl (pH 7.4), 180 µM NADPH, BSA (0.2 mg/mL), Tween 20 [0.01 % (v/v)], and cell lysate (5 µl). The reaction was started by the addition of 2 µL 2,6-dichlorophenolindophenol (DCPIP, 20 mM stock in DMSO). Reduction of DCPIP was measured at room temperature for 1 min at 600 nm (ε = 21 × 103 M−1cm−1) with or without 20 µM dicoumarol. The dicoumarol-inhibitable part of DCPIP reduction was used to calculate NQO1 activity expressed as nmol DCPIP/mg protein/min. A minimum of triplicate cultures were assayed.
2.11. Detection of intracellular oxidative stress by flow cytometric analysis
Induction of intracellular oxidative stress by DCPIP was analyzed by flow cytometry using 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) as a sensitive non-fluorescent precursor dye according to a published standard procedure [14]. Human A375 melanoma cells were treated with DCPIP (10, 20, and 40 µM, 24 h) followed by DCFH-DA loading. To this end, cells were incubated for 60 min in the dark (37°C, 5% CO2) with culture medium containing DCFH-DA (5 µg/mL final concentration). Cells were then harvested, washed with PBS, resuspended in 300 µl PBS and immediately analyzed by flow cytometry.
2.11.Determination of total cellular glutathione content
Pharmacological modulation of intracellular glutathione content was analyzed using the photometric HT Glutathione Assay Kit (Trevigen, Gaithersburg, MD, USA) performed in 96 well format. This kinetic assay is based on the enzymatic recycling method involving glutathione reductase and DTNB (5,5’-dithiobis-2-nitrobenzoic acid, Ellman’s reagent) to produce yellow colored 5-thio-2-nitrobenzoic acid (TNB) that absorbs at 405 nm. Human A375 melanoma (1 × 106) cells were exposed to DCPIP (40 µM, 6 h) and harvested by trypsinization followed by sample processing according to the manufacturer’s instructions. Oxidized glutathione was determined separately after 4-vinylpyridine-derivatization. Glutathione content of total cellular extracts was normalized to protein content determined using the BCA assay (Pierce, Rockford, IL, USA).
2.12.siRNA transfection and NQO1 expression analysis by real time RT-PCR
G361 cells were transiently transfected with a 100 nMol pool of four siRNA oligonucleotides targeting NQO1 or a 100 nMol pool of four non-targeting siRNA oligonucleotides using the DharmaFECT 1 transfection reagent (Dharmacon RNA Technologies,Lafayette,CO, USA). The sequences of siGENOME NQO1 SMARTpool (NQO1 siRNA) [GenBank: NM 001025434] were GAAAGGACAUCACAGGUAA; GAAGGACCCUGCGAACUUU; GCAAGUCCAUCCCAACUGA; and CCGACUCUGUUCUGGCUUA. The oligos were resuspended in the Dharmacon 1X siRNA buffer and incubated in serum free media for 5 minutes. DharmaFECT 1 was also incubated in serum free media for 5 minutes prior to the addition of the siRNA oligos. The oligos were incubated with the transfection reagent for 20 minutes prior to cellular treatment. Complete media was added to the siRNA oligo mixture and the cells were incubated with the siRNAs in appropriate cell culture conditions for 72 hours. Cells were than re-transfected with another 100 nMol pool of four siRNA oligonucleotides targeting NQO1 or a 100 nMol pool of four non-targeting siRNA oligonucleotides. Twenty-four hours after the second transfection, cells were either harvested for expression analysis or plated for subsequent DCPIP treatment. For NQO1 expression analysis by real time RT-PCR, total cellular RNA (3×106 cells) was prepared using the RNEasy kit from Qiagen (Valencia, CA, USA). Reverse transcription was performed using TaqMan Reverse Transcription Reagents (Roche Molecular Systems, Branchburg, NJ, USA) and 200 ng of total RNA in a 50-µl reaction. Reverse transcription was primed with random hexamers and incubated at 25 °C for 10 minutes followed by 48 °C for 30 minutes, 95°C for 5 minutes, and a chill at 4 °C. Each PCR reaction consisted of 3.75 µl of cDNA added to 12.5 µl of TaqMan Universal PCR Master Mix (Roche Molecular Systems), 1.25 µl of gene-specific primer/probe mix (Assays-by-Design; Applied Biosystems, Foster City, CA) and 7.5 µl of PCR water. PCR conditions were: 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 15 seconds, alternating with 60 °C for 1 minute using an Applied Biosystems 7000 SDS and Applied Biosystems’ Assays On Demand primers specific to NQO1 (assay ID Hs00168547_m1) and GAPDH (assay ID Hs99999905_m1). Gene-specific product was normalized to GAPDH and quantified using the comparative (ΔΔCt) Ct method as described in the ABI Prism 7000 sequence detection system user guide [11]. Expression values were averaged across three independent experiments, and standard deviation was calculated for graphing.
2.13. Human stress and toxicity pathfinder™ RT2 profiler™ PCR expression array
After pharmacological exposure, total cellular RNA (3×106 A375 melanoma cells) was prepared according to a standard procedure using the RNeasy kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed using the RT2 First Strand kit (Superarray, Frederick, MD, USA) and 5 µg total RNA. The RT2 Human Stress and Toxicity Pathfinder™ PCR Expression Array (SuperArray) profiling the expression of 84 stress-related genes was run using the following PCR conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 sec alternating with 60 °C for 1 min (Applied Biosystems 7000 SDS). Gene-specific product was normalized to GAPDH and quantified using the comparative (ΔΔCt) Ct method as described in the ABI Prism 7000 sequence detection system user guide. Expression values were averaged across three independent array experiments, and standard deviation was calculated for graphing.
2.14. Statistical Analysis
Unless indicated differently, the results are presented as means ± S.D. of at least three independent experiments. They were analyzed using the two-sided Student’s t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
3. Results
3.1. DCPIP induces apoptosis with procaspase-3 and PARP cleavage in cultured human A375 melanoma cells
First, the dose-response of induction of A375 melanoma cell death by exposure to DCPIP was assessed by flow cytometric analysis of annexinV-FITC/propidium iodide-stained cells (Fig. 2A). Exposure to DCPIP at concentrations higher than 10 µM for 24 h induced pronounced apoptosis that could be suppressed significantly by pretreatment (1 h) with the pancaspase inhibitor zVAD-fmk added before DCPIP exposure suggesting a caspase-dependent mode of DCPIP-induced cell death. This finding was confirmed, when cleavage of procaspase-3 and PARP, important markers of induction of apoptosis, were examined in A375 cells exposed to increasing doses of DCPIP (10, 20, and 40 µM, 24 h exposure) (Fig. 2B and C, respectively). At 20 and 40 µM DCPIP (24 h exposure) pronounced cleavage of procaspase 3 was detected (Fig. 2B), and complete PARP cleavage was observed (40 µM, 24 h) (Fig. 2C).
Figure 2. Exposure to DCPIP dose-dependently induces apoptosis with cleavage of procaspase-3 and PARP in human A375 melanoma cells.
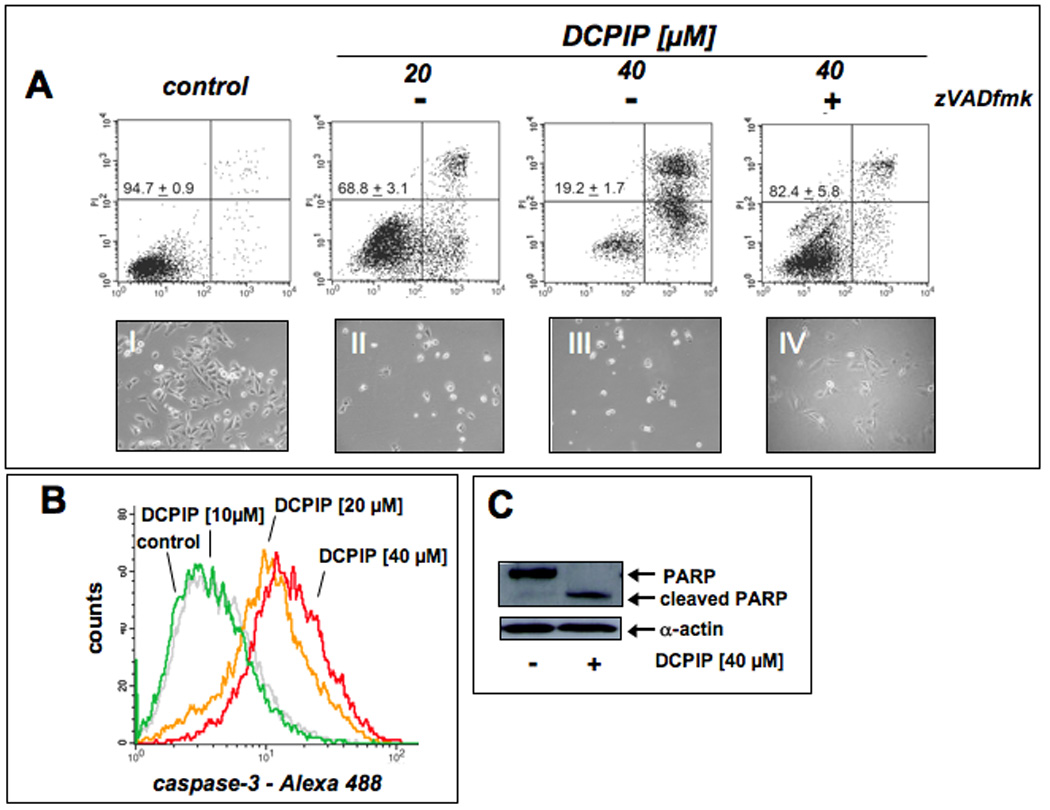
(A) Induction of cell death by exposure to increasing doses of DCPIP (20 and 40 µM, 24 h) in the absence or presence of the pancaspase inhibitor zVADfmk was assessed by flow cytometric analysis of annexinV-FITC/propidium iodide-stained cells. The numbers indicate viable cells (AV−, PI−, lower left quadrant) in percent of total gated cells (mean ± SD, n=3). Representative light microscopy pictures taken after 24 h exposure to DCPIP are shown in panels I-IV. (B) DCPIP-induced (10, 20, and 40 µM, 24 h) caspase-3 activation was examined by flow cytometric detection using an Alexa Fluor 488-conjugated monoclonal antibody against cleaved procaspase-3. One representative experiment of three similar repeats is shown. (C) DCPIP-induced (40 µM, 24 h) PARP cleavage was examined by immunoblot analysis.
Taken together, these findings demonstrate that DCPIP treatment at lower micromolar concentrations induces apoptotic cell death in A375 melanoma cells.
3.2. Exposure to the combined action of DCPIP and the NQO1 inhibitor dicoumarol induces apoptosis in cultured human G361 melanoma cells
In contrast to A375 melanoma cells, human G361 metastatic melanoma cells displayed a marked resistance to induction of apoptosis by DCPIP as assessed by flow cytometric analysis of annexinV-FITC/propidium iodide-stained cells after 24 h exposure to up to 40 µM DCPIP (Fig. 3A, upper panels). Similar results that support preferential DCPIP-sensitivity of A375 versus G361 melanoma cells were obtained when inhibition of cell proliferation in response to DCPIP treatment [IC50 (µM): 1.9 ± 0.5 (A375); 19.3 ± 3.1 (G361), 72 h continuous exposure] was assessed (data not shown).
Figure 3. Dicoumarol treatment sensitizes human G361 melanoma cells to DCPIP-induced apoptosis.
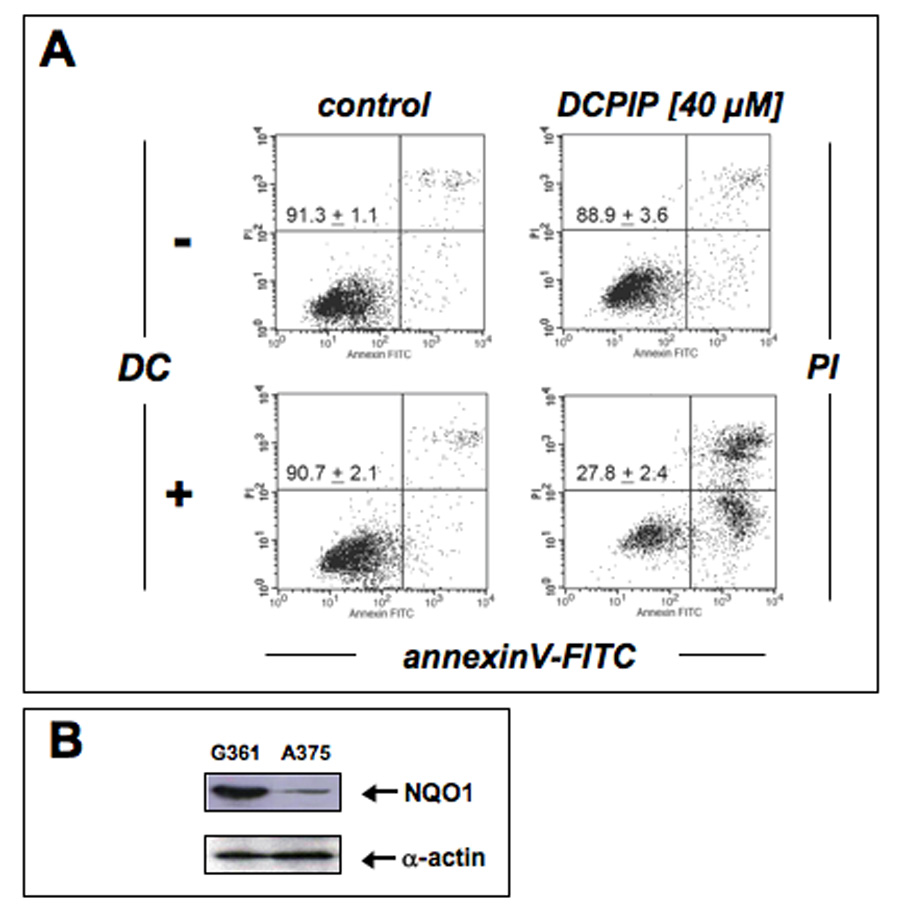
(A) Induction of cell death by exposure to DCPIP (40 µM, 24 h) in the absence or presence of the NQO1 inhibitor dicoumarol (DC, 60 µM) was assessed by flow cytometric analysis of annexinV-FITC/propidium iodide-stained cells. The numbers indicate viable cells (AV−, PI−, lower left quadrant) in percent of total gated cells (mean ± SD, n=3). (B) NQO1 protein levels in G361 and A375 cells were compared by immunoblot analysis.
Based on the established reductive metabolism of DCPIP by NQO1 [23], a flavoprotein that catalyzes the two-electron reduction of various quinones and quinone imines using NAD(P)H, thereby preventing semiquinone-dependent redox cycling and facilitating conjugation and systemic excretion [22, 33], we tested the hypothesis that NQO1-inhibition would sensitize G361 melanoma cells to DCPIP treatment. Indeed, pronounced induction of apoptosis was achieved in G361 cells exposed to the combined action of the NQO1-inhibitor dicoumarol (DC) and DCPIP (Fig. 3A, lower panels). Importantly, exposure to dicoumarol only (up to 60 µM, 24 h) was not associated with any cytotoxicity. Consistent with our earlier findings demonstrating high specific enzymatic activity of NQO1 in G361 cells (2403 ± 215 nmol DCPIP/mg protein/min, n=3) and low specific enzymatic activity in A375 cells (283 ± 37 nmol DCPIP/mg protein/min, n=3) [14], Western blot analysis confirmed differential expression levels of NQO1 in these two metastatic melanoma cell lines (Fig. 3B).
Taken together, these findings suggest that DCPIP cytotoxicity may be modulated by cellular expression and enzymatic activity of the quinone-detoxifying enzyme NQO1, i.e. differential sensitivity to DCPIP apoptogenicity as observed in A375 and G361 melanoma cells may result from differential expression levels of NQO1 and may be modulated by pharmacological NQO1 inhibition.
3.3. Genetic antagonism of NQO1 sensitizes G361 human melanoma cells to DCPIP apoptogenic activity
Based on our experiments using the pharmacological inhibitor dicoumarol as a molecular probe for the involvement of NQO1 enzymatic activity in the reductive detoxification of the quinoneimine DCPIP, we further examined the role of NQO1 in DCPIP chemoresistance using a specific genetic approach based on siRNA-mediated target modulation (Fig. 4). Knockdown of NQO1 gene expression was confirmed by real time RT-PCR analysis indicating downregulation of NQO1 mRNA by approximately 5 fold when compared to control siRNA transfected A375 cells (Fig 4B). Western-analysis further confirmed the substantial downregulation of NQO1 protein levels (Fig. 4C) that was also apparent from an approximately fourfold reduction of NQO1 specific enzymatic activity detected in cell protein extracts prepared from siNQO1- (24.5 ± 0.6 % of wildtype activity, n =3, p < 0.001) versus siControl-treated (110.7 ± 3.7 % of wildtype, n =3, p < 0.05) and untreated wildtype cells (100.0 ± 3.5 %, n =3). Remarkably, NQO1 knockdown (Fig. 4A, lower panels), but not treatment with siControl (Fig. 4A, middle panels), resulted in pronounced DCPIP-chemosensitization of G361 melanoma cells (Fig. 4A, upper panels) confirming the results obtained with the pharmacological NQO1-inhibitor dicoumarol (Fig. 3A).
Figure 4. Genetic downregulation of NQO1 expression sensitizes human G361 melanoma cells to DCPIP-induced apoptosis.
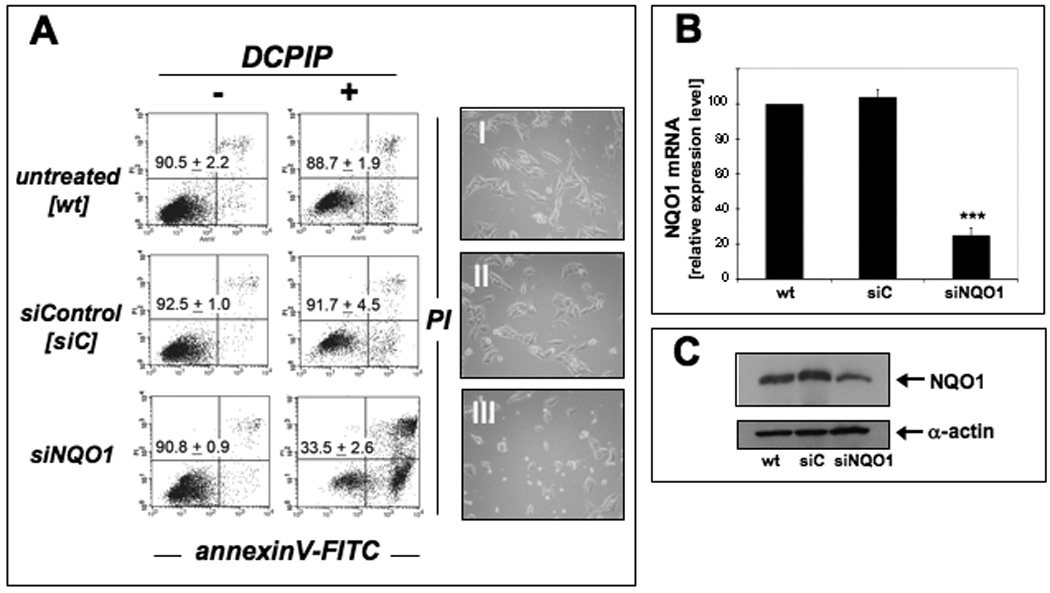
(A) Induction of cell death by exposure to DCPIP (40 µM, 24 h) was examined in G361 wild type cells (untreated, wt) and after control siRNA treatment (siControl, siC) and NQO1 siRNA knockdown (siNQO1). The numbers indicate viable cells (AV−, PI−, lower left quadrant) in percent of total gated cells (mean ± SD, n=3). Representative light microscopy pictures taken after 24 h exposure to DCPIP are shown in panels I-III. (B) NQO1-Knockdown was confirmed by expression analysis using quantitative RT-PCR (mean ± SD, n=3) and (C) immunoblot analysis as specified in Materials and Methods.
In summary, our data obtained from pharmacological and genetic target modulation studies strongly suggest that DCPIP apoptogenic potency observed in human A375 and G361 melanoma cells is a function of cellular NQO1 expression levels.
3.4. DCPIP induces oxidative stress with rapid depletion of cellular glutathione in A375 and NQO1-modulated G361 melanoma cells
Based on earlier work that has demonstrated the involvement of oxidative stress in mediating the cytotoxic effects of redox quinones [8, 22, 30, 34, 35], we examined the role of cellular oxidative stress and redox alterations in DCPIP apoptogenicity (Fig. 5). First, a dose-dependent elevation of intracellular oxidative stress could be observed in A375 melanoma cells exposed to DCPIP (10, 20, and 40 µM, 24 h) as assessed by 2’,7’-dichloro-dihydrofluorescein diacetate detection of intracellular peroxides using flow cytometry (Fig. 5A). Consistent with a mechanistic involvement of oxidative stress in the induction of apoptosis by DCPIP, preincubation (24 h) of A375 cells with the antioxidant and glutathione precursor Nα-acetyl-L-cysteine (NAC, 10 mM) strongly suppressed DCPIP-induced procaspase 3 cleavage (Fig. 5B). In addition, modulation of cellular glutathione levels in A375 and G361 melanoma cells exposed to DCPIP (40 µM) was examined at an early time point (6 h) in order to assess treatment-induced glutathione depletion that occurs independent of plasma membrane permeabilization observed at later time points during advanced stages of apoptosis (Fig. 5C and D). In A375 cells, DCPIP treatment reduced cellular glutathione levels by approximately 30%, whereas in G361 cells DCPIP-induced GSH depletion did not reach the level of statistical significance. In contrast, G361 glutathione levels were strongly reduced by approximately 70% when DCPIP treatment occurred in the presence of the NQO1 inhibitor dicoumarol (Fig. 5C). Importantly, no glutathione depletion occurred upon exposure to dicoumarol only. Genetic antagonism of NQO1 employing the siRNA methodology described above (Fig. 4) strongly sensitized G361 melanoma cells to the glutathione-depleting effects of DCPIP (Fig. 5D) consistent with a protective role of NQO1 against DCPIP cytotoxicity and glutathione depletion.
Figure 5. DCPIP induces oxidative stress in human melanoma cell lines.
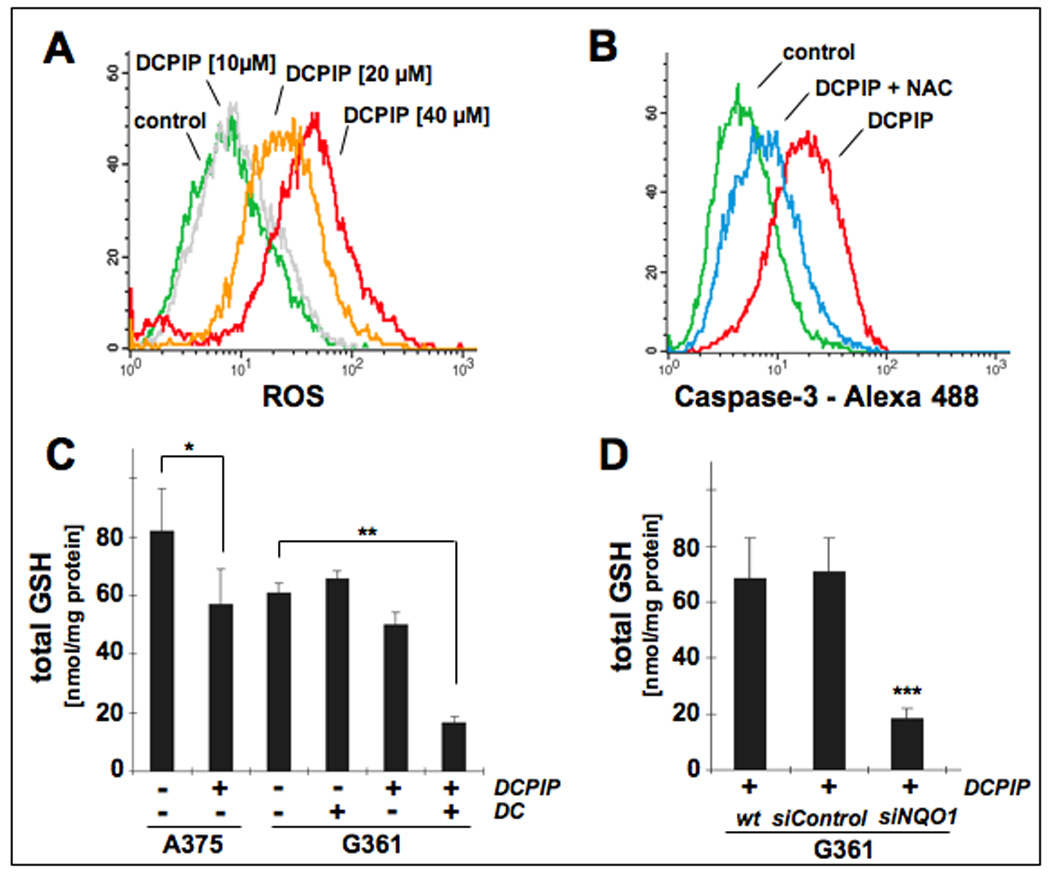
(A) Induction of intracellular oxidative stress in human A375 melanoma cells by treatment with DCPIP. Cells were exposed to DCPIP (10, 20, 40 µM, 24 h) and intracellular oxidative stress was assessed by 2’,7’-dichloro-dihydrofluorescein diacetate staining followed by flow cytometric analysis. One representative experiment of three similar repeats is shown. (B) Antioxidant protection against DCPIP-induced caspase-3 activation. Cells were pretreated with NAC (10 mM, 24 h) or left untreated. After medium change, DCPIP (40 µM) was added and caspase-3 activation was examined after another 24 h by flow cytometric detection using an Alexa Fluor 488-conjugated monoclonal antibody against cleaved procaspase-3. One representative experiment of three similar repeats is shown. (C) Modulation of intracellular glutathione content in A375 and G361 melanoma cells exposed to DCPIP (40 µM, 6h) in the absence or presence of DC (60 µM). Total glutathione content was normalized to protein content. (D) For NQO1 knockdown, G361 melanoma cells were treated with siNQO1- or siControl or left untreated as described in Materials and Methods. Intracellular glutathione content was then determined in G361 melanoma cells exposed to DCPIP (40 µM, 6h). Total glutathione content was normalized to protein content (mean ± SD, n=3).
Taken together these data suggest that induction of oxidative stress, as evidenced by upregulation of cellular peroxide levels combined with glutathione depletion and antioxidant suppression of DCPIP-induced caspase 3 activation, is associated with DCPIP-induced cytotoxicity in human A375 cells. In G361 cells DCPIP-induced glutathione depletion was observed only after pharmacological or genetic antagonism of NQO1 enzymatic activity.
3.5. DCPIP induces a broad oxidative stress response in human A375 melanoma cells
After demonstrating apoptogenic and prooxidant activity of DCPIP in simple melanoma cell culture models, modulation of stress and toxicity response gene expression was examined in A375 human melanoma cells exposed to DCPIP. The RT2 Human Stress and Toxicity Profiler™ PCR Expression Array technology (SuperArray, Frederick, MD) was applied to A375 cells exposed to DCPIP (40 µM, 24 h exposure) to assess expression of 84 stress-related genes contained on the array (Fig. 6A, left panel). DCPIP-induced gene expression changes in A375 human melanoma cells affected 15 genes on the array by at least three-fold over untreated control cells as summarized in Fig. 6A (right panel). Genes that were more than tenfold upregulated encoded the oxidative stress responsive heat shock proteins Hsp70B’ (HSPA6; 306-fold) and Hsp70 (HSPA1A; 16-fold) [36], the heat shock protein and antioxidant enzyme heme oxygenase-1 (HMOX1; 42-fold), the oxidative stress-responsive transcription factor and tumor suppressor early growth response protein 1 (EGR1; 14-fold) [37], and lymphotoxin-alpha (LTA; 14-fold), an apoptogenic member of the tumor necrosis factor family [38]. Moreover, the stress- and DNA-damage response genes DNA-damage-inducible transcript 3 (DDIT3) and growth arrest and DNA-damage-inducible alpha (GADD45A) were upregulated by approximately 4 fold in response to DCPIP exposure [39, 40].
Figure 6. DCPIP induces oxidative stress and heat shock response gene expression in human A375 melanoma cells.
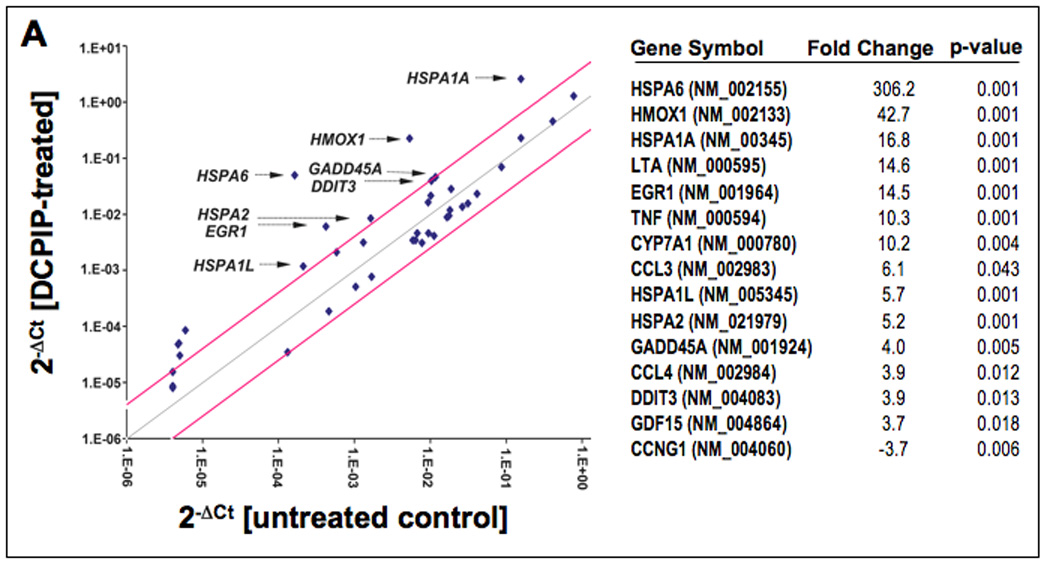
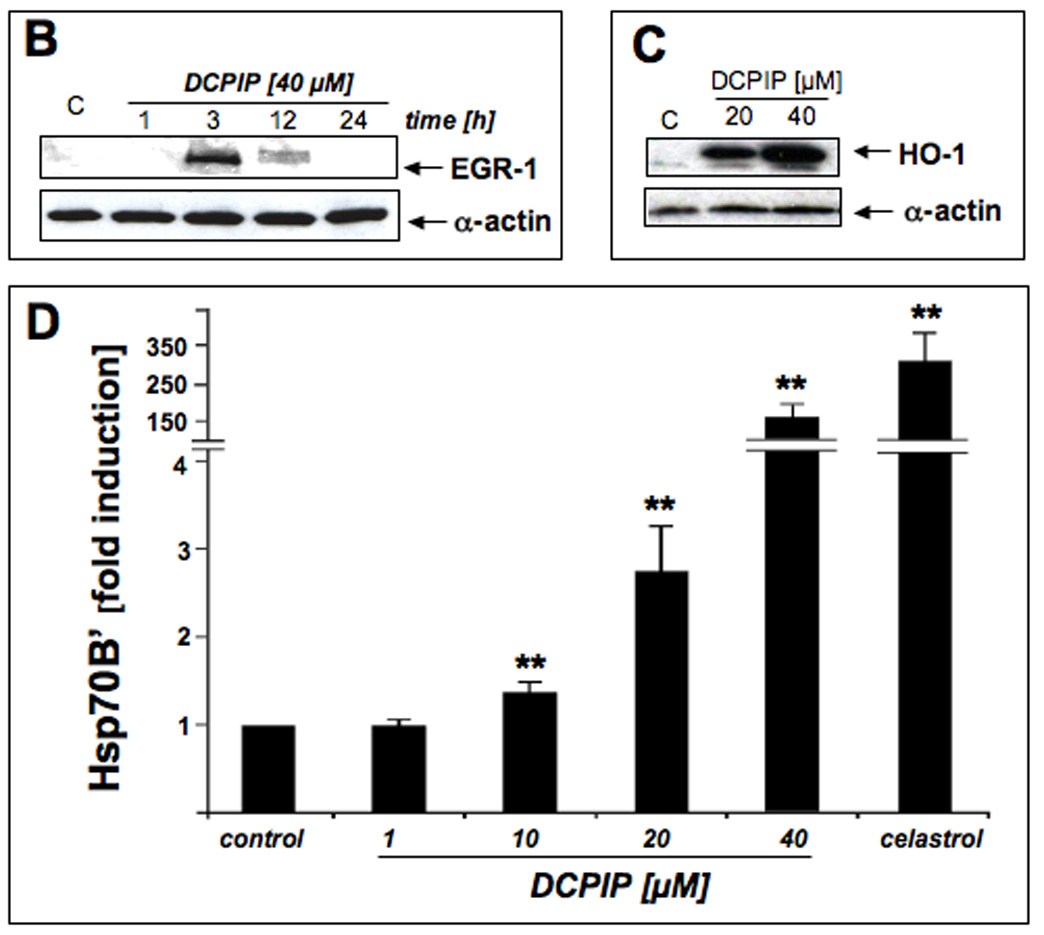
(A) DCPIP-induced gene expression changes in A375 human melanoma cells. The scatter blot (left panel) depicts differential gene expression as detected by the RT2 Human Stress and Toxicity Profiler™ PCR Expression Array technology profiling the expression of 84 (oxidative) stress- and toxicity related genes after DCPIP treatment (40 µM, 24 h). Upper and lower lines represent the cut-off indicating three fold up- or down-regulated expression, respectively. Arrows specify selected genes with at least 4 fold up-regulated expression versus untreated controls. Expression array analysis was performed in three independent repeats and analyzed using the two-sided Student’s t test. The table (right panel) summarizes statistically significant expression changes by at least three fold (p < 0.05). (C) The time course of DCPIP-modulation (40 µM) of early growth response protein 1 (EGR1) levels was examined by immunoblot analysis of total cellular protein extracts. Detection of α-actin expression served as a loading control. (C) DCPIP-modulation (20 and 40 µM, 24 h exposure) of cellular heme oxygenase-1 (HO-1) protein levels were examined in total cellular protein extracts by immunoblot analysis. (D) Modulation of cellular Hsp70B’ protein levels by DCPIP (1, 10, 20, and 40 µM, 24 h) was examined in total cellular protein extracts followed by ELISA analysis as specified in Materials and Methods (mean ± SD, n=3). Treatment with celastrol (2 µM, 24 h exposure) was used as a positive control for pharmacological Hsp70B’ upregulation.
Next, DCPIP-induced upregulation of cellular HMOX1, EGR1, and HSPA6 gene expression was examined at the protein level by immunoblot or ELISA analysis, respectively (Fig. 6B–D). Consistent with a role in early redox stress response, pronounced upregulation of cellular EGR1 protein levels was detected within 3h exposure to DCPIP, but returned to very low background levels over the next 9h (Fig. 6B). Within 24 h, cellular heme oxygenase-1 protein levels were strongly upregulated upon exposure to DCPIP (20 and 40 µM, Fig. 6C). Protein levels of Hsp70B’, the HSPA6 gene product, were also strongly upregulated as assessed by ELISA analysis (Fig. 6D). Significant induction was observed starting at 10 µM DCPIP, and higher than 100-fold upregulated protein expression occurred in response to 40 µM DCPIP over untreated controls, with cells exposed to celastrol (2 µM, 24 h) serving as a positive control [41].
3.6. Intraperitoneal administration of DCPIP impairs growth of A375 human melanoma xenografts in SCID mice
The significant apoptogenic and prooxidant stress-inducing activity against A375 melanoma cells combined with a low acute systemic toxicity documented earlier in mice lead us to test DCPIP as a potential inhibitor of tumor growth in a human melanoma SCID-mouse xenograft model (Fig. 7).
Figure 7. DCPIP inhibits tumor growth in a human A375 melanoma SCID-mouse xenograft model.
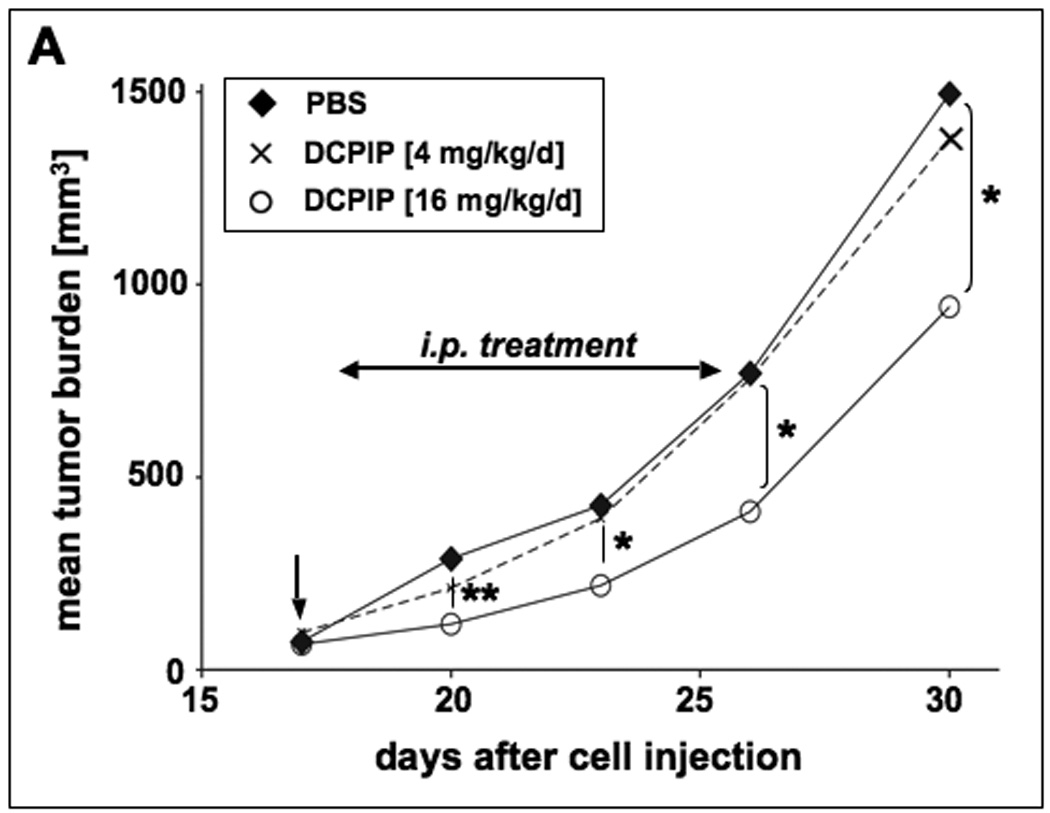
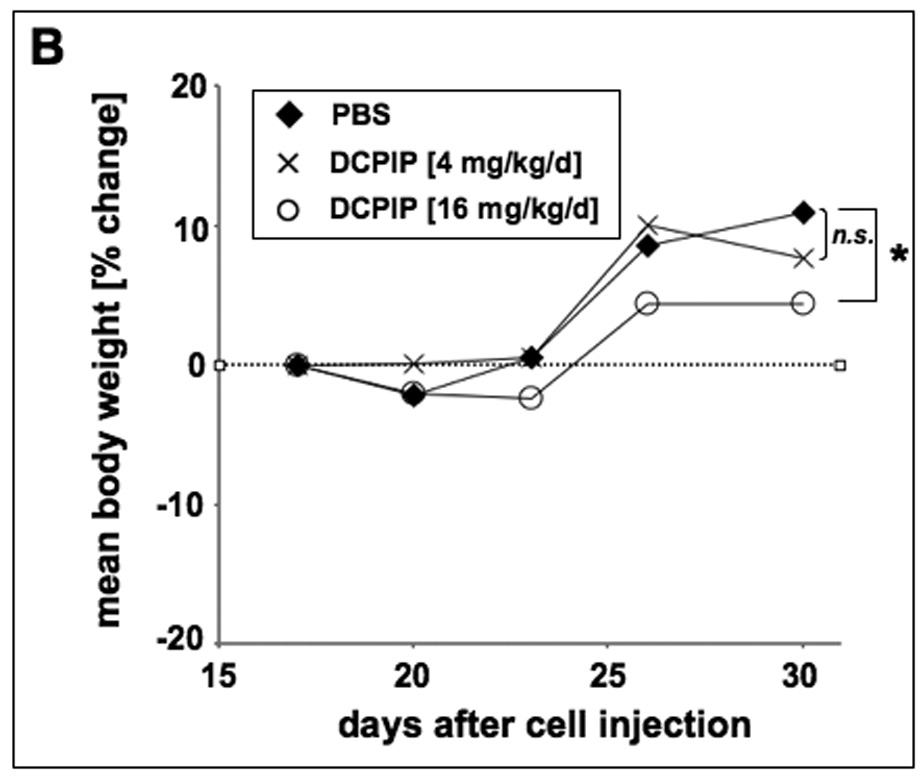
Human A375 melanoma cells (10 × 106) were implanted s.c. into the right flank of SCID mice. 17 days after cell injection animals were pair-matched (65 mm3 average tumor size) and one day later (vertical arrow) daily treatment (DCPIP: low dose group: 4 mg/kg/d, 100 µl, q.d., n=12; high dose group: 16 mg/kg/d, 200 µl, b.i.d., n=11) was initiated by intraperitoneal injection as specified in Materials and Methods. Control animals (n=12) received PBS only. (A) Tumor growth curves were obtained by determining average tumor volumes until day 30 after cell injection. Data points are depicted as means ± SEM and statistical comparison between individual data points was performed using the two-sided Student’s t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001). (B) Mean body weight was monitored during the duration of the experiment and expressed as % change from the average weight obtained on the day of pairmatching.
Daily intraperitoneal DCPIP treatment (16 mg/kg/d) of human A375 melanoma xenograft bearing SCID mice induced a significant suppression of tumor growth that reached the level of statistical significance (p < 0.05 versus PBS treated control) between days 18 to 30 after cell injection, where average tumor weights of DCPIP-treated animals were up to 45% lower than that of carrier-treated controls (Fig. 7A). These data are consistent with impairment of tumor cell proliferative capacity in xenograft-bearing mice treated with DCPIP. However, administration of DCPIP at lower doses (4 mg/kg/d) did not achieve a significant reduction of average tumor weights (Fig. 7A). During DCPIP treatment, no compound-related adverse reactions or statistically significant average weight loss were observed suggesting the safety of administration of high doses of DCPIP during the duration of the experiment (Fig. 7B). Moreover, no indication of organ toxicity as revealed by necropsy was obtained (data not shown). However, between days twenty-three and thirty increase in body average weight was slightly delayed in tumor bearing animals receiving DCPIP-treatment at high (16 mg/kg/d) but not at low (4 mg/kg/d) doses.
Taken together, these findings document for the first time feasibility of antimelanoma intervention by intraperitoneal administration of DCPIP achieved in a mouse xenograft model.
4. Discussion
Metastatic melanoma displays a notorious resistance to conventional chemotherapy, and the current dearth of treatment options creates an urgent need for the development of more efficacious antimelanoma chemotherapeutics [5, 42, 43]. Based on the well documented preferential sensitivity of melanoma cells to small molecule prooxidant intervention [5, 9, 14, 16, 17, 20], we tested the feasibility of using the redox-active halogenated benzoquinoneimine-dye DCPIP for melanoma treatment in vitro and in vivo.
First, we observed that DCPIP induces apoptosis in metastatic human A375, but not in G361 melanoma cells (Fig. 2 and Fig. 3). Based on our earlier work that demonstrated differential NQO1 specific enzymatic activity in A375 and G361 cells [14], we then tested the hypothesis that elevated NQO1 expression levels may render G361 melanoma cells resistant to the apoptogenic activity of the prooxidant NQO1-substrate DCPIP. Using the pharmacological NQO1-inhibitor dicoumarol and genetic target modulation by NQO1-siRNA we then obtained strong evidence for the causative role of NQO1 expression in DCPIP-chemoresistance exhibited by G361 human melanoma cells (Fig. 3 and Fig. 4). Next, DCPIP-induction of cellular oxidative stress was demonstrated in A375 and G361 melanoma cells assessing cellular peroxide and glutathione levels (Fig. 5). In A375 melanoma cells, intracellular ROS were elevated dose dependently (Fig. 5A) and glutathione levels were lowered significantly within 6 h of exposure (Fig. 5C). Moreover, DCPIP-induced procaspase 3 cleavage could be antagonized by 24 h preincubation of A375 melanoma cells with the thiol antioxidant and glutathione-precursor NAC (Fig. 5B), suggesting the crucial involvement of oxidative stress in DCPIP-induced apoptosis. As seen before, pharmacological or genetic antagonism of NQO1 strongly sensitized G361 cells to DCPIP-induced glutathione depletion (Fig. 5C–D). Consistent with induction of cellular stress by DCPIP treatment, upregulated expression of established oxidative and heat shock stress response genes including HSPA6, HSPA1A, HMOX1, GADD45A, DDIT3, and EGR1 was detected by detailed array analysis (Fig. 6A) [44]. DCPIP-induced upregulation of HSPA6, HMOX1, and EGR1 expression was then confirmed at the protein level using ELISA and Western-analysis, respectively (Fig. 6B–D). Remarkably, earlier expression array analysis examining activity of the investigational antimelanoma prooxidant elesclomol against human Hs294T melanoma cells has revealed a similar degree of upregulation of HSPA6 (more than 300fold) encoding Hsp70B’ [17], an Hsp70 subtype not constitutively expressed and induced only under conditions of extreme cellular stress [36]. Moreover, it is tempting to speculate that DCPIP-induced upregulation of the redox sensitive transcription factor EGR1, known to activate expression of tumor suppressor genes including PTEN and TP53, plays a functional role in mediating DCPIP-antimelanoma effects, a hypothesis to be tested by future experiments [37].
Based on these results we then conducted a melanoma xenograft experiment to test DCPIP antimelanoma activity in a relevant animal model of the disease [45]. Daily intraperitoneal administration of DCPIP (16 mg/kg/d) resulted in a significant suppression of tumor growth (Fig. 7A) in a standard A375 human melanoma xenograft SCID mouse model. Importantly, DCPIP administration was well tolerated without induction of significant weight loss over the duration of the experiment (Fig. 7B) or indication of organ toxicity as revealed by necropsy (data not shown). To the best of our knowledge, this is the first report documenting antimelanoma activity of DCPIP in vivo.
Numerous prooxidant quinone-derivatives including benzoquinones, naphthoquinones, and tyrosinase-derived acetaminophen-metabolites display anticancer activity that involves induction of oxidative stress [8, 30, 34, 35]. The structure activity relationship of prooxidant quinone-pharmacophores is complex and thought to involve ROS formation via redox cycling and thiol-adduction of protein- and glutathione-cysteine residues [22, 46, 47]. For example, it is well established that 1,4-benzoquinone, a toxicologically relevant metabolite of benzene, induces oxidative stress in selected target cells including renal proximal tubular epithelial cells through Michael adduction of proteins and glutathione followed by redox cycling of quinol-thioether-adducts [34, 48, 49]. A considerable body of prior research has examined redox biochemistry and thiol-adduction associated with DCPIP [27, 50, 51]. However, the molecular mechanism of DCPIP-induced oxidative stress, evidenced by ROS formation (Fig. 5A), antioxidant protection (Fig. 5B), glutathione depletion (Fig. 5C and D), and upregulated expression of redox sensitive genes (Fig. 6), and its causative role in antimelanoma activity observed in vivo remains to be further explored by future experiments. In this context it is interesting to note that the dehalogenated DCPIP-analogue phenolindophenol [4-(4-hydroxyphenyl)-iminocyclohexa-2,5-dien-1-one], when tested by us at equal concentrations as DCPIP, did not display any apoptogenic, caspase 3-activating, or prooxidant activity, and expression of HSPA6, the major stress response gene induced by DCPIP in A375 cells (Fig. 6A), was unchanged (data not shown), suggesting that 2,6-dichloro-substitution is a structural requirement for DCPIP apoptogenicity. Future studies will examine the detailed structure activity relationship between DCPIP-associated cancer cell apoptogenicity and structural parameters including standard reduction potential and electrophilicity providing a basis for rational lead optimization by medicinal chemistry.
In our experiments examining DCPIP apoptogenicity in human A375 and G361 melanoma cells, sensitivity to this treatment was inversely correlated with NQO1 expression levels consistent with the established reductive metabolism of DCPIP and similar quinones by NQO1 known to prevent semiquinone-dependent redox cycling and ROS formation [22, 33]. Indeed, considerable chemosensitization of G361 cells displaying high constitutive NQO1 expression levels could be achieved by combining DCPIP treatment with pharmacological or genetic NQO1 inhibition. Similar results were observed in other cancer cell lines previously shown to differ by expression of enzymatically active NQO1: e.g., MIA-PaCa-2 pancreas carcinoma cells with high specific NQO1 activity displayed pronounced DCPIP chemoresistance, whereas MDA-MB231 breast carcinoma cells, known to display the NQO1*2 genotype resulting in negligible NQO1 specific enzymatic activity [14, 33], were highly sensitive to DCPIP apoptogenicity (data not shown). Taken together, these data suggest that expression of active NQO1 protects against DCPIP-quinoneimine associated oxidative stress and cytotoxicity and is therefore an important determinant and predictor of DCPIP chemosensitivity of cancer cells.
Only limited information is currently available on NQO1 expression and activity levels in human metastatic melanoma. In our earlier experiments performed in cultured human metastatic melanoma cell lines (G361, LOX, A375) [14], high NQO1 activity was observed in G361 and LOX cells, whereas low activity was detected in A375 cells, a cell line widely employed in many standard xenograft models for the identification of potential antimelanoma agents [45]. In another study, a spectrum of NQO1 activities ranging from high to very low was reported (in that order) for SKMEL5, WM266.4, SKMEL 28, and SKMEL 2 human melanoma cells [52]. In conjunctival melanomas and primary acquired melanosis immunohistochemical detection of NQO1 was performed recently, but no enzymatic activity was assessed [53].
Importantly, constitutive NQO1 overexpression leading to high specific enzymatic activity is associated with various human malignancies including non-small cell lung cancer, pancreas carcinoma, and colon adenocarcinoma [54, 55], and it has long been known that tumors that display high NQO1 enzymatic activity can be targeted by NQO1-dependent bioreductively activated anticancer agents including alkylating agents such as mitomycin C and aziridinylbenzoquinones [33]. In contrast, other tumors, including glioblastoma [56], leukemia [57], lymphoma [58], and breast carcinoma [59], display impaired expression of functional NQO1 resulting in very low or absent NQO1 activity [33]. For example, it has recently been demonstrated that a homozygous common missense genotype [NQO1(*)2, NM_000903.2:c.558C>T] encoding the functionally impaired NQO1 P187S protein, rapidly degraded via the ubiquitin proteasomal pathway, strongly predicts poor survival among women with breast cancer [59]. Remarkably, the NQO1*2 missense variant (NP_000894:p.187P4S) is homozygous in 4–20% of the human population. Moreover, response to the anthracycline tumor antibiotic epirubicin is impaired in NQO1(*)2-homozygous breast carcinoma cells in vitro, suggesting that this NQO1-genotype is a prognostic and predictive marker for breast cancer chemotherapy.
Our results that demonstrate feasibility of DCPIP-based chemotherapy of A375 melanoma suggest that DCPIP or improved derivatives may target human melanoma or other tumors (including breast carcinoma and lymphoma) that display low NQO1 enzymatic activity, a hypothesis to be tested by future experiments. Moreover, feasibility of sensitization of G361 melanoma cells to DCPIP cytotoxicity by pharmacological or genetic NQO1 antagonism as demonstrated in this study suggests that combination therapy employing a member of the rapidly expanding group of non-cytotoxic NQO1 inhibitors together with a DCPIP-like molecule may provide therapeutic efficacy against tumors that display high NQO1 enzymatic activity [60].
Based on the significant antimelanoma activity of DCPIP observed here at well-tolerated intraperitoneal doses, future preclinical studies will further examine the mostly unexplored anticancer and safety profile of this experimental redox chemotherapeutic.
Acknowledgements
Supported in part by grants from the National Institutes of Health [R01CA122484, ES007091, ES06694, Arizona Cancer Center Support Grant CA023074], and from the Arizona Biomedical Research Commission (ABRC 0721). Animal experimentation was performed at the AZCC experimental mouse shared service (EMSS).
Abbreviations
- AV
annexinV
- DC
dicoumarol
- DCPIP
2,6-dichlorophenolindophenol
- DDIT3
DNA-damage-inducible transcript 3
- EGR1
early growth response gene 1
- FITC
fluorescein isothiocyanate
- GADD45A
growth arrest and DNA-damage-inducible alpha
- GSH
glutathione
- Hsp
heat shock protein
- HMOX1
heme oxygenase-1
- NAC
Nα-acetyl-L-cysteine
- NQO1
NAD(P)H:quinone oxidoreductase 1
- PARP
poly (ADP-ribose) polymerase
- PI
propidium iodide
- q.d.
quaque die/every day
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecylsulfate polyacrylamide gel electrophoresis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Laurent A, Nicco C, Chereau C, Goulvestre C, Alexandre J, Alves A, et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005;65:948–956. [PubMed] [Google Scholar]
- 2.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 3.Cabello CM, Bair WB, Wondrak GT., 3rd Experimental therapeutics: Targeting the redox Achilles heel of cancer. Curr Opin Investig Drugs. 2007;8:1022–1037. [PubMed] [Google Scholar]
- 4.Fruehauf JP, Meyskens FL., Jr Reactive oxygen species: a breath of life or death? Clin Cancer Res. 2007;13:789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 5.Fruehauf JP, Trapp V. Reactive oxygen species: an Achilles' heel of melanoma? Expert Rev Anticancer Ther. 2008;8:1751–1757. doi: 10.1586/14737140.8.11.1751. [DOI] [PubMed] [Google Scholar]
- 6.Efferth T. Mechanistic perspectives for 1,2,4-trioxanes in anti-cancer therapy. Drug Resist Updat. 2005;8:85–97. doi: 10.1016/j.drup.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Efferth T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. Curr Drug Targets. 2006;7:407–421. doi: 10.2174/138945006776359412. [DOI] [PubMed] [Google Scholar]
- 8.Verrax J, Stockis J, Tison A, Taper HS, Calderon PB. Oxidative stress by ascorbate/menadione association kills K562 human chronic myelogenous leukaemia cells and inhibits its tumour growth in nude mice. Biochem Pharmacol. 2006;72:671–680. doi: 10.1016/j.bcp.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 9.Loewe R, Valero T, Kremling S, Pratscher B, Kunstfeld R, Pehamberger H, et al. Dimethylfumarate impairs melanoma growth and metastasis. Cancer Res. 2006;66:11888–11896. doi: 10.1158/0008-5472.CAN-06-2397. [DOI] [PubMed] [Google Scholar]
- 10.Wang CC, Chiang YM, Sung SC, Hsu YL, Chang JK, Kuo PL. Plumbagin induces cell cycle arrest and apoptosis through reactive oxygen species/c-Jun N-terminal kinase pathways in human melanoma A375.S2 cells. Cancer Lett. 2008;259:82–98. doi: 10.1016/j.canlet.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Cabello CM, Bair WB, Lamore SD, 3rd, Bause AS, Azimian S, Wondrak GT. The cinnamon-derived Michael acceptor cinnamic aldehyde impairs melanoma cell proliferation, invasiveness, and tumor growth. Free Radic Biol Med. 2009;46:220–231. doi: 10.1016/j.freeradbiomed.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fry FH, Holme AL, Giles NM, Giles GI, Collins C, Holt K, et al. Multifunctional redox catalysts as selective enhancers of oxidative stress. Org Biomol Chem. 2005;3:2579–2587. doi: 10.1039/b502197a. [DOI] [PubMed] [Google Scholar]
- 13.Magda D, Miller RA. Motexafin gadolinium: a novel redox active drug for cancer therapy. Semin Cancer Biol. 2006;16:466–476. doi: 10.1016/j.semcancer.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Wondrak GT. NQO1-activated phenothiazinium redox cyclers for the targeted bioreductive induction of cancer cell apoptosis. Free Radic Biol Med. 2007;43:178–190. doi: 10.1016/j.freeradbiomed.2007.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger TG, Dieckmann D, Efferth T, Schultz ES, Funk JO, Baur A, et al. Artesunate in the treatment of metastatic uveal melanoma--first experiences. Oncol Rep. 2005;14:1599–1603. [PubMed] [Google Scholar]
- 16.Cen D, Brayton D, Shahandeh B, Meyskens FL, Jr, Farmer PJ. Disulfiram facilitates intracellular Cu uptake and induces apoptosis in human melanoma cells. J Med Chem. 2004;47:6914–6920. doi: 10.1021/jm049568z. [DOI] [PubMed] [Google Scholar]
- 17.Kirshner JR, He S, Balasubramanyam V, Kepros J, Yang CY, Zhang M, et al. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. 2008;7:2319–2327. doi: 10.1158/1535-7163.MCT-08-0298. [DOI] [PubMed] [Google Scholar]
- 18.Tuma RS. Reactive oxygen species may have antitumor activity in metastatic melanoma. J Natl Cancer Inst. 2008;100:11–12. doi: 10.1093/jnci/djm299. [DOI] [PubMed] [Google Scholar]
- 19.Brar SS, Kennedy TP, Sturrock AB, Huecksteadt TP, Quinn MT, Whorton AR, et al. An NAD(P)H oxidase regulates growth and transcription in melanoma cells. Am J Physiol Cell Physiol. 2002;282:C1212–C1224. doi: 10.1152/ajpcell.00496.2001. [DOI] [PubMed] [Google Scholar]
- 20.Wittgen HG, van Kempen LC. Reactive oxygen species in melanoma and its therapeutic implications. Melanoma Res. 2007;17:400–409. doi: 10.1097/CMR.0b013e3282f1d312. [DOI] [PubMed] [Google Scholar]
- 21.Fried L, Arbiser JL. The reactive oxygen-driven tumor: relevance to melanoma. Pigment Cell Melanoma Res. 2008;21:117–122. doi: 10.1111/j.1755-148X.2008.00451.x. [DOI] [PubMed] [Google Scholar]
- 22.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, et al. Menadione-induced Reactive Oxygen Species Generation via Redox Cycling Promotes Apoptosis of Murine Pancreatic Acinar Cells. J Biol Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 23.Ernster L. DT-diaphorase. Methods Enzymol. 1967;10:309–317. [Google Scholar]
- 24.Wilson GS. Determination of oxidation-reduction potentials. Methods Enzymol. 1978;54:396–410. doi: 10.1016/s0076-6879(78)54025-1. [DOI] [PubMed] [Google Scholar]
- 25.VanderJagt DJ, Garry PJ, Hunt WC. Ascorbate in plasma as measured by liquid chromatography and by dichlorophenolindophenol colorimetry. Clin Chem. 1986;32:1004–1006. [PubMed] [Google Scholar]
- 26.Yamashita T, Butler WL. Photoreduction and photophosphorylation with tris-washed chloroplasts. Plant Physiol. 1968;43:1978–1986. doi: 10.1104/pp.43.12.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayer D, Naumann R, Edler L, Bannasch P. Investigation by amperometric methods of intracellular reduction of 2,6-dichlorophenolindophenol in normal and transformed hepatocytes in the presence of different inhibitors of cellular metabolism. Biochim Biophys Acta. 1990;1015:258–263. doi: 10.1016/0005-2728(90)90029-4. [DOI] [PubMed] [Google Scholar]
- 28.Kumar S, Acharya SK. 2,6-Dichloro-phenol indophenol prevents switch-over of electrons between the cyanide-sensitive and -insensitive pathway of the mitochondrial electron transport chain in the presence of inhibitors. Anal Biochem. 1999;268:89–93. doi: 10.1006/abio.1998.3009. [DOI] [PubMed] [Google Scholar]
- 29.Wolchok JD, Williams L, Pinto JT, Fleisher M, Krown SE, Hwu WJ, et al. Phase I trial of high dose paracetamol and carmustine in patients with metastatic melanoma. Melanoma Res. 2003;13:189–196. doi: 10.1097/00008390-200304000-00013. [DOI] [PubMed] [Google Scholar]
- 30.Vad NM, Yount G, Moore D, Weidanz J, Moridani MY. Biochemical mechanism of acetaminophen (APAP) induced toxicity in melanoma cell lines. J Pharm Sci. 2008 doi: 10.1002/jps.21505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MSDS. Sigma-Aldrich Version, 1.5. 2006. 31 01, 2,6-Dichloroindophenol sodium salt (CAS#620-45-1. [Google Scholar]
- 32.Wondrak GT, Jacobson MK, Jacobson EL. Antimelanoma activity of apoptogenic carbonyl scavengers. J Pharmacol Exp Ther. 2006;316:805–814. doi: 10.1124/jpet.105.094953. [DOI] [PubMed] [Google Scholar]
- 33.Ross D, Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol. 2004;382:115–144. doi: 10.1016/S0076-6879(04)82008-1. [DOI] [PubMed] [Google Scholar]
- 34.Baigi MG, Brault L, Neguesque A, Beley M, Hilali RE, Gauzere F, et al. Apoptosis/necrosis switch in two different cancer cell lines: influence of benzoquinone- and hydrogen peroxide-induced oxidative stress intensity, and glutathione. Toxicol In Vitro. 2008;22:1547–1554. doi: 10.1016/j.tiv.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Fukuyo Y, Inoue M, Nakajima T, Higashikubo R, Horikoshi NT, Hunt C, et al. Oxidative stress plays a critical role in inactivating mutant BRAF by geldanamycin derivatives. Cancer Res. 2008;68:6324–6330. doi: 10.1158/0008-5472.CAN-07-6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noonan E, Giardina C, Hightower L. Hsp70B' and Hsp72 form a complex in stressed human colon cells and each contributes to cytoprotection. Exp Cell Res. 2008;314:2468–2476. doi: 10.1016/j.yexcr.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 37.Baron V, Adamson ED, Calogero A, Ragona G, Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006;13:115–124. doi: 10.1038/sj.cgt.7700896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen Y, Wang J, Yang T, Li Y, Jiang W, Guan Z, et al. Platinums sensitize human epithelial tumor cells to lymphotoxin alpha by inhibiting NFkappaB-dependent transcription. Cancer Biol Ther. 2008;7:1407–1414. doi: 10.4161/cbt.7.9.6429. [DOI] [PubMed] [Google Scholar]
- 39.Sarkar D, Su ZZ, Lebedeva IV, Sauane M, Gopalkrishnan RV, Valerie K, et al. mda-7 (IL-24) Mediates selective apoptosis in human melanoma cells by inducing the coordinated overexpression of the GADD family of genes by means of p38 MAPK. Proc Natl Acad Sci U S A. 2002;99:10054–10059. doi: 10.1073/pnas.152327199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott DW, Loo G. Curcumin-induced GADD153 gene up-regulation in human colon cancer cells. Carcinogenesis. 2004;25:2155–2164. doi: 10.1093/carcin/bgh239. [DOI] [PubMed] [Google Scholar]
- 41.Chow AM, Brown IR. Induction of heat shock proteins in differentiated human and rodent neurons by celastrol. Cell Stress Chaperones. 2007;12:237–244. doi: 10.1379/CSC-269.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wondrak GT, Jacobson MK, Jacobson EL. An Emerging Molecular Target in Melanoma: Cellular Carbonyl Stress and the Inhibition of Mitochondrial Survival Pathways by Carbonyl Scavenger Agents. Curr Cancer Ther Rev. 2005;1:271–276. [Google Scholar]
- 43.Hocker TL, Singh MK, Tsao H. Melanoma genetics and therapeutic approaches in the 21st century: moving from the benchside to the bedside. J Invest Dermatol. 2008;128:2575–2595. doi: 10.1038/jid.2008.226. [DOI] [PubMed] [Google Scholar]
- 44.Han ES, Muller FL, Perez V, Qi W, Liang H, Xi L, et al. The in vivo Gene Expression Signature of Oxidative Stress. Physiol Genomics. 2008 doi: 10.1152/physiolgenomics.00239.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paine-Murrieta GD, Taylor CW, Curtis RA, Lopez MH, Dorr RT, Johnson CS, et al. Human tumor models in the severe combined immune deficient (scid) mouse. Cancer Chemother Pharmacol. 1997;40:209–214. doi: 10.1007/s002800050648. [DOI] [PubMed] [Google Scholar]
- 46.Abdelmohsen K, Gerber PA, von Montfort C, Sies H, Klotz LO. Epidermal growth factor receptor is a common mediator of quinone-induced signaling leading to phosphorylation of connexin-43: role of glutathione and tyrosine phosphatases. J Biol Chem. 2003;278:38360–38367. doi: 10.1074/jbc.M306785200. [DOI] [PubMed] [Google Scholar]
- 47.Schmieder PK, Tapper MA, Kolanczyk RC, Hammermeister DE, Sheedy BR, Denny JS. Discriminating redox cycling and arylation pathways of reactive chemical toxicity in trout hepatocytes. Toxicol Sci. 2003;72:66–76. doi: 10.1093/toxsci/kfg016. [DOI] [PubMed] [Google Scholar]
- 48.Lau SS, Hill BA, Highet RJ, Monks TJ. Sequential oxidation and glutathione addition to 1,4-benzoquinone: correlation of toxicity with increased glutathione substitution. Mol Pharmacol. 1988;34:829–836. [PubMed] [Google Scholar]
- 49.Bratton SB, Lau SS, Monks TJ. The putative benzene metabolite 2,3, 5-tris(glutathion-S-yl)hydroquinone depletes glutathione, stimulates sphingomyelin turnover, and induces apoptosis in HL-60 cells. Chem Res Toxicol. 2000;13:550–556. doi: 10.1021/tx0000015. [DOI] [PubMed] [Google Scholar]
- 50.Hadler HI, Alt SK, Falcone AB. Conjugation of 2,6-dichloroindophenol with mitochondrial thiol groups. J Biol Chem. 1966;241:2886–2890. [PubMed] [Google Scholar]
- 51.Dupuy C, Kaniewski J, Ohayon R, Deme D, Virion A, Pommier J. Nonenzymatic NADPH-dependent reduction of 2,6-dichlorophenol-indophenol. Anal Biochem. 1990;191:16–20. doi: 10.1016/0003-2697(90)90379-n. [DOI] [PubMed] [Google Scholar]
- 52.Sharp SY, Boxall K, Rowlands M, Prodromou C, Roe SM, Maloney A, et al. In vitro biological characterization of a novel, synthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors. Cancer Res. 2007;67:2206–2216. doi: 10.1158/0008-5472.CAN-06-3473. [DOI] [PubMed] [Google Scholar]
- 53.Wilson MW, Schelonka LP, Siegel D, Meininger A, Ross D. Immunohistochemical localization of NAD(P)H:quinone oxidoreductase in conjunctival melanomas and primary acquired melanosis. Curr Eye Res. 2001;22:348–352. doi: 10.1076/ceyr.22.5.348.5495. [DOI] [PubMed] [Google Scholar]
- 54.Danson S, Ward TH, Butler J, Ranson M. DT-diaphorase: a target for new anticancer drugs. Cancer Treat Rev. 2004;30:437–449. doi: 10.1016/j.ctrv.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 55.Lewis AM, Ough M, Hinkhouse MM, Tsao MS, Oberley LW, Cullen JJ. Targeting NAD(P)H:quinone oxidoreductase (NQO1) in pancreatic cancer. Mol Carcinog. 2005;43:215–224. doi: 10.1002/mc.20107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seng S, Avraham HK, Birrane G, Jiang S, Li H, Katz G, et al. NRP/B mutations impair Nrf2-dependent NQO1 induction in human primary brain tumors. Oncogene. 2009;28:378–389. doi: 10.1038/onc.2008.396. [DOI] [PubMed] [Google Scholar]
- 57.Wiemels JL, Pagnamenta A, Taylor GM, Eden OB, Alexander FE, Greaves MF. A lack of a functional NAD(P)H:quinone oxidoreductase allele is selectively associated with pediatric leukemias that have MLL fusions. United Kingdom Childhood Cancer Study Investigators. Cancer Res. 1999;59:4095–4099. [PubMed] [Google Scholar]
- 58.Bruge F, Virgili S, Cacciamani T, Principi F, Tiano L, Littarru GP. NAD(P)H:quinone oxidoreductase (NQO1) loss of function in Burkitt's lymphoma cell lines. Biofactors. 2008;32:71–81. doi: 10.1002/biof.5520320109. [DOI] [PubMed] [Google Scholar]
- 59.Fagerholm R, Hofstetter B, Tommiska J, Aaltonen K, Vrtel R, Syrjakoski K, et al. NAD(P)H:quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nat Genet. 2008;40:844–853. doi: 10.1038/ng.155. [DOI] [PubMed] [Google Scholar]
- 60.Nolan KA, Timson DJ, Stratford IJ, Bryce RA. In silico identification and biochemical characterization of novel inhibitors of NQO1. Bioorg Med Chem Lett. 2006;16:6246–6254. doi: 10.1016/j.bmcl.2006.09.015. [DOI] [PubMed] [Google Scholar]