

Abstract
The aim of this study was to characterize the pathological and functional consequences of the G1961E mutant allele in the Stargardt disease gene ABCA4. Data from 15 patients were retrospectively reviewed and all the patients had at least one G1961E mutation. Comprehensive ophthalmic examination, full-field and pattern electroretinograms, and fundus autofluorescence (FAF) imaging were performed on all patients. Microperimetry, spectral-domain optical coherence tomography (OCT), and fluorescein angiography were performed in selected cases. Genetic screening was performed using the ABCR400 micro-array that currently detects 496 disctinct ABCA4 variants. All patients had normal full-field scotopic and photopic electroretinograms (ERGs) and abnormal pattern electroretinograms (PERGs) performed on both eyes, and all the fundi had bull’s eye maculopathy without retinal flecks on FAF. On OCT, one patient had disorganization of photoreceptor outer segment, two had outer nuclear layer (ONL) thinning likely due to photoreceptor atrophy proximal to the foveal center, and three had additional retinal pigment epithelium (RPE) atrophy. On microperimetry, six patients had eccentric superior fixation and amongst this group, five had an absolute scotoma in the foveal area. DNA analysis revealed that three patients were homozygous G1961E/G1961E and the rest were compound heterozygotes for G1961E and other ABCA4 mutations. The G1961E allele in either homozygosity or heterozygosity is associated with anatomical and functional pathologies limited to the parafoveal region and a trend to delayed onset of symptoms, relative to other manifestations of ABCA4 mutations. Our observations support the hypothesis that the G1961E allele contributes to localized macular changes rather than generalized retinal dysfunction, and is a cause of bull’s eye maculopathy in either the homozygosity or heterozygosity state. In addition, genetic testing provides precise diagnosis of the underlying maculopathy, and current non-invasive imaging techniques could be used to detect photoreceptor damage at the earliest clinical onset of the disease.
Keywords: Stargardt, mutation, maculopathy, retinal degeneration, dystrophy
1. Introduction
ABCA4, a member of the ATP-binding cassette (ABC) transporter superfamily, is localized in the outer segments of rod and cone photoreceptors and is involved in retinal metabolism (Allikmets et al., 1997; Molday et al., 2000; Sun and Nathans, 1997). ABCA4 deficiency leads to accumulation of lipofuscin in the retinal pigment epithelium (RPE), presumably causing RPE and subsequent photoreceptor degeneration. N-retinylidene-N-retinylethanolamine (A2E) and other bisretinoid components of lipofuscin (Kim et al., 2007) can be visualized as autofluorescence in patients diagnosed with Stargardt disease (Lois et al., 2004). A2E forms via condensation of all-trans-retinal and phosphatidylethanolamine to yield N-retinylidene-phosphatidylethanolamine (APE), which reacts with a second molecule of all-trans-retinal to form the intermediate dihydro-phosphatidyl-pyridinium bisretinoid (Kim et al., 2007) that finally undergoes spontaneous oxidation to form the phosphatidyl-pyridinium bisretinoid (A2-PE) (Ben-Shabat et al., 2002; Liu et al., 2000). Hydrolysis of the phosphoester bond in A2-PE yields A2E (Ben-Shabat et al., 2002; Liu et al., 2000; Sparrow and Boulton, 2005). It has been suggested that A2E damages RPE by initiating photooxidative processes (Jang et al., 2005; Sparrow et al., 2000, 2002), inhibiting lysosomal hydrolysis (Holz et al., 1999), and at high concentrations, by acting as a detergent (A2E is a salt) that solubilizes membranes (Eldred and Lasky, 1993; Sparrow et al., 1999, 2006). Damage to RPE cells is thought to cause secondary degeneration of photoreceptors.
Allelic heterogeneity in the ABCA4 gene (>500 possible disease-associated variants have been described) has been associated with five distinct phenotypes, including autosomal recessive Stargardt disease/fundus flavimaculatus (STGD1/FFM), bull’s eye maculopathy, cone-rod dystrophy (CRD), cone dystrophy, and age-related macular degeneration (AMD) (Klevering et al., 2005; Michaelides et al., 2007). Clinically, STGD1 can be classified according to the fundus appearance (Fishman et al., 1999). As the morphology of retinal atrophy and flecks could change over time and are not well correlated to retinal function (Aaberg, 1986), patients could also be classified into three groups based on the results of electroretinography (Lois et al., 2001): (1) full-field scotopic and photopic electroretinograms (ERG) are normal and pattern-ERG (PERG) is markedly decreased; (2) the photopic ERG is decreased and PERG is markedly decreased; and (3) photopic and scotopic full-field ERGs and PERGs are all abnormal. Despite the availability of clinical classifications, phenotype-genotype relationships between STGD1 patients and ABCA4 variants have yet to be established. The prevalence of ABCA4 allelic heterogeneity poses a significant challenge to establishing precise genotype-phenotype correlations (Gerth et al., 2002; Hargitai et al., 2005; Klevering et al., 2005; Lewis et al., 1999; Simonelli et al., 2005).
Increased deposition of lipofuscin in RPE cells has been considered the first clinically detectable pathophysiological change (Cideciyan et al., 2004) and fundus autofluorescence (FAF) imaging could be used to detect and quantify lipofuscin fluorophore accumulation (Delori et al., 1995; Robson et al., 2003).
Our aim in this study is to characterize the pathological and functional consequences of the G1961E allele in the ABCA4 gene, and we report an association between group 1 STGD1 patients with bull’s eye maculopathy and homozygous or heterozygous G1961E mutations. We also describe in detail (for the first time in the literature to our knowledge) the phenotype of homozygous G1961E mutation as observed in three patients. We found that the G1961E mutation is associated with early foveal disorganization of photoreceptor outer segment that appear before clinically evident RPE damage.
2. Material and Methods
2.1. Subjects
Clinical records of 15 patients (from 11 unrelated families) with the G1961E allele and bull’s eye maculopathy based on fundus autofluorescence (FAF) were examined. Patients were enrolled with the approval of the Institutional Review Board at Columbia University, and all research procedures were performed in accordance with the tenets of the Declaration of Helsinki. Informed consent was obtained from all study subjects before their enrollment.
The age of onset and duration of the disease were recorded for all patients. The age of onset was defined as the age at which decreased visual acuity was first noted, or as the age of first ophthalmic consultation. Disease duration is defined as the time interval between the age of onset and the age at which patients were examined at Columbia University.
Full medical history, best-corrected visual acuity, and dilated ophthalmic examination performed by at least one retina consultant were obtained from every patient. In addition, all patients received slit lamp biomicroscopy, FAF, and full-field scotopic and photopic ERGs. Blood samples were taken for genetic screening. Spectral-domain optical coherence tomography and microperimetry were performed on selected patients.
2.2. Fundus autofluorescence
FAF imaging was performed with a confocal scanning laser ophthalmoscope (cSLO, Heidelberg Retina Angiograph 2; Heidelberg Engineering, Dossenheim, Germany) after pupil dilation with topical tropicamide and phenylephrine. FAF imaging was performed using a 30° field of view at a resolution of 1536 × 1536 pixels. An optically pumped solid-state laser (488 nm) was used for excitation and a 495 nm barrier filter was used to modulate the blue argon excitation light. Standard procedure was followed for the acquisition of FAF images, including focus of the retinal image in the infrared reflection mode at 820 nm, sensitivity adjustment at 488 nm, and acquisition of 9 single 30° × 30° FAF images encompassing the entire macular area with at least a portion of the optic disc. The 9 single images were computationally averaged to produce a single frame with improved signal-to-noise ratio. Based on FAF findings, the bull’s eye maculopathy patients were further divided into three subgroups (Kurz-Levin et al., 2002): (A) presence of a ring of increased autofluorescence surrounding an area of decreased autofluorescence; (B) decreased foveal autofluorescence without a surrounding ring of increased autofluorescence; and (C) speckled macular appearance.
2.3. Optical coherence tomography
Spectral-domain optical coherence tomography (SD-OCT, Cirrus™ HD-OCT, Carl Zeiss, Dublin, CA, USA) was performed on 6 patients bilaterally. The acquisition protocol consisted of a five-line raster scan and a macular cube 512 × 128 scan pattern in which a 6 × 6 mm region of the retina was scanned (a total of 65,536 sampled points) within a scan time of 2.4 seconds. After image acquisition, those with a signal strength ≤ 8 were excluded. The morphology of the inner/outer segment junction, a band of high-reflectance inner to the RPE layer (Ko et al., 2005), was evaluated. The thickness of the parafoveal outer nuclear layer (ONL) was also recorded. Horizontal scans through the macular area were repeated three times and images were registered and averaged.
2.4. Electroretinogram
Ganzfeld full-field ERGs (Diagnosys LLC, Lowell, USA) were recorded from both eyes with DTL electrodes according to the International Society for Clinical Electrophysiology of Vision (ISCEV) standard (Marmor et al., 2004), in both scotopic and photopic states to assess retinal function. The amplitudes and implicit times obtained from both eyes of each patient were compared to age-matched normal values. ISCEV standard PERGs were obtained before mydriasis and the P50 component was used as a measure of macular function.
Based on the ERG findings, patients were classified into the following three groups (Lois et al., 2001): (1) Normal full-field scotopic and photopic ERG, with decreased PERG amplitude; (2) normal scotopic ERG, and decreased photopic 30-Hz flicker response, photopic B wave amplitudes and PERG; and (3) decreased scotopic, photopic ERG amplitudes and PERG.
2.5. Preferred Retinal Location (PRL) and microperimetry
The PRL was evaluated in 18 eyes of 9 patients using the MP-1 (MP-1 Microperimeter, Nidek Technologies Inc., Padova, Italy). Following 30 minutes of mesopic light levels adaptation, the location and fixation stability of the PRL were measured. Patients were asked to maintain fixation on a red cross (2° in diameter) for 30 seconds. The non-tested eye was occluded throughout the procedure. Microperimetry testing was performed subsequently and the sensitivity of the central visual field was obtained using a 10–2 program. “White” test lights (stimulus size Goldmann III, duration 200 milliseconds) were presented on a dim “white” background (1.27 cd/m2) using a 4–2 threshold. For the 10–2 program, 68 samplings covering an area of 20° in diameter were tested. An absolute scotoma was defined by the failure to detect the stimulus presented at 0 dB (400 apostilbs). The results of the fixation and microperimetry tests were displayed on color digital photographs acquired by the MP-1 color camera.
2.6. Screening for G1961E mutations
The screening for mutations in the ABCA4 gene was performed using the ABCR400 micro-array (Jaakson et al., 2003) that detects all currently described disease-associated variants in the ABCA4 gene (496), and confirmed by direct sequencing. Every case listed in Table 1 was screened for ABCA4 mutations. DNA was extracted from white blood cells, and all coding exons of ABCA4 were PCR-amplified as described (Asper Biotech, Tartu, Estonia) (Jaakson et al., 2003; Zernant et al., 2005). In the amplification mix, 20% of the dTTP was substituted with dUTP (Jaakson et al., 2003; Zernant et al., 2005) and subsequent treatment with uracil n-glycosylase fragments the amplification product.
Table 1.
Summary of clinical and genetic data of patients with both homozygous and heterozygous G1961E mutation
| Case #, sex | Age of onset | Duration (years) | Visual acuity (OD, OS) | Allele 2 | Bull’s eye type (FAF) | SD-OCT | MP-1 |
|---|---|---|---|---|---|---|---|
| 1, f | 20 | 1 | 20/25, 20/40 | G1961E (homozygous) | B | Not tested | Not tested |
| 2, f | 49 | 13 | 20/200, 20/150 | G1961E (homozygous) | B | Photoreceptor loss, thinner ONL and RPE atrophy | Absolute scotoma in the central 4 degrees OD and in the central 6 degrees OS, eccentric PRL (superior retina) |
| 3, m | 19 | 13 | 20/70, 20/70 | G1961E (homozygous) | A | Not tested | Absolute scotoma in the central 6 degrees in both eyes, eccentric PRL (superior retina) |
| 4.1, f | 17 | 30 | 20/200, 20/200 | L541P/A1038V | B | Not tested | Not tested |
| 4.2, m | 28 | 2 | 20/25, 20/30 | L541P/A1038V | B | Not tested | Decreased sensitivity by 6 dB in the central 2 degrees in both eyes, foveal fixation |
| 4.3, m | 28 | 2 | 20/30, 20/40 | L541P/A1038V | B | Not tested | Decreased sensitivity by 9 dB OD and 11 dB OS in the central 2 degrees, foveal fixation |
| 5.1, f | 14 | 5 | 20/200, 20/400 | L541P/A1038V | C | Photoreceptor loss (foveal optical gap), thinner ONL and normal RPE | Decreased sensitivity by 8 dB in the central 2 degrees in both eyes, eccentric PRL (superior retina) |
| 5.2, f | 14 | 1 | 20/20, 20/25 | L541P/A1038V | A | Photoreceptor disorganization, normal ONL and normal RPE | Decreased sensitivity by 6 dB in the central 2 degrees in both eyes, foveal fixation |
| 6.1, f | 17 | 5 | 20/100, 20/100 | IVS20+5 G→A | C | Photoreceptor loss, thinner ONL and RPE atrophy | Absolute scotoma in the central 2 degrees in both eyes, eccentric PRL (superior retina) |
| 6.2, m | 14 | 3 | 20/40, 20/25 | IVS20+5 G→A | A | Photoreceptor loss (foveal optical gap), thinner ONL and normal RPE | Absolute scotoma in the central 2 degrees OD and decreased sensitivity by 18 dB in the central 2 degrees OS, eccentric PRL (superior retina) |
| 7, m | 28 | 12 | 20/200, 20/150 | Q636H | B | Photoreceptor loss, thinner ONL and RPE atrophy | Not tested |
| 8, f | 25 | 9 | 20/80, 20/25 | R2077W | B | Not tested | Not tested |
| 9, m | 67 | 2 | 20/800, 20/60 | T1253M | B | Not tested | Not tested |
| 10, f | 26 | 10 | 20/80, 20/80 | C54Y | B | Not tested | Not tested |
| 11, f | 44 | 20 | 20/400, 20/60 | D1532N | C | Not tested | Absolute scotoma in the central 8–10 degrees OD and absolute scotoma in the central 8 degrees OS, eccentric PRL (superior retina) |
Abbreviations: m, male; f, female; OD, right eye; OS, left eye; FAF, fundus autofluorescence; bull’s eye type A, presence of a ring of increase autofluorescence surrounding decreased autofluorescence; bull’s eye type B, decreased fovea autofluorescence without a surrounding ring of increase autofluorescence; bull’s eye type C, speckled macular appearance with slightly increased surround autofluorescence; SD-OCT, spectral-domain optical coherence tomography; ONL, outer nuclear layer; MP-1, microperimetry; PRL, preferred retinal location.
One-sixth of the amplification product was utilized in a primer extension reaction on the ABCA4 array. Each Arrayed Primer Extension reaction consists of a fragmented and denatured PCR product, 4 units of ThermoSequenase DNA Polymerase (Amersham Biosciences), 1× reaction buffer and 1.4 μM final concentration of each fluorescence-labeled ddNTP: Texas Red-ddATP, fluorescein-ddGTP (Amersham Biosciences), Cy3-ddCTP, and Cy5-ddUTP (NEN). The reaction mixture was applied to the microarray slide for 15 minutes at 58° C and terminated by washing the slide at 95° C in filtered water (Millipore, Milli-Q) (Jaakson et al., 2003; Zernant et al., 2005). Microarry imaging was performed on the Genorama™ QuattroImager (Asper Biotech, Ltd.) and sequence variants were identified using the Genorama™ Genotyping Software (Jaakson et al., 2003; Zernant et al., 2005). Patients were classified as either G1961E/G1961E homozygous, or compound heterozygous (G1961E and another mutant allele).
3. Results
Fifteen patients from 11 unrelated families were enrolled in the study. Six patients were male and nine were female. The age of disease onset ranged between 14 to 67 years, and the disease duration ranged between 1 to 30 years. Visual acuity ranged from 20/20 to 20/800. Three patients were homozygous for the G1961E allele and 12 were compound heterozygotes. Of the compound heterozygous group, 5 patients from 2 families had the complex mutation L541P/A1038V, 2 patients (siblings) had the splicing mutation IVS20+5 G→A, and 5 patients had missense mutations Q636H, R2077W, T1253M, C54Y and D1532N (Table 1).
3.1. Fundus autofluorescence imaging, optical coherence tomography and electroretinogram
On FAF imaging, bull’s eye maculopathy was predominantly type B. This was observed in 9 of 15 patients. Type A maculopathy was observed in 3 patients and type C in the remaining 3 patients (examples are shown in Figs 1 and 2). Relatively intact RPE autofluorescence was observed in patients 1, 4.2 and 5.2. (Figs 1, 2 and 4).
Figure 1.

Bull’s eye maculopathy in patient 1 with homozygous G1961E/G1961E mutation. 1A, Fundus image showing abnormal foveal granularity and absence of yellow-whitish flecks. 1B, Fundus autofluorescence showing decreased foveal autofluorescence without a surrounding ring of increased autofluorescent (type B bull’s eye maculopathy); 1C, Fluorescein angiography showing discrete areas of RPE atrophy (transmission window defect) partially surrounding the fovea, without dark choroid; 1D, Normal full-field ERG and abnormal pattern electroretinogram showing residual activity in both eyes of less than 0.9 microvolts, which was markedly delayed to 57 milliseconds and with the N95 component proportionately reduced to less than 1 microvolt.
Figure 2.
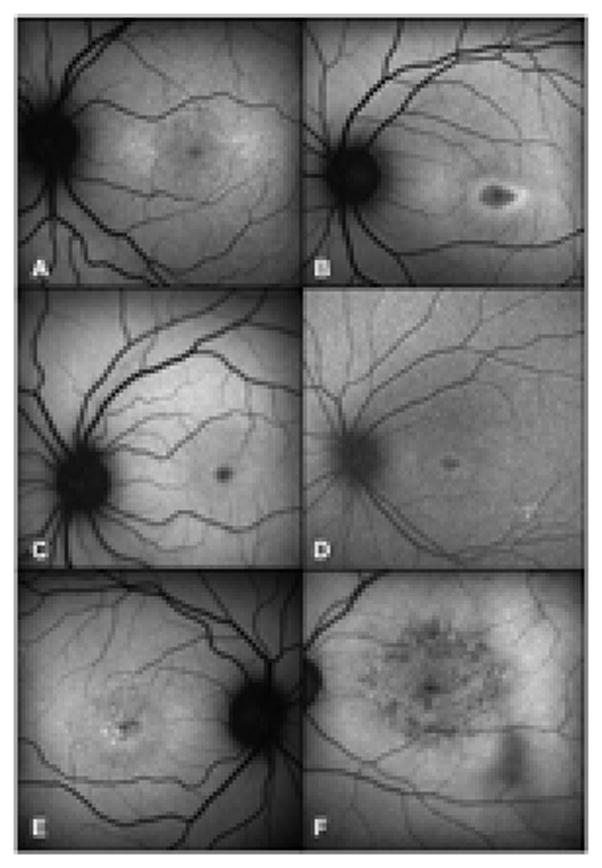
Bull’s eye maculopathy phenotypes associated with G1961E mutations. 2A and 2B, type A maculopathy with foveal decrease in autofluorescence surrounded by a continuous ring of increased autofluoresence (patients 5.2 and 6.2, respectively); 2C and 2D, type B maculopathy with decrease in foveal autofluorescence and absence of surrounding ring (patients 4.2 and 8, respectively); 2E and 2F, type C maculopathy with speckled macular appearance and a ring of slightly increased autofluorescence (patients 5.1 and 9, respectively).
Figure 4.

Bull’s eye maculopathy associated with homozygous G1961E/G1961E mutation. 4A and 4B (top and middle lines), Patients 1 and 2, respectively, with type B bull’s eye maculopathy, characterized by decreased autofluorescence in the fovea on autofluorescence; 4C (bottom line), Patient 3 with type A bull’s eye maculopathy, featuring an additional ring of increased autofluorescence surrounding the fovea.
SD-OCT was obtained in 12 eyes of 6 patients and revealed different stages of photoreceptor damage (Fig 3). Patient 5.2 had 1 year of disease duration and presented with early photoreceptor disorganization within the foveal area, and no RPE or ONL changes. Patients 6.2 and 5.1 had 3 and 5 years of disease duration, respectively, and both presented with an absence of photoreceptor outer segments in the fovea, and thinning of the ONL near to the region of photoreceptor loss when compared to the parafoveal area, but no RPE damage was apparent. Patient 6.1 (5 years of disease duration), patient 7 (12 years of disease duration) and patient 2.1 (13 years of disease duration) presented with extensive photoreceptor loss, RPE atrophy, and thinner ONL in the parafoveal area.
Figure 3.
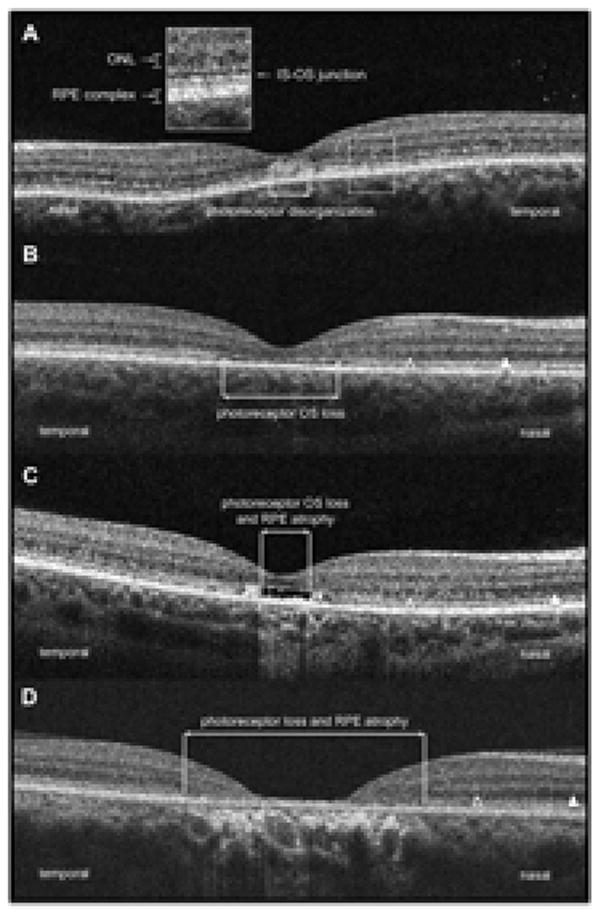
SD-OCT of patients carrying the G1961E mutation. 3A, Patient 5.2 (1 year of disease duration) showing photoreceptor disorganization in the fovea with normal appearing ONL and RPE layers (box in detail: ONL, outer nuclear layer; RPE, retinal pigment epithelium; IS-OS, inner segment-outer segment junction); 3B, Patient 6.2 (3 years of disease duration) showing photoreceptor outer segment loss represented by a optical gap in the foveal area, thinner ONL (open arrowhead) closer to the fovea compared to the extrafoveal area (arrowhead) and normal appearing RPE; 3C, Patient 5.1 (5 years of disease duration) showing photoreceptor outer segment loss, thinner ONL closer to fovea and RPE atrophy; 3D, Patient 7 (12 years of disease duration) showing extensive photoreceptor loss, RPE atrophy and thinner ONL closer to fovea.
Full-field scotopic and photopic ERG amplitudes were within the normal range for all 15 patients and all had decreased PERG P50 amplitude (sample results for Patient 1 are shown in Fig 1). All patients were classified as STGD1 group 1.
3.2. Microperimetry and PRL
Microperimetry and measurements of the preferred retinal location(s) (PRL) were obtained on 18 eyes of 9 patients. Of the 18 eyes, 6 had foveal fixation, and 12 had eccentric fixation with PRLs located superior to the fovea. Absolute scotoma in the foveal area was recorded in 10 of the 12 eyes with eccentric fixation and ranged in diameter from 2–10 degrees (Table 1).
3.3. Genetic analysis
For further analysis, we grouped patients according to G1961E segregation. Three of the patients were homozygotes, and 12 were compound heterozygotes.
3.3.1. Homozygous G1961E/G1961E mutation
Phenotypic consequences for homozygous G1961E/G1961E have yet to be reported, despite the fact that has been considered one of the most frequent variants in AMD and Stargardt (Lewis et al., 1999). G1961E/G1961E causes bull’s eye maculopathy in these unrelated patients with varying disease onset age and duration. Type B bull’s eye maculopathy was found in 2 patients and type A in one (Fig 4). Patient 1 had a mild phenotype with visual acuity of 20/25 in the right eye and 20/40 in the left eye. Discrete abnormal RPE granularity corresponding to reduced foveal autofluorescence was observed (type B). On fluorescein angiography, transmission defects around the fovea were seen, and the PERG P50 amplitude was decreased (Figs 1 and 4A).
Patient 2 had a longer disease duration (13 years) and poorer visual acuity (20/200 in the right eye and 20/150 in the left eye). She presented with type B maculopathy with decreased foveal FAF (Fig 4B). Microperimetry revealed an eccentric PRL in the superior retina and an absolute scotoma in the foveal and perifoveal areas in both eyes. There was interruption of the inner-outer segment junction and RPE atrophy was present in the central macula on SD-OCT scans.
Patient 3 had type A bull’s eye maculopathy, with a well-defined hyperautofluorescent ring surrounding the hypoautofluorescent fovea (Fig 4C). The patient maintained a visual acuity of 20/70 in both eyes despite foveal atrophy and eccentric PRL.
3.3.2. Compound heterozygous G1961E mutation
Twelve patients were ABCA4 compound heterozygous with the G1961E allele. Five patients from 2 unrelated families carried the complex mutation L541P/A1038V in addition to the G1961E allele. Different types of bull’s eye maculopathy were present among these patients, with type B present in patients from family 4, and type A and C in patients from family 5. Patients from both families 4 and 5 had decreased visual sensitivity of 5–12 dB (compared to normals) in the central 2 degrees on microperimetry analysis. In addition, SD-OCT of patient 5.1 revealed photoreceptor outer segment loss greater than foveal RPE atrophy, and juxtafoveolar ONL thinning. Patient 5.2 showed fragmentation of the inner-outer segment junction and photoreceptor disorganization in the foveal area. RPE atrophy was not observed in both patients.
Two patients from family 6 had the G1961E allele paired with cryptic splice site mutation IVS20+5 G→A, resulting in early-onset, rapid progression, and moderately-to-severely impaired visual acuity. Patient 6.1 had a visual acuity of 20/100 in both eyes after 5 years of disease duration with type C bull’s eye maculopathy, and photoreceptor and RPE damage on SD-OCT. Patient 6.2 had visual acuity of 20/40 in the right eye and 20/25 in the left eye after 3 years of disease duration. Type A bull’s eye maculopathy was noted and SD-OCT showed photoreceptor damage without RPE atrophy (Table 1).
In 5 patients (patients 7 to 11), missense mutations Q636H, R2077W, T1253M, C54Y and D1532N were found in addition to the G1961E allele, respectively. All 5 patients had anatomical and physiological changes limited to the macula and presented with maculopathy type A, B or C. They had late-onset maculopathy (i.e., after the age of 20 years) and levels of visual acuity were not associated with the disease duration, i.e., visual acuity ranged from 20/25 to 20/400 with disease duration from 2 to 20 years.
4. Discussion
G1961E is one of the most frequently observed mutant ABCA4 alleles (Allikmets et al., 1997; Gerth et al., 2002; Simonelli et al., 2005). It is predicted to alter protein function by decreasing ATP binding and ATPase activity (Sun et al., 2000). This variant is found in up to 10% of STGD1 patients of European descent, while its frequency in the general population of the same origin is ~0.2% (Allikmets, 2000).
Our study demonstrates that the G1961E allele in either the homozygous or heterozygous state is associated with bull’s eye maculopathy and early central photoreceptor disorganization, resulting in reduced pattern electroretinogram (PERG) P50 responses. As our G1961E patients have normal full-field ERGs and abnormal PERG, we hypothesize that this mutant allele is associated with retinal dysfunction restricted to the macula, and it is not associated with generalized retinal dysfunction.
All patients were diagnosed with bull’s eye maculopathy according to the FAF parameters established by Kurz-Levin et al (2002). FAF is used to estimate the pattern of distribution and accumulation of lipofuscin within the RPE. In normal subjects, macular pigment can mask FAF in the central macula (Delori et al., 1995; Robson et al., 2006). Abnormal FAF has been suggested as the earliest pathological sign in ABCA4-related maculopathy, although in milder cases flecks or lipofuscin accumulation could be absent (Cideciyan et al., 2004). All patients in our series had altered macular autofluorescence, indicating abnormal accumulation of lipofuscin in the fovea. Nine out of 15 patients had type B maculopathy with only a central decrease in macular autofluorescence. Three patients had type A maculopathy, characterized by a ring of increased autofluorescence around the hypoautofluorescent fovea, and 3 patients had type C maculopathy, characterized by a speckled macular appearance. It is interesting to note that, in some type C patients, a faint ring of increased autofluorescence was observed as a variant to the type C described by Kurz-Levin et al (2002).
Lipofuscin accumulation leads to photoreceptor damage (Sparrow and Boulton, 2005) and our findings suggest that damage to the photoreceptor outer segment architecture is among the earliest signs of pathology in patients carrying the G1961E mutation. Photoreceptor outer segment disorganization was present in one patient (case 5.2) with recent disease onset (1 year) and good visual acuity (20/20 in the right eye and 20/25 in the left eye). Another 2 patients (cases 5.1 and 6.2) had central photoreceptor loss with an optical gap in the outer segment layer, in addition to thinning of ONL proximal to regions of photoreceptor loss. Changes to ONL thickness in association with photoreceptor inner-outer segment junction disruption in a Stargardt patient have been previously described using high-speed ultrahigh-resolution OCT (Srinivasan et al., 2006). As some retinal structures imaged with ultrahigh-resolution technology are known to accurately correspond to those obtained using lower resolution devices such as the Stratus OCT (Ko et al., 2005), we believe our observations represent early ONL damage.
Upon subgroup analysis based on ABCA4 allelic combinations, we found that the 3 homozygous G1961E/G1961E patients had retinal changes limited to the central macula. Manifestation of bull’s eye maculopathy in these patients suggests that G1961E is a disease-causing mutation rather than a neutral polymorphism (Guymer et al., 2001). The missense mutation in exon 42 results in a glycine to glutamine mutation that is predicted to lie outside of ABCA4 functional domains (Lewis et al., 1999). G1961E decreases ATP binding and ATPase activity (Sun and Nathans, 1997) and is considered a pathologic mutation that cosegregates with STGD1 (Lewis et al., 1999). G1961E is associated with milder phenotypes in homozygous (Fishman et al., 1999) or compound heterozygous states (Simonelli et al., 2005). In other words, patients who are homozygous or compound heterozygous for the G1961E allele usually have milder fundus changes with the full-field scotopic and photopic ERG being normal or borderline normal, despite abnormal macular cone mediated responses on the multifocal electroretinogram (mfERG) (Gerth et al., 2002).
In the compound heterozygous group, 5 patients had the complex mutation allele L541P/A1038V, in addition to G1961E. Poor visual acuity and several juxtafoveal higher density autofluorescence deposits were noted in both patient 4.1 (with type B maculopathy) and in patient 5.1 (with type C maculopathy). Patient 4.1 had longer disease duration than patient 5.1 who had a more rapid disease progression. The other members of family 4 and 5 had shorter disease duration and maintained visual acuity levels ranging from 20/20 to 20/40. The L541P/A1038V mutation has been described in patients with more favorable clinical prognosis (Hargitai et al., 2005). Similarly, although G1961E is considered a “mild-to-moderate” allele, it has been reported in bull’s eye maculopathy with more severe phenotype such as extensive atrophic RPE changes (Gerth et al., 2002; Simonelli et al., 2005). Since either mutation could severely reduce the ATPase activity of ABCA4 (Sun and Nathans, 1997), the actual disease manifestation could be altered by the other paired mutant allele (Gerth et al., 2002). In addition, currently unknown modifying genetic and environmental factors may play a role in the phenotypic expression. These factors are of particular importance to the analysis of the other compound heterozygous patients in our series. The 2 siblings from family 6 have the splicing mutation IVS20+5G→A that changes the highly conserved guanine base at the +5 splice site. This splice mutation has been reported to cause maculopathy before the age of 30 (Rivera et al., 2000) and our patients had early-onset disease with rapid progression, and one patient had severely impaired visual acuity with longer disease duration. Visual acuity level related to disease duration was also observed in patients 7, 10 and 11 with missense mutations Q636H, C54Y and D1532N. These missense mutations result in amino acid changes outside of the nucleotide-binding domain of ABCA4 (Lewis et al., 1999; Sun et al., 2000). An exception was patient 9 with the missense mutation T1253M that imparts an amino acid change outside of ABCA4 functional domain and presented with poor visual acuity but uncertain disease duration. Despite disease duration being what may appear to be an easily quantifiable measurement, it relies on subjective information and has been demonstrated to vary independently of disease severity (Lois et al., 1999). Finally, patient 8 had the missense mutation R2077W in addition to the G1961E allele and a mild-to-moderate phenotype, with asymmetrical visual acuity and discrete autofluorescence changes. Although the R2077W mutation occurs within the second nucleotide-binding domain of ABCA4 (Lewis et al., 1999), it did not appear to cause severe disease phenotype.
Our study confirms that the G1961E allele in either homozygosity or compound heterozygosity causes bull’s eye maculopathy featuring photoreceptor outer segment disruption as the earliest detectable finding. Even in the presence of a normal full-field ERG, central cone photoreceptor dysfunction can be detected on mfERG (Gerth et al., 2002), PERG (Lois et al., 2001) or microperimetry. Nine patients in our series had decreased visual sensitivities on microperimetry and of this group, 4 had decreased thickness in the photoreceptor outer nuclear layer.
The clinical implications of our findings are that precise and early diagnosis of the underlying maculopathy is essential in patient prognostic and treatment counseling. Vitamin A supplementation should be avoided because it can potentially accelerate disease progression in ABCA4-related bull’s eye maculopathy (Radu et al., 2008). In addition, patients carrying ABCA4 mutations should be advised to limit excessive light exposure that is known to increase A2E concentration in RPE cells (Radu et al., 2004; Radu et al., 2003).
In conclusion, the G1961E mutant allele in either homozygosity or heterozygosity is associated with localized macular changes rather than generalized retinal dysfunction (Fishman et al., 1999; Lewis et al., 1999; Lois et al., 2004). Consistent with these observations, all patients in our series had normal full-field ERG with abnormal PERG and presented with bull’s eye maculopathy (mainly characterized by a decrease in foveal autofluorescence, or type B), absence of retinal flecks and a relatively slow disease progression.
Acknowledgments
Grant information: Supported in part by: NIH Grants RO1-EY02115 (VG) EY013435 (RA), R01EY018213 (SHT), unrestricted funds from Research to Prevent Blindness, New York, NY, the Foundation Fighting Blindness, Schneeweiss Stargardt Fund, and The Starr Foundation. SHT is a Fellow of the Burroughs-Wellcome Program in Biomedical Sciences, and has been supported by the Bernard Becker-Association of University Professors in Ophthalmology-Research to Prevent Blindness Award and Foundation Fighting Blindness. Columbia Retinal Phenomics Program is supported by Dennis W. Jahnigen Award of the American Geriatrics Society, Joel Hoffman Fund, Gale and Richard Siegel Stem Cell Fund, Charles Culpeper Scholarship, Schneeweiss Stem Cell Fund, Irma T. Hirschl Charitable Trust, and Bernard and Anne Spitzer Stem Cell Fund, Barbara & Donald Jonas Family Fund, and Eye Surgery Fund.
We appreciate the assistance of Chai Lin Chou for his participation in preliminary experiments and reading of the manuscript, and members of Bernard & Shirlee Brown and Allikmets laboratories for the sharing of ideas, and equipment. We are also grateful for the Photography Department at the Edward Harkness Eye Institute for support, especially Rachel Revilla and Helen Marie Havnaer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aaberg TM. Stargardt’s disease and fundus flavimaculatus: evaluation of morphologic progression and intrafamilial co-existence. Trans Am Ophthalmol Soc. 1986;84:453–487. [PMC free article] [PubMed] [Google Scholar]
- Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet. 2000;67:487–491. doi: 10.1086/303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature Genetics. 1997;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- Ben-Shabat S, Parish CA, Vollmer HR, Itagaki Y, Fishkin N, Nakanishi K, Sparrow JR. Biosynthetic studies of A2E, a major fluorophore of retinal pigment epithelial lipofuscin. J Biol Chem. 2002;277:7183–7190. doi: 10.1074/jbc.M108981200. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Swider M, Schwartz SB, Steinberg JD, Brucker AJ, Maguire AM, Bennett J, Stone EM, Jacobson SG. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet. 2004;13:525–534. doi: 10.1093/hmg/ddh048. [DOI] [PubMed] [Google Scholar]
- Delori FC, Staurenghi G, Arend O, Dorey CK, Goger DG, Weiter JJ. In vivo measurement of lipofuscin in Stargardt’s disease-fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1995;36:2327–2331. [PubMed] [Google Scholar]
- Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993;361:724–726. doi: 10.1038/361724a0. [DOI] [PubMed] [Google Scholar]
- Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol. 1999;117:504–510. doi: 10.1001/archopht.117.4.504. [DOI] [PubMed] [Google Scholar]
- Gerth C, Andrassi-Darida M, Bock M, Preising MN, Weber BH, Lorenz B. Phenotypes of 16 Stargardt macular dystrophy/fundus flavimaculatus patients with known ABCA4 mutations and evaluation of genotype-phenotype correlation. Graefe’s Arch Clin Exp Ophthalmol. 2002;240:628–638. doi: 10.1007/s00417-002-0502-y. [DOI] [PubMed] [Google Scholar]
- Guymer RH, Heon E, Lotery AJ, Munier FL, Schorderet DF, Baird PN, McNeil RJ, Haines H, Sheffield VC, Stone EM. Variation of codons 1961 and 2177 of the Stargardt disease gene is not associated with age-related macular degeneration. Arch Ophthalmol. 2001;119:745–751. doi: 10.1001/archopht.119.5.745. [DOI] [PubMed] [Google Scholar]
- Hargitai J, Zernant J, Somfai GM, Vamos R, Farkas A, Salacz G, Allikmets R. Correlation of clinical and genetic findings in Hungarian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2005;46:4402–4408. doi: 10.1167/iovs.05-0504. [DOI] [PubMed] [Google Scholar]
- Holz FG, Schutt F, Kopitz J, Eldred GE, Kruse FE, Volcker HE, Cantz M. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 1999;40:737–743. [PubMed] [Google Scholar]
- Jaakson K, Zernant J, Kulm M, Hutchinson A, Tonisson N, Glavac D, Ravnik-Glavac M, Hawlina M, Meltzer MR, Caruso RC, Testa F, Maugeri A, Hoyng CB, Gouras P, Simonelli F, Lewis RA, Lupski JR, Cremers FP, Allikmets R. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- Jang YP, Matsuda H, Itagaki Y, Nakanishi K, Sparrow JR. Characterization of peroxy-A2E and furan-A2E photooxidation products and detection in human and mouse retinal pigment epithelial cell lipofuscin. J Biol Chem. 2005;280:39732–39739. doi: 10.1074/jbc.M504933200. [DOI] [PubMed] [Google Scholar]
- Kim SR, Jang YP, Jockusch S, Fishkin NE, Turro NJ, Sparrow JR. The all-trans-retinal dimer series of lipofuscin pigments in retinal pigment epithelial cells in a recessive Stargardt disease model. Proc Natl Acad Sci USA. 2007;104:19273–19278. doi: 10.1073/pnas.0708714104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Hoyng CB. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefe’s Arch Clin Exp Ophthalmol. 2005;243:90–100. doi: 10.1007/s00417-004-1079-4. [DOI] [PubMed] [Google Scholar]
- Ko TH, Fujimoto JG, Schuman JS, Paunescu LA, Kowalevicz AM, Hartl I, Drexler W, Wollstein G, Ishikawa H, Duker JS. Comparison of ultrahigh- and standard-resolution optical coherence tomography for imaging macular pathology. Ophthalmology. 2005;112:1922.e1–15. doi: 10.1016/j.ophtha.2005.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz-Levin MM, Halfyard AS, Bunce C, Bird AC, Holder GE. Clinical variations in assessment of bull’s-eye maculopathy. Arch Ophthalmol. 2002;120:567–575. doi: 10.1001/archopht.120.5.567. [DOI] [PubMed] [Google Scholar]
- Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, Lupski JR, Leppert M, Dean M. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64:422–434. doi: 10.1086/302251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Itagaki Y, Ben-Shabat S, Nakanishi K, Sparrow JR. The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J Biol Chem. 2000;275:29354–29360. doi: 10.1074/jbc.M910191199. [DOI] [PubMed] [Google Scholar]
- Lois N, Halfyard A, Bird A, Holder G, Fitzke F. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol. 2004;138:55–63. doi: 10.1016/j.ajo.2004.02.056. [DOI] [PubMed] [Google Scholar]
- Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119:359–369. doi: 10.1001/archopht.119.3.359. [DOI] [PubMed] [Google Scholar]
- Lois N, Holder GE, Fitzke FW, Plant C, Bird AC. Intrafamilial variation of phenotype in Stargardt macular dystrophy-fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1999;40:2668–2675. [PubMed] [Google Scholar]
- Marmor MF, Holder GE, Seeliger MW, Yamamoto S International Society for Clinical Electrophysiology of Vision. Standard for clinical electroretinography (2004 update) Doc Ophthalmol. 2004;108:107–114. doi: 10.1023/b:doop.0000036793.44912.45. [DOI] [PubMed] [Google Scholar]
- Michaelides M, Chen LL, Brantley MA, Jr, Andorf JL, Isaak EM, Jenkins SA, Holder GE, Bird AC, Stone EM, Webster AR. ABCA4 mutations and discordant ABCA4 alleles in patients and siblings with bull’s-eye maculopathy. Br J Ophthalmol. 2007;91:1650–1655. doi: 10.1136/bjo.2007.118356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Am J Ophthalmol. 2000;130:689. doi: 10.1016/s0002-9394(00)00756-x. [DOI] [PubMed] [Google Scholar]
- Radu RA, Mata NL, Bagla A, Travis GH. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt’s macular degeneration. Proc Natl Acad Sci USA. 2004;101:5928–5933. doi: 10.1073/pnas.0308302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci USA. 2003;100:4742–4747. doi: 10.1073/pnas.0737855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu RA, Yuan Q, Hu J, Peng JH, Lloyd M, Nusinowitz S, Bok D, Travis GH. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008;49:3821–3829. doi: 10.1167/iovs.07-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera A, White K, Stohr H, Steiner K, Hemmrich N, Grimm T, Jurklies B, Lorenz B, Scholl HP, Apfelstedt-Sylla E, Weber BH. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet. 2000;67:800–813. doi: 10.1086/303090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson AG, Saihan Z, Jenkins SA, Fitzke FW, Bird A, Webster AR, Holder GE. Functional characterisation and serial imaging of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Br J Ophthalmol. 2006;90:472–479. doi: 10.1136/bjo.2005.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson AG, Moreland JD, Pauleikhoff D, Morrissey T, Holder GE, Fitzke FW, Bird AC, van Kuijk FJ. Macular pigment density and distribution: comparison of fundus autofluorescence with minimum motion photometry. Vision Res. 2003;43:1765–1775. doi: 10.1016/s0042-6989(03)00280-3. [DOI] [PubMed] [Google Scholar]
- Simonelli F, Testa F, Zernant J, Nesti A, Rossi S, Allikmets R, Rinaldi E. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res. 2005;37:159–167. doi: 10.1159/000086073. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Cai B, Jang YP, Zhou J, Nakanishi K. A2E, a fluorophore of RPE lipofuscin, can destabilize membrane. Adv Exp Med Biol. 2006;572:63–68. doi: 10.1007/0-387-32442-9_10. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res. 2005;80:595–606. doi: 10.1016/j.exer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Nakanishi K, Parish CA. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci. 2000;41:1981–1989. [PubMed] [Google Scholar]
- Sparrow JR, Parish CA, Hashimoto M, Nakanishi K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest Ophthalmol Vis Sci. 1999;40:2988–2995. [PubMed] [Google Scholar]
- Sparrow JR, Zhou J, Ben-Shabat S, Vollmer H, Itagaki Y, Nakanishi K. Involvement of oxidative mechanisms in blue-light-induced damage to A2E-laden RPE. Invest Ophthalmol Vis Sci. 2002;43:1222–1227. [PubMed] [Google Scholar]
- Srinivasan VJ, Wojtkowski M, Witkin AJ, Duker JS, Ko TH, Carvalho M, Schuman JS, Kowalczyk A, Fujimoto JG. High-definition and 3-dimensional imaging of macular pathologies with high-speed ultrahigh-resolution optical coherence tomography. Ophthalmology. 2006;113:2054.e1–14. doi: 10.1016/j.ophtha.2006.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Nathans J. Stargardt’s ABCR is localized to the disc membrane of retinal rod outer segments. Nat Genet. 1997;17:15–16. doi: 10.1038/ng0997-15. [DOI] [PubMed] [Google Scholar]
- Sun H, Smallwood PM, Nathans J. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet. 2000;26:242–246. doi: 10.1038/79994. [DOI] [PubMed] [Google Scholar]
- Zernant J, Kulm M, Dharmaraj S, den Hollander AI, Perrault I, Preising MN, Lorenz B, Kaplan J, Cremers FP, Maumenee I, Koenekoop RK, Allikmets R. Genotyping microarray (disease chip) for Leber congenital amaurosis: detection of modifier alleles. Invest Ophthalmol Vis Sci. 2005;46:3052–3059. doi: 10.1167/iovs.05-0111. [DOI] [PubMed] [Google Scholar]