

Abstract
An enzyme with sarcosine dimethylglycine methyltransferase activity has been identified in the thermophilic eukaryote, Galdieria sulphuraria. The crystal structure of the enzyme, solved to a resolution of 1.95 Å, revealed a fold highly similar to that of mycolic acid synthases. The kcat and apparent KM values were 64.3 min-1 and 2.0 mM for sarcosine and 85.6 min-1 and 2.8 mM for dimethylglycine. Apparent KM values for S-adenosylmethionine were 144 and 150 μM for sarcosine and dimethylglycine respectively, and the enzyme melting temperature was 61.1 °C. Modeling of cofactor binding in the active site based on the structure of methoxy mycolic acid synthase 2 revealed a number of conserved interactions within the active site.
Keywords: Betaine synthesis, structural genomics, thermophile, osmoregulation
INTRODUCTION
Betaine serves a variety of metabolic roles. In mammals, it is involved in the regulation of cell volume and serves as a methyl donor during the synthesis of methionine from homocysteine 1,2. In many plants and in bacteria, betaine has been shown to maintain cellular turgor and to stabilize the structure and function of certain macromolecules during stress conditions such as exposure to high salt concentration or low temperature 3-5. In the main pathway for betaine biosynthesis, the precursor choline is converted to betaine aldehyde either by choline dehydrogenase or by choline monoxygenase, which is in turn converted to betaine by betaine aldehyde dehydrogenase. In some bacteria, both reactions are carried out by choline oxidase 5.
A distinct pathway for betaine biosynthesis has been observed in several bacteria that thrive in high salt environments. These organisms are capable of the successive methylation of the amine of glycine to give sarcosine, dimethylglycine, and finally betaine using S-adenosyl methionine (SAM) as a cofactor (Figure 1). These bacteria include Synechococcus sp. WH8102, Aphanothece halophytica, Actinopolyspora halophila, Ectothiorhodospira halochloris, and Methanohalophilus portucalensis 6-9. In both Synechococcus and E. halochloris, two enzymes were identified in this biosynthetic pathway. One enzyme catalyzed the methylation of either glycine or sarcosine, while the other catalyzed the methylation of either sarcosine or dimethylglycine 6,8. Two similar enzymes were also identified in A. halophytica; however, the second enzyme exclusively used dimethylglycine as the substrate 7. In both A. halophila and M. portucalensis, a single large, multi-domain enzyme was shown to carry out the entire reaction 8,9. This class of enzymes has generated biotechnological interest for their ability to help reduce environmental stress in plants and industrial bacteria 10.
Figure 1.
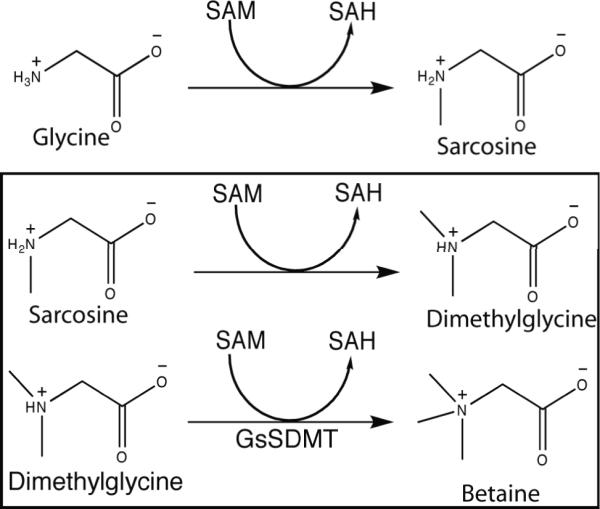
Betaine synthesis. Reactions carried out by sarcosine dimethylglycine methyltransferase are bordered by black lines.
Galdieria sulphuraria is an acidophilic and moderately thermophilic eukaryote that can live at a pH of 0 and at temperatures as high as 57 °C 11. This red micro-alga grows on more than 50 different carbon sources, demonstrates resistance to high concentrations of toxic metals, and can survive in environments with up to 10% (w/v) salinity 12-15. Expressed sequence tag analysis of G. sulphuraria previously identified a gene with 32% identity to a Na+/H+ symporter from Deinococcus radiodurans, that may be involved in the excretion of Na+ ions into the environment 16.
A number of structural genomics centers use extremophilic prokaryotes as protein sources due to their expected enhanced stability 17-19. CESG is interested in G. sulphuraria as a possible surrogate source for eukaryotic gene families and is currently investigating whether its proteins may have favorable properties for structural studies as has been previously established for proteins from thermophilic bacteria.
In this work, we report on the cloning, expression, purification, and X-ray crystal structure of the G. sulphuraria protein derived from gene Gs07580.1. The protein structure coordinates were deposited into the Protein Data Bank eight months after initial gene selection. The structure revealed homology with other bacterial sarcosine dimethylglycine methyltransferases (SDMT) and SAM-dependent methoxy mycolic acid synthase 2 from mycobacteria, despite low overall primary sequence identity. Steady-state kinetic analysis showed that the Galdieria sulphuraria protein was a SAM-dependent methyltransferase capable of reacting with either sarcosine or dimethylglycine, but not with glycine. Considering the high salt environment this organism occupies, G. sulphuraria sarcosine dimethylglycine methyltransferase (GsSDMT) may thus have a role in maintaining osmotic homeostasis by contributing to a two-step pathway for betaine biosynthesis.
MATERIALS AND METHODS
Reagents
All reagents were purchased through Sigma-Aldrich unless otherwise noted.
Cloning, Expression, and Purification
The gene encoding GsSDMT was cloned, and the selenomethionine-labeled protein was expressed and purified using protocols and bioinformatics management tools developed at the Center for Eukaryotic Structural Genomics 20. In summary, the cDNA encoding GsSDMT was cloned into expression vector pVP16 containing an N-terminal His8-maltose binding protein tag with a tobacco etch virus (TEV) protease recognition site in the linker region 21. The fusion protein was expressed using Escherichia coli B834 in 2 L of factorial evolved auto-induction medium 22. The cells were harvested by centrifugation, and the resulting 14.3 g of cell pellet were resuspended in a sonication/wash solution (500 mM sodium chloride, 20 % ethylene glycol, 35 mM imidazole, 0.3 mM tris(2-carboxyethyl)phosphine HCl, 20 mM sodium phosphate monobasic (pH 7.5), and a protease inhibitor cocktail). The sonicated cell suspension was clarified by centrifugation and the fusion protein was purified from the supernatant using Ni IMAC chromatography 23. The His8-MBP fusion protein was treated with His7-TEV protease and the liberated GsSDMT was separated from the His8-MBP fusion and His7-TEV protease by subtractive Ni IMAC chromatography 24. The GsSDMT was further purified using Superdex gel filtration chromatography, concentrated to 10 mg/mL, and drop-frozen in liquid N2 in 5 mM bis(2-hydroxyethyl)-imino-tris(hydroxymethyl)-methane, pH 7.0, containing 50 mM sodium chloride, 3.1 mM sodium azide, and 0.3 mM tris(2-carboxyethyl)phosphine HCl. The three-step purification gave ~150 mg of recombinant GsSDMT with 92% selenomethionine incorporation as determined by mass spectrometry.
Identification of Enzyme Products
Individual reactions of GsSDMT with SAM and the substrates glycine, sarcosine or dimethylglycine were performed as follows. A 100 μL reaction mixture containing 10 μM enzyme in 50 mM 3-(N-morpholino)propanesulfonic acid (pH 7.5) was incubated with 100 μM of SAM and 20 mM of substrate at 37 °C for 15 min. Then, 20 μL of the reaction was spotted onto a Whatman Partisil LK6DF silica TLC plate (Whatman Inc., Florham Park, NJ) and separated in a 4:1 phenol:H2O solvent mixture. After treatment of the TLC plate with iodine, the reaction products were compared to known standards of glycine, sarcosine, dimethylglycine, betaine, SAM, and S-adenosyl homocysteine (SAH).
Steady-state Kinetics
Reactions to determine the pH optimum were performed at 37 °C in 100 mM 2-(N-morpholino)ethanesulfonic acid, 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 100 mM N-tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid from a pH range of 6 to 8.5, or. The assay buffer also contained 100 μM MgCl2. Briefly, 150 μM SAM and either 20 mM sarcosine or dimethylglycine were combined in a total volume of 1000 μL of reaction buffer with a coupled enzyme system consisting of 40 nM adenosylhomocysteine nucleosidase and 4 nM adenine deaminase (G-Biosciences, St. Louis, MO). After 10 min of incubation at 37 °C, GsSDMT was added to the mixture to a final concentration of 0.62 μM. The reaction was mixed thoroughly and added by pipette into a quartz cuvette. The reaction was monitored using an 8435A UV-Visible spectrophotometer (Agilent, Santa Clara, CA) at 1s intervals for a total of 1 min. Initial reaction rates were determined from the change in absorbance given by the deamination of adenine to hypoxanthine by the coupled enzyme system (Δε265= 6.7 mM-1 cm-1) 25. Control reactions were performed at each pH to verify that the activity of the coupled enzyme system was not rate limiting. Absorbance corrections were made for residual SAH present in the SAM preparation as determined by controls lacking either sarcosine or dimethylglycine.
The apparent kcat and KM for each substrate were determined using the coupled enzyme system at the optimal pH. Reactions with sarcosine were performed in 100 mM HEPES (pH 8.0), while reactions with dimethylglycine were performed in 100 mM HEPES (pH 7.5). The apparent kinetic parameters for sarcosine and dimethylglycine were determined with SAM fixed at 150 μM. The kinetic parameters for SAM were determined with either sarcosine or dimethylglycine fixed at 20 mM. Reactions of GsSDMT with 20 mM of either glycine or alanine were also tested using the coupled enzyme system in 100 mM HEPES (pH 7.5) with 150 μM SAM.
Thermal Stability
CD measurements were carried out in a Jasco J-175 spectropolarimeter with a Jasco PTC-348WI peltier-effect temperature controller. GsSDMT (0.1 mg/mL) in 10 mM sodium phosphate, pH 7.5, containing 50 mM NaCl was placed in a 0.1 cm quartz cuvette. Samples were monitored at 195 nm and 222 nm and were taken at 3 °C intervals. Measurements were taken 5 min after the cell temperature had reached the desired level.
Crystallization
Initial screening was performed with an in-house, 192 condition crystallization screen. Optimized GsSDMT crystals were grown by the hanging drop vapor-diffusion method. The reservoir solution contained 23% (w/v) or methyl ether polyethylene glycol 5000, 30 mM sarcosine, and 100 mM (pH 7.0). The hanging drop consisted of 2 μL of thawed protein solution mixed with 2 μL of reservoir solution. Crystallization trays were stored at 20 °C. GsSDMT crystals took 2 weeks to reach full size (300 × 300 × 300 μm). Crystals were soaked in increasing concentrations of ethylene glycol (up to 15%) and flash-frozen in a stream of liquid N2.
Data Collection, Reduction, and Structure Solution
Diffraction data were collected at wavelengths of 0.9792 Å and 0.9640 Å at the Advanced Photon Source on General Medicine and Cancer Institutes Collaborative Access Team beamline 23-ID-D. Data were collected with 1° oscillations per frame, an exposure time of 3 s, and 200-fold attenuation of the incident beam at a temperature of 93 K. Reflections were indexed, integrated, and scaled using the HKL2000 package 26.
A total of 12 out of 12 potential selenium sites were identified with HySS 27,28. The data were phased via multi-wavelength anomalous diffraction using autoSHARP with the help of auxiliary programs from the CCP4 suite 29,30. The structure was then completed through multiple rounds of model building with Coot and refinement with REFMAC 31,32. The quality of the final structure was assessed with ProCheck and Molprobity 33,34.
RESULTS
Purification of GsSDMT
The G. sulphuraria protein was highly expressed in E. coli B834 as fusion to His8-MBP, of which approximately 35% was soluble. After Ni IMAC purification, the fusion protein was cleaved by treatment with TEV protease with 95 - 100% efficiency. The liberated GsSDMT had Mr of ~34,000 Da in denaturing electrophoresis experiments, and an apparent Mr of ~67,700 Da in gel filtration, suggesting that GsSDMT had a dimeric quaternary structure in solution.
Structure Determination
Table I summarizes the crystallographic statistics. Crystals grown from methyl ether polyethylene glycol 5000 were from space group C2221. The GsSDMT model was refined to a nominal resolution of 1.95 Å, with Rcryst of 16.8% and Rfree of 22.1%. The asymmetric unit contained 8744 protein atoms and 1042 ordered water molecules. The protein atoms in the asymmetric unit were organized in four polypeptide chains, labeled A, B, C, and D. The amino terminal of each chain could not be modeled due to insufficient electron density in the map. This included the first 15 residues of chain A, 24 residues of chain B, 25 residues of chain C, and 29 residues of chain D. Likewise, the residues Ile256, Ala257, and Ser258 found in a surface loop of chain D could not be modeled.
Table I.
A summary of crystal parameters, data collection, and refinement statistics for GsSDMT. The values in parentheses are for the highest resolution shell.
| Peak | Remote | |
|---|---|---|
| Spacegroup | C2221 | |
| Unit Cell Parameters (Å) | a=97.5 b=291.4 c=86.3 | |
| Collection Statistics | ||
| Wavelength | 0.97923 | 0.96400 |
| Resolution Range (Å) | 48.77 - 1.95 (2.02-1.95) | 48.77 -1.95 (2.02-1.95) |
| Measured Reflections (Unique) | 646950 (89709) | 1306077 (89078) |
| Completeness (%) | 99.4 (95.0) | 99.9 (99.4) |
| Rmerge a | 0.110 (0.597) | 0.092 (0.605) |
| Redundancy | 7.2 (5.1) | 14.7 (11.7) |
| Mean I/σ | 14.69 (2.06) | 28.86 (3.5) |
| Phasing Statistics | ||
| Phasing Power | 1.622 | |
| Mean FOM centric (acentric) | 0.303 (0.475) | |
| Rcullis | 0.673 | |
| Refinement Statistics | ||
| Resolution Range (Å) | 48.77-1.95 | |
| Total Reflections (test) | 89679 (4502) | |
| Rcrystb / Rfreec | 0.166 / 0.221 | |
| Rmsd bonds (Å) | 0.017 | |
| Rmsd angles (°) | 1.452 | |
| ESU from Rfree | 0.149 | |
| No. of atoms | 9790 | |
| Mean B-factor protein / water | 19.6 / 38.0 | |
| Ramachandran (%) | ||
| Core region | 94.1 | |
| Allowed region | 5.9 |
Rmerge = ΣhΣi|Ii(h) - <I(h)>|/ ΣhΣiIi(h), where Ii(h) is the intensity of an individual measurement of the reflection and <I(h)> is the mean intensity of the reflection.
Rcryst = Σh||Fobs|-|Fcalc||/Σh|Fobs|, where Fobs and Fcalc are the observed and calculated structure-factor amplitudes, respectively.
Rfree was calculated as Rcryst using 5.0% of the randomly selected unique reflections that were omitted from structure refinement.
Description of Fold
Figure 2 shows that GsSDMT has a fold that is similar to the class I methyltransferases, namely an NAD(P)-binding Rossman fold in which the central β-sheet contains seven β-strands 35. Except for the β-strand at the carboxy terminal of the protein, the β-strands have a parallel arrangement. The core β-sheet is flanked by 14 helices.
Figure 2.

A cartoon representation of the fold of a monomer of GsSDMT. The color scheme changes from blue to green to represent regions predicted to interact with the SAM cofactor. The helices are labeled H1 through H14, and the β-strands are labeled A through G.
The alpha carbons of chains B, C, and D align with those of chain A with rmsd-values of 0.593 Å, 0.471 Å, and 1.10 Å, respectively. Likewise the rmsd-values between chains B and C, B and D, and C and D are 0.781 Å, 1.32 Å, and 1.10 Å, respectively. Most of the disorder, particularly in chain D, occurs in helices H1 and H14 surrounding the active site. The overall mean B value for all atoms is 21.56, and all phi/psi angles were in acceptable regions of the Ramachandran plot with 94.1% being in the most favored regions.
The four protein chains are arranged as two homodimers in the asymmetric unit (Figure 3). The surface area buried within the homodimer interface is approximately 980 Å236. The majority of this interface occurs between the penultimate F β-strands of the two chains, resulting in the formation of a 14-stranded β-sheet extending through the core of the protein. However, there are some additional interactions between the loop prior to helix H3 (residues 50-52) and the carboxy terminal of helix H4 (residues 73-78) provided by both members of the homodimer. Between chains A and B, these interactions provide a total of 16 hydrogen bonds and 4 salt bridges in the dimer interface. There are only weak contacts between the dimer units in the asymmetric unit.
Figure 3.
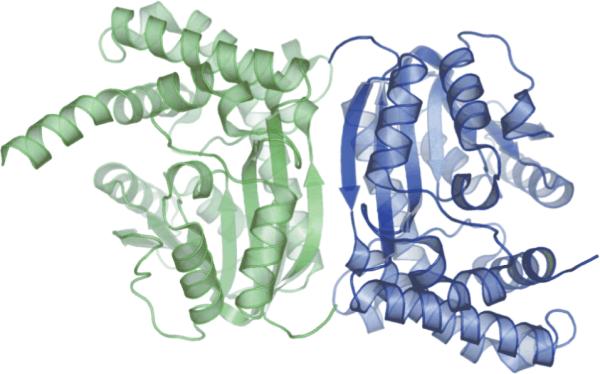
An illustration of the GsSDMT dimer interface, with the subunits of the dimer colored green and blue.
Comparison of SDMT Sequences
Figure 4 shows a partial sequence alignment of known SDMT enzymes. Overall, GsSDMT displays between 38 to 40% identity to SDMT from Synechococcus sp. WH8102 and E. halochloris, the dimethylglycine methyltransferase from A. halophytica, and to one domain of the multidomain A. halophila enzyme. This alignment also contains methoxymycolic acid synthase 2 (MMAS2), whose structure has been solved in complex with SAH. Five distinct regions corresponding to residues that line the cofactor-binding site in MMAS2 and the corresponding residues predicted for the cofactor-binding site in GsSDMT are shown.
Figure 4.

Partial sequence alignment between GsSDMT and other experimentally verified SDMTs. Conserved residues are capitalized. Conserved residues near the predicted substrate-binding site are designated with an asterisk. The residues colored blue surround the predicted site of the cofactor.
Steady-State Kinetic Characterization
GsSDMT had an optimal pH of 8 for methylation of sarcosine, while pH 7.5 was optimal for methylation of dimethylglycine (Figure 5). Table II shows the steady-state kinetic parameters determined for GsSDMT at these optimal pH values. The apparent kinetic parameters for SAM were determined with either sarcosine or dimethylglycine in fixed, large excess (20 mM), while kinetic studies of sarcosine and dimethylglycine were carried out using a fixed concentration of 150 μM SAM. The apparent kcat and KM values for sarcosine and dimethylglycine were comparable at fixed SAM: 86 min-1 and 3 mM for dimethylglycine and 64 min-1 and 2 mM for sarcosine. The apparent KM value for SAM was similar for both substrates at 150 μM for dimethylglycine and 144 μM for sarcosine. Due to the high absorbance at 265 nm in the presence of high SAM concentration, the apparent kcat of 77.2 min-1 and 51.8 min-1 were estimated by non-linear least squares fitting of the lower concentration data for dimethylglycine and sarcosine respectively.
Figure 5.
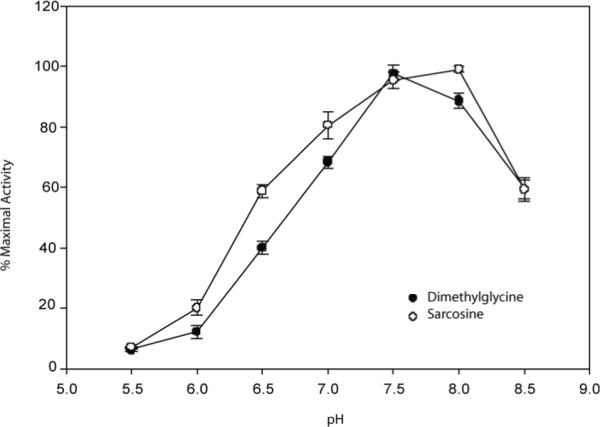
A plot of the pH vs. % relative activity of SDMT. Reactions were performed from pH 5.5-8.5 in 100 mM MES, 100 mM HEPES, 100 mM TAPS and 100 μM MgCl2 at 37 °C. Activity levels of dimethylglycine (•) and sarcosine (°) are normalized to the pH of maximal activity. Reactions at each pH were performed in triplicate.
Table II.
Kinetic Parameters of GsSDMT Catalysis.
| Varied Substrate (Constant) | kcat (min-1) | KM |
|---|---|---|
| Dimethylglycine (SAM 150 μM) | 85.6 (5.1) | 2.8 (.6) mM |
| SAM (dimethylglycine 20 mM) | 77.2 (4.8) | 150 (38) μM |
| Sarcosine (SAM 150 μM) | 64.3 (4.2) | 2.0 (.7) mM |
| SAM (Sarcosine 20 mM) | 51.8 (4.5) | 144 (44) μM |
Specificity for Different Methyl Acceptors
The activity of GsSDMT towards structurally related glycine was approximately 1 min-1, which was less than 2% of the activity observed with either sarcosine or dimethylglycine. There was no detectable activity with alanine. There was also no detectable activity for the reverse reaction, the transfer of a methyl group from betaine back to SAH.
Thermal Stability
The melting profile of GsSDMT is depicted in Figure 6. The melting temperature of the enzyme was 61.1 (0.5) °C. The melting profile of the enzyme with 200 μM SAH and 1 mM betaine was virtually identical to that of the substrate-free enzyme.
Figure 6.
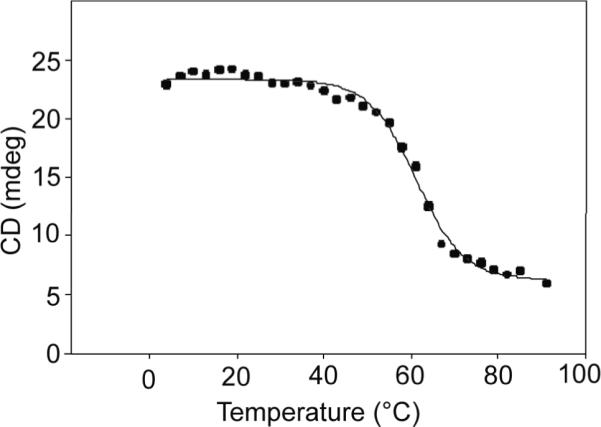
Melting temperature profile for GsSDMT.
DISCUSSION
Functional Annotation
This work has provided a conclusive functional identity for the 34 kDa protein encoded by G. sulphuraria gene Gs07580.1. This enzyme is a SAM-dependent sarcosine and dimethylglycine transferase. This enzyme fits into a pathway for betaine biosynthesis previously identified in halophilic bacteria, but now also clearly associated with hyperacidophilic, thermophilic eukaryotes. Since the enzyme has no appreciable reactivity with glycine or alanine, completion of betaine biosynthesis by this pathway in G. sulphuraria requires the participation of one other methyltransferase for conversion of glycine to sarcosine. Analysis of the G. sulphuraria genome to identify the glycine methyltransferase has so far been unsuccessful.
GsSDMT is abundantly expressed in E. coli as a fusion to His8-MBP, and the liberated protein is relatively soluble, easily purified, and handled. Independent of substrates, GsSDMT has a melting temperature of ~60 °C, which is reasonable considering the mesophilic growth temperature of G. sulphuraria.
Catalytic Specificity
The observed specificity of GsSDMT is consistent with that of other SDMT enzymes, which typically have high specificity for one or two of the successive products in the betaine biosynthetic pathway, but not for all three substrates (Figure 1). This specificity may help to avoid unproductive methylation of other amino acids in vivo, but also provides possibilities for multiple flux regulation points through the biosynthetic pathway.
GsSDMT can methylate both sarcosine and dimethylglycine. The kcat values for these two substrates (~65 to 85 min-1, Table II) are comparable to other known SDMT enzymes 10. Additionally, the apparent Km values for sarcosine (2.0 mM) and dimethylglycine (2.8 mM) are within the range of the previously characterized SDMT enzymes, providing compelling evidence that methyl-group transfer for betaine synthesis is the physiological role of this enzyme in G. sulphuraria 10.
Similarity to other Structures
We searched the Protein Data Bank using the FFAS03 server for other proteins with either structure homology or sequence similarity to GsSDMT 37. The strongest FFAS03 scores were from MMAS2 and methoxymycolic acid synthase 4 (PDB IDs 1tpy and 2fk7) and mycolic acid cyclopropane synthase (PDB ID 1kp9) from Mycobacterium tuberculosis 38,39. However, pairwise alignments of GsSDMT with these sequences showed less than 20% identity. Despite the low sequence identity, the alpha carbon rmsd between 238 comparable residues of SDMT chain A and 2fk7 was 2.08 Å, was 1.83 Å for 1kp9 chain A (232 comparable residues), and was 1.82 Å for 1tpy (240 comparable residues).
Unlike GsSDMT, each of the M. tuberculosis enzymes crystallized as a monomer. GsSDMT contains insertions of a few residues in the loops corresponding to residues 50 through 52 and 73 through 78 relative to that of the M. tuberculosis proteins. These loops interact with each other in the GsSDMT homodimer, providing an additional four hydrogen bonds, two salt bridges, and 190 Å2 of buried surface area. This may play a role in the difference in oligomerization in these structurally related enzymes.
Predicted Cofactor Binding Site
MMAS2 was crystallized in the presence of SAH and six out of the ten hydrogen-bonding interactions between MMAS2 and SAH involved backbone nitrogen and oxygen atoms. The backbone atoms of GsSDMT closely match the MMAS2 backbone in this region with an alpha carbon rmsd of 0.92 (between GsSDMT residues 44-46, 88 - 96, 112-117, 140-142, and 156-163 and the homologous residues in MMAS2). This strongly suggests that the cofactor may bind in a similar manner in GsSDMT (Figure 7). In MMAS2, the side chain atoms of Ser34, Thr94, Gln99, and Glu124 provide four interactions that do not involve the backbone atoms. In GsSDMT, Gln117 and Glu143 are substituted for Gln99 and Glu124. Furthermore, Ser34 is replaced with His46, and Thr94 is replaced with Asn112. These two latter substitutions may also provide interactions with SAM or SAH, albeit with small conformational adjustments in the backbone and side chain positions. The active site cavities of the enzymes were also very similar. The cavity volume of MMAS2 was 1510 Å3 while that of GsSDMT (chain B) was 2064 Å3 as calculated via CASTp 40. The variation in the cavity size appeared to be caused by conformational changes in the loop between helices H10 and H11 and the incomplete N-terminal of GsSDMT.
Figure 7.

Stereoview of GsSDMT and MMAS2 structural alignment. GsSDMT (blue backbone) was aligned with MMAS2 (green backbone) with SAH cofactor in the active site. Conserved functional groups within the active site include 6 of 10 backbone nitrogen and oxygen atoms and Gln117 and Glu124. In addition, N112 and H46 appear to be potential residues capable of interaction with SAH.
Multiple attempts were made to solve the structure in the presence of cofactor or substrate. The crystals studied here were obtained from a crystallization solution that contained 30 mM sarcosine, yet no recognizable density for sarcosine was observed. In contrast, cocrystallization experiments containing SAM or SAH failed to produce crystals.
Attempts to soak SAM into GsSDMT crystals at concentrations ranging from 5 mM to 50 mM failed to produce any observable electron density for the cofactor within the resulting maps. In the methoxymycolic acid synthase 4 structure 38, the helix comparable to the short helix H7 from GsSDMT (Figure 2) formed only upon binding of SAM. In contrast, the GsSDMT helix H7 was formed in the crystals without the presence of SAM and may thus prevent the necessary induced fit motions required for binding of the cofactor.
Comparison to Glycine N-Methyltransferase
Glycine N-methyltransferase (GNMT) catalyzes the transfer of a methyl group from SAM to glycine resulting in the formation of SAH and sarcosine 41. This reaction is believed to help regulate the ratio of SAM to SAH in the cell 42. The crystal structure of recombinant GNMT from rat liver bound with SAM has been solved (PDB ID 1xva, 43). Unlike dimeric GsSDMT, GNMT forms a tetramer where the amino terminal of each monomer fits into the active site of an adjacent monomer. A comparison of the GsSDMT and GNMT monomers highlights the differences between the two folds (Figure 8). With the exception of a few inserted loops, the backbones of the two structures follow each other fairly closely up to residue 190 of GsSDMT. After this, GNMT deviates to form a secondary 3-stranded β-sheet that closes one side of the active site. As a consequence of this change in secondary structure, the nucleotide and sugar portions of the SAM cofactor bind to GNMT in a different location from that predicted for GsSDMT (Figure 8). However, despite this difference in binding for the majority of the cofactor, the sulfur atoms of the two cofactors reside within 3 Å of each other upon overlaying the two monomers.
Figure 8.

Ribbon diagrams of GsSDMT (above) and GNMT (below) illustrating the difference in cofactor binding between the two enzymes. Color is used to define the structurally similar amino terminals of the proteins (green) as well as the structurally divergent carboxy terminals (blue in GsSDMT and red in GNMT). The placement of the cofactor (carbon:gray, sulfur:yellow, nitrogen:blue, oxygen:red) in GsSDMT is predicted based on the structure of MMAS2.
Comparison of SDMT Sequences
A number of other conserved residues whose side chains are near to the predicted substrate-binding site are also shown in Figure 7. These include Asp33, Asp44, His46, Asn117, Asp158, and His162. Of these, His162 is positioned 4.9 Å from the predicted position of the SAH sulfur atom, and thus appears to be positioned for orienting the carboxyl group of sarcosine or dimethylglycine. Rotation of the Asp158 side chain places the carboxyl oxygen within 3 Å of the SAH sulfur group, which could potentially interact with the positively charged amine group of the substrate.
In a previous study, the P171Q, M172R, and P171Q/M172R mutants of the A. halophytica dimethylglycine methyltransferase were constructed and tested for activity 7. The M172R and P171Q/M172R mutations gave an inactive enzyme, while the P171Q mutation reduced the kcat to 27% of the wild-type enzyme. In addition, the apparent KM for dimethylglycine was increased 15-fold, while the apparent KM for SAM increased 2-fold. These results were interpreted to indicate that Pro171 and Met172 might be involved in substrate binding. However, their equivalents in GsSDMT, Pro188 and Met189, are positioned greater than 8 Å away from the active site, such that it is unlikely that they can be directly involved in the binding of dimethylglycine. They are however positioned near the helix containing residues Asp158 and His162. Assuming the fold of the A. halophytica dimethylglycine methyltransferase is similar to that of GsSDMT, it seems likely these mutations may result in misalignment of the dimethylglycine methyltransferase equivalents of Asp158 and His162 (Asp141 and His145), rather than directly interacting with the substrate.
CONCLUSION
Our kinetic analysis of the product of G. sulphuraria gene Gs07580.1 shows that the enzyme functions as a sarcosine dimethylglycine methyltransferase. This represents the first instance of this activity being observed outside of certain salt-tolerant prokaryotic organisms, and is also the first example of this enzyme activity from a eukaryote. The structural similarity of the enzyme to mycolic acid synthases allows us to predict the binding site of both the cofactor, SAM, as well as that residues involved in substrate recognition.
ACKNOWLEDGEMENTS
We thank the members of the Center for Eukaryotic Structural Genomics team. Special thanks are also offered to the personnel of the Galdieria sulphuraria Genome Project at Michigan State University for their assistance with the gene models. National Institutes of Health, Protein Structure Initiative grant U54 GM074901 (John L. Markley, PI; G.N.P., Co-Investigator; B.G.F.; Co-Investigator) supported this work. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. W-31-109-ENG-38. General Medicine and Cancer Institutes Collaborative Access Team has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). J.G.M. is supported by an NHGRI training grant to the Genomic Sciences Training Program (5T32HG002760). L.J.B. is supported by National Science Foundation grant MCB-0316232 to B.G.F.
REFERENCES
- 1.Schliess F, Haussinger D. The cellular hydration state: a critical determinant for cell death and survival. Biol Chem. 2002;383(3-4):577–583. doi: 10.1515/BC.2002.059. [DOI] [PubMed] [Google Scholar]
- 2.Finkelstein JD, Martin JJ, Harris BJ, Kyle WE. Regulation of hepatic betaine-homocysteine methyltransferase by dietary betaine. J Nutr. 1983;113(3):519–521. doi: 10.1093/jn/113.3.519. [DOI] [PubMed] [Google Scholar]
- 3.Santoro MM, Liu Y, Khan SM, Hou LX, Bolen DW. Increased thermal stability of proteins in the presence of naturally occurring osmolytes. Biochemistry. 1992;31(23):5278–5283. doi: 10.1021/bi00138a006. [DOI] [PubMed] [Google Scholar]
- 4.Ladas NP, Papageorgiou GC. Cell turgor: A critical factor for the proliferation of cyanobacteria at unfavorable salinity. Photosynth Res. 2000;65(2):155–164. doi: 10.1023/A:1006423221150. [DOI] [PubMed] [Google Scholar]
- 5.Sakamoto A, Murata N. The role of glycine betaine in the protection of plants from stress: clues from transgenic plants. Plant Cell Environ. 2002;25(2):163–171. doi: 10.1046/j.0016-8025.2001.00790.x. [DOI] [PubMed] [Google Scholar]
- 6.Lu WD, Chi ZM, Su CD. Identification of glycine betaine as compatible solute in Synechococcus sp. WH8102 and characterization of its N-methyltransferase genes involved in betaine synthesis. Arch Microbiol. 2006;186(6):495–506. doi: 10.1007/s00203-006-0167-8. [DOI] [PubMed] [Google Scholar]
- 7.Waditee R, Tanaka Y, Aoki K, Hibino T, Jikuya H, Takano J, Takabe T, Takabe T. Isolation and functional characterization of N-methyltransferases that catalyze betaine synthesis from glycine in a halotolerant photosynthetic organism Aphanothece halophytica. J Biol Chem. 2003;278(7):4932–4942. doi: 10.1074/jbc.M210970200. [DOI] [PubMed] [Google Scholar]
- 8.Nyyssola A, Kerovuo J, Kaukinen P, von Weymarn N, Reinikainen T. Extreme halophiles synthesize betaine from glycine by methylation. J Biol Chem. 2000;275(29):22196–22201. doi: 10.1074/jbc.M910111199. [DOI] [PubMed] [Google Scholar]
- 9.Lai MC, Wang CC, Chuang MJ, Wu YC, Lee YC. Effects of substrate and potassium on the betaine-synthesizing enzyme glycine sarcosine dimethylglycine N-methyltransferase from a halophilic methanoarchaeon Methanohalophilus portucalensis. Res Microbiol. 2006;157(10):948–955. doi: 10.1016/j.resmic.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Nyyssola A, Reinikainen T, Leisola M. Characterization of glycine sarcosine N-methyltransferase and sarcosine dimethylglycine N-methyltransferase. Appl Environ Microbiol. 2001;67(5):2044–2050. doi: 10.1128/AEM.67.5.2044-2050.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brock TD, University of Wisconsin--Madison . Thermophilic microorganisms and life at high temperatures. Springer-Verlag; New York: 1978. p. xi.p. 465. Libraries. [Google Scholar]
- 12.Rigano C, Fuggi A, Di Martino Rigano V, Aliotta G. Studies on utilization of 2-ketoglutarate, glutamate and other amino acids by the unicellular alga Cyanidium caldarium. Arch Microbiol. 1976;107(2):133–138. doi: 10.1007/BF00446832. [DOI] [PubMed] [Google Scholar]
- 13.Gross W, Schnarrenberger C. Heterotrophic growth of two strains of the acido- thermophilic red alga Galdieria sulphuraria. Plant and Cell Physiology. 1995;36(4):633–638. [Google Scholar]
- 14.Oesterhelt C, Schnarrenberger C, Gross W. Characterization of a sugar/polyol-uptake system in the red alga Galdiera sulphuraria. Eur J Phycol. 1999;34:271–277. [Google Scholar]
- 15.Albertano P, Ciniglia C, Pinto G, Pollio A. The taxonomic position of Cyanidium, Cyanidioschyzon, and Galdieria: an update. Hydrobiologia. 2000;433:137–143. [Google Scholar]
- 16.Weber AP, Oesterhelt C, Gross W, Brautigam A, Imboden LA, Krassovskaya I, Linka N, Truchina J, Schneidereit J, Voll H, Voll LM, Zimmermann M, Jamai A, Riekhof WR, Yu B, Garavito RM, Benning C. EST-analysis of the thermo-acidophilic red microalga Galdieria sulphuraria reveals potential for lipid A biosynthesis and unveils the pathway of carbon export from rhodoplasts. Plant Mol Biol. 2004;55(1):17–32. doi: 10.1007/s11103-004-0376-y. [DOI] [PubMed] [Google Scholar]
- 17.Jenney FE, Adams MW. The impact of extremophiles on structural genomics (and vice versa) Extremophiles. 2007 doi: 10.1007/s00792-007-0087-9. [DOI] [PubMed] [Google Scholar]
- 18.DiDonato M, Deacon AM, Klock HE, McMullan D, Lesley SA. A scaleable and integrated crystallization pipeline applied to mining the Thermotoga maritima proteome. J Struct Funct Genomics. 2004;5(1-2):133–146. doi: 10.1023/B:JSFG.0000029194.04443.50. [DOI] [PubMed] [Google Scholar]
- 19.Lesley SA, Kuhn P, Godzik A, Deacon AM, Mathews I, Kreusch A, Spraggon G, Klock HE, McMullan D, Shin T, Vincent J, Robb A, Brinen LS, Miller MD, McPhillips TM, Miller MA, Scheibe D, Canaves JM, Guda C, Jaroszewski L, Selby TL, Elsliger MA, Wooley J, Taylor SS, Hodgson KO, Wilson IA, Schultz PG, Stevens RC. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc Natl Acad Sci U S A. 2002;99(18):11664–11669. doi: 10.1073/pnas.142413399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zolnai Z, Lee PT, Li J, Chapman MR, Newman CS, Phillips GN, Jr., Rayment I, Ulrich EL, Volkman BF, Markley JL. Project management system for structural and functional proteomics: Sesame. J Struct Funct Genomics. 2003;4(1):11–23. doi: 10.1023/a:1024684404761. [DOI] [PubMed] [Google Scholar]
- 21.Thao S, Zhao Q, Kimball T, Steffen E, Blommel PG, Riters M, Newman CS, Fox BG, Wrobel RL. Results from high-throughput DNA cloning of Arabidopsis thaliana target genes using site-specific recombination. J Struct Funct Genomics. 2004;5(4):267–276. doi: 10.1007/s10969-004-7148-4. [DOI] [PubMed] [Google Scholar]
- 22.Sreenath HK, Bingman CA, Buchan BW, Seder KD, Burns BT, Geetha HV, Jeon WB, Vojtik FC, Aceti DJ, Frederick RO, Phillips GN, Jr., Fox BG. Protocols for production of selenomethionine-labeled proteins in 2-L polyethylene terephthalate bottles using auto-induction medium. Protein Expr Purif. 2005;40(2):256–267. doi: 10.1016/j.pep.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 23.Jeon WB, Aceti DJ, Bingman CA, Vojtik FC, Olson AC, Ellefson JM, McCombs JE, Sreenath HK, Blommel PG, Seder KD, Burns BT, Geetha HV, Harms AC, Sabat G, Sussman MR, Fox BG, Phillips GN., Jr. High-throughput purification and quality assurance of Arabidopsis thaliana proteins for eukaryotic structural genomics. J Struct Funct Genomics. 2005;6(2-3):143–147. doi: 10.1007/s10969-005-1908-7. [DOI] [PubMed] [Google Scholar]
- 24.Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif. 2007;55(1):53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorgan KM, Wooderchak WL, Wynn DP, Karschner EL, Alfaro JF, Cui Y, Zhou ZS, Hevel JM. An enzyme-coupled continuous spectrophotometric assay for S-adenosylmethionine-dependent methyltransferases. Anal Biochem. 2006;350(2):249–255. doi: 10.1016/j.ab.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276(A):307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 11):1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 28.Weeks CM, Adams PD, Berendzen J, Brunger AT, Dodson EJ, Grosse-Kunstleve RW, Schneider TR, Sheldrick GM, Terwilliger TC, Turkenburg MG, Uson I. Automatic solution of heavy-atom substructures. Methods Enzym. 2003;374:37–83. doi: 10.1016/S0076-6879(03)74003-8. [DOI] [PubMed] [Google Scholar]
- 29.de la Fortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple insomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzym. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project N The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DM, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystalogr. 1993;26:283–291. [Google Scholar]
- 34.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28(6):329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci U S A. 1996;93(1):13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A. FFAS03: a server for profile--profile sequence alignments. Nucleic Acids Res. 2005;33:W284–288. doi: 10.1093/nar/gki418. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boissier F, Bardou F, Guillet V, Uttenweiler-Joseph S, Daffe M, Quemard A, Mourey L. Further insight into S-adenosylmethionine-dependent methyltransferases: structural characterization of Hma, an enzyme essential for the biosynthesis of oxygenated mycolic acids in Mycobacterium tuberculosis. J Biol Chem. 2006;281(7):4434–4445. doi: 10.1074/jbc.M510250200. [DOI] [PubMed] [Google Scholar]
- 39.Huang CC, Smith CV, Glickman MS, Jacobs WR, Jr., Sacchettini JC. Crystal structures of mycolic acid cyclopropane synthases from Mycobacterium tuberculosis. J Biol Chem. 2002;277(13):11559–11569. doi: 10.1074/jbc.M111698200. [DOI] [PubMed] [Google Scholar]
- 40.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–118. doi: 10.1093/nar/gkl282. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heady JE, Kerr SJ. Purification and characterization of glycine N-methyltransferase. J Biol Chem. 1973;248(1):69–72. [PubMed] [Google Scholar]
- 42.Balaghi M, Horne DW, Wagner C. Hepatic one-carbon metabolism in early folate deficiency in rats. Biochem J. 1993;291(Pt 1):145–149. doi: 10.1042/bj2910145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu Z, Hu Y, Konishi K, Takata Y, Ogawa H, Gomi T, Fujioka M, Takusagawa F. Crystal structure of glycine N-methyltransferase from rat liver. Biochemistry. 1996;35(37):11985–11993. doi: 10.1021/bi961068n. [DOI] [PubMed] [Google Scholar]