

Abstract
While bacterial iterative type I polyketide synthases are now known to participate in the biosynthesis of a small set of diverse natural products, the subsequent downstream modification of the resulting polyketide products remains poorly understood. Toward this goal, we report the X-ray structure determination at 2.5 Å resolution and preliminary characterization of the putative orsellenic acid P450 oxidase (CalO2) involved in calicheamicin biosynthesis. These studies represent the first crystal structure for a P450 involved in modifying a bacterial iterative type I polyketide product and suggest the CalO2-catalyzed step may occur after CalO3-catalyzed iodination and may also require a coenzyme A- (CoA) or acyl carrier protein- (ACP) bound substrate. Docking studies also reveal a putative docking site within CalO2 for the CLM orsellinic acid synthase (CalO5) ACP domain which involves a well-ordered helix along the CalO2 active site cavity that is unique compared to other P450 structures.
Keywords: ACP, type I PKS, natural product, biosynthesis, thioester
INTRODUCTION
Due to their remarkable molecular architectures, spectacular biological activities and therapeutic value, the enediynes are often considered among some of the most notorious natural products known to man. There exist two naturally-occurring enediyne structural subfamilies - the chromoprotein (or 9-membered) enediynes and 10-membered enediynes.1,2 The chromoprotein enediynes contain a bicyclo[7.3.0]enediyne 9-membered core and typically require a specific protein for enediyne stabilization. The 10-membered enediynes share a common bicyclo[7.3.1]enediyne core and, while the 10-membered enediynes lack protein stabilizers, some producing organisms of this family rely upon a novel `self-sacrifice' resistance protein.3,4 All enediynes are highly efficient DNA/RNA-damaging agents. As exemplified by mechanistic studies of calicheamicin (CLM) γ1I (Fig. 1, 5), a prominent member of the 10- membered enediynes, the CLM aryltetrasaccharide docks in the minor groove of the target DNA to facilitate an efficiently precise, and lethal, oxidative DNA-strand scission event - initiated via a highly reactive diradical intermediate of enediyne cycloaromatization.5-8 The incredible in vitro and in vivo potency of CLM also paved the way for CLM-based therapeutics with the CLM-CD33 antibody conjugate (Mylotarg) emerging as the first approved antibody-cytotoxin based drug in 2000.9-11 This demonstrated clinical success continues to fuel both synthetic and biosynthetic efforts toward the generation of enediyne analogs with optimized therapeutic properties.12-15
Fig. 1.
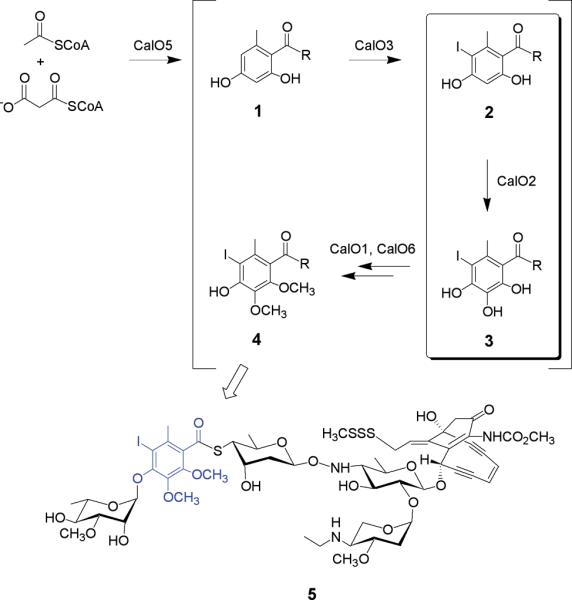
The proposed pathway for orsellinic acid biosynthesis and modification en route to calicheamicin (5) construction. The putative reaction catalyzed by CalO2 is highlighted within the box and the mature orsellinate progeny in the context of calicheamicin is emphasized in blue. Based upon the ligand-binding studies described, `R' is anticipated to be an ACP or CoA thioester.
Early metabolic labeling studies suggested the 9- and 10-membered enediynes derive from distinct biosynthetic pathways.16-18 In contrast, the recent cloning and characterization of gene clusters encoding both 9-membered and 10-membered enediynes revealed an enediyne polyketide synthase (PKSE) gene common to all enediyne loci and thereby established the first unified, divergent polyketide paradigm for enediyne core biosynthesis.19-25 In addition to the novel enediyne core, most enediynes also contain additional constituents of polyketide origin as exemplified by the CLM orsellenic acid moiety. The biosynthesis of such enediyne substructures (Fig. 1, 1) are catalyzed by a unique set of iterative type I polyketide synthases (PKSs) reminiscent of the fungal 6-methylsalicyclic acid synthase (6-MSAS)26 and are subsequently modified via enzymatic oxidation, halogenation and/or methylation (Fig. 1, 2→4). While bacterial iterative type I PKSs are now known to participate in the biosynthesis of at least five diverse natural products - avilamycin,27 CLM,19 neocarzinostatin,23,28 chlorothricin29,30 and maduropeptin24 - the subsequent downstream modification of the resulting polyketide products remains poorly understood. Toward this goal, we report the X-ray structure determination of the putative CLM orsellenic acid P450 oxidase (CalO2) and subsequent ligand-binding studies in the presence of various substrate analogs. These studies represent the first crystal structure for a P450 involved in modifying a bacterial iterative type I polyketide product and suggest the CalO2-catalyzed step may occur after CalO3-catalyzed iodination and may also require a coenzyme A- (CoA) or acyl carrier protein- (ACP) bound substrate.
MATERIALS AND METHODS
Protein purification
The calO2 gene was amplified from M. echinospora genomic DNA using the primer pair calO2-forward (5'-ggaaggggcaccatatgctggtcgatgc-3') and calO2-reverse (gctcatgtcgagatctcctcctgctcg) and ligated into a Ndel/Smal-digested pUC-based plasmid. Upon confirmation via DNA sequencing, the Ndel/Smal calO2-containing fragment was subcloned into the E.coli-Streptomyces expression shuttle vector pPW50 for expression of the N-His6-CalO2 fusion protein (referred to as CalO2 throughout this manuscript) in Streptomyces. For constitutive expression in Streptomyces lividans a single transformant was used to inoculate starter cultures of 75 mL YEME broth containing 25 mg/mL thiostrepton in 250 mL baffled flasks with 4 g of glass beads. Cultures were grown at 28 °C until they reached a pinkish color, generally 2 to 3 days. Aliquots (500 μL) from the starter culture were subsequently used to inoculate expression cultures (75 mL YEME broth, 25 mg/mL thiostrepton in 250 mL baffled flask containing 4 g of glass beads). The expression cultures were grown 2 to 3 days, at which time they were supplemented with 1 mM 5-aminolevulinic acid and 750 μL of a trace elements solution (0.9 mM EDTA, 38.0 mM ZnSO4-7H2O; 18.0 mM FeSO4-7H2O; 1.9 mM CoCl2, pH 8.0). Cultures were then grown for an additional 4 to 5 days and cells were harvested via centrifugation (3,000 xg, 40 min) and stored at -80 °C. Following this protocol, the typical cell yield was 180 gm/L.
All purification steps were carried out at 4 °C. The cell pellet (~ 180 gm) was resuspended in 120 mL of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 1 mg/mL lysozyme pH 8.0) and incubated on ice for 30 minutes after which time the cells were lysed by sonication (Virsonic 475; Virtis, Gardiner, NY; 100 W, 8 × 45 sec pulses, 1 min between pulses, 0 °C). The cell debris was removed by centrifugation (3,000 xg, 40 min) and the recovered supernatant was combined with 2 mL Ni-NTA resin (Qiagen) and the mixture incubated on an orbital mixer (Reliable Scientific, location, 8 rpm, 4 °C) for 1 hr. The mixture was loaded onto a disposable 5 mL column, the resin washed with 10 mL wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) and CalO2 eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0) until the red color was no longer visible on the resin (~ 5 mL). The CalO2-containing eluent was concentrated to 2.5 mL via diafiltration (Vivaspin 15 mL column with Hydrosart membrane, 30 KDa MWCO, 3000 xg). UV-vis analysis of protein at this stage revealed a typical absorbance at 423 nm (data not shown), indicative of an inhibitor-bound heme. The concentrated sample was exchanged with assay/storage buffer (50 mM potassium phosphate buffer, pH 8.0) using a PD-10 column (Amersham Biosciences) and the eluent concentrated to a protein concentration of approximately 10 mg/mL as determined by the Bradford Assay (Bio-Rad, Hercules, CA), flash frozen with liquid nitrogen, and stored at -80 °C. Upon buffer exchange, the CalO2 heme Soret peak shifted to a single peak at 418 nm, indicative of low spin iron bound to water. The reduced carbon monoxide bound form of N-His6- CalO2 displayed a typical P450 Soret peak at 450 nm (Fig. 2A) with a small amount of the inactive P420 form which was found to increase over time (Fig. 2B).
Fig. 2.

(A) Spectrum of oxidized CalO2 with CO difference spectrum (inset). (B) Increase in the inactive P420 enzyme species over time (0 to 15 min).
Ligand binding
An array of benzoic acid analogs (Fig. 3A) were selected as possible CalO2 substrates or substrate mimics. The selected commercially available aromatic acids were converted to their sodium salts by dissolving 100 mg of acid in methanol to which a molar equivalent of 2 M NaOH was added. The corresponding mixture was stirred at room temperature for 1 hr, at which time the solvents were removed under vacuum. The N-acetyl cysteamine (SNAc) derivatives 11-15 for this study were synthesized from the commercially available acids using standard procedures.31 Specifically, to a solution carboxylic acid (1 eq) and 1-hydroxybenzotriazole (1.2 eq) in THF (10 mL) was added dicyclohexyl carbodiimide (1.2 eq in THF), followed by N-acetylcysteamine (1.0 - 5.0 eq). The reaction was stirred for 1 hr at 24 °C and potassium carbonate (1.0 eq) was added. The reaction was stirred for an additional 2 hr, filtered and concentrated by rotary evaporation. The solid residue was dissolved in EtOAc, washed with 10% NaHCO3 and H2O, and the organics dried (MgSO4), concentrated, and purified by silica gel column chromatography (2 ~ 4% methanol in chloroform) to give the desired products in 18- 62% yield. SNAc 11: 28% yield, 1H NMR (CD3OD) δ 6.18 (d, 2H), 3.41 (dd, 2H), 3.18 (t, 2H), 2.22 (s, 3H), 1.97 (s, 3H), ESI-MS m/z calculated for C12H15NO4S [M-H], 268.1, observed 268.0; SNAc 12 - 45% yield, 1H NMR (CDCl3) δ 10.91 (s, 1H), 8.12 (d, 1H), 7.69 (dd, 1H), 6.78 (d, 1H), 6.42 (br s, 1H), 3.53 (dd, 2H), 3.27 (t, 2H), 1.98 (s, 3H), ESI-MS m/z calculated for C11H12INO3S [M+H], 364.9, observed 365.9; SNAC 13 - 41% yield, 1H NMR (CDCl3) δ 8.23 (t, 1H), 7.92 (m, 2H), 7.19 (t, 1H), 6.65 (br s, 1H), 3.49 (dd, 2H), 3.23 (t, 2H), 1.98 (s, 3H), ESI-MS m/z calculated for C11H12INO2S [M+H], 349.0, observed 349.9; characterization data for 14 - 62% yield, 1H NMR (CD3OD) δ 7.82 (d, 1H), 7.41 (dd, 1H), 6.83 (m, 2H), 3.52 (dd, 2H), 3.19 (t, 2H), 1.96 (s, 3H); ESI-MS m/z calculated for C11H13NO3S [M+H], 240.1, observed 240.0; SNAc 15 - 18% yield, 1H NMR (CD3OD) δ 7.77 (d, 1H), 6.35 (dd, 1H), 6.22 (d, 1H), 3.42 (dd, 2H), 3.18 (t, 2H), 1.92 (s, 3H), ESI-MS m/z calculated for C11H13NO4S [M+H], 255.1, observed 256.0. For both the acid salts and SNAc derivatives, stock solutions (250 mM) were prepared in 50 mM potassium phosphate buffer, pH 8.0 for the studies described.
Fig. 3.

CalO2 ligand binding. (A) Putative ligands derived from orsellinic acid (6 and 11), 5-iodosalicylic acid (7 and 12), 3-iodobenzoic acid (8 and 13), salicylic acid (9 and 14) and 2,4-dihydroxybenzoic acid (10 and 15). Representative difference spectra of CalO2 titrated with 11 (B, 56.2, 112.5, and 168.8 μM, respectively), 12 (C, 56.2, 230, and 460 μM, respectively), 13 (D, 56.2, 112.5, 168.8, 225, 337.5, and 450 μM, respectively), and 14 (E, 110, 230, 340, and 450 μM, respectively).
Binding assays were conducted with 15 mM CalO2 in 400 μL 50 mM potassium phosphate buffer (pH 7.2) at 25 °C in a 1 mL quartz cuvette using a Beckman-Coulter, DU800. Assays were initiated by titrating ligands into the assay mixture over a range of 0.06 - 8 mM and the difference spectra (blanked against 15 mM CalO2 in 400 μL 50 mM potassium phosphate buffer, pH 7.2 and corrected for ligand absorbance) were recorded after equilibration (typically 5 min). Although ligand solubility prohibited ligand saturation in most cases, Kd values were estimated using difference spectra at five different ligand concentrations. Fig. 3 illustrates representative difference spectra from this study.
Protein crystallization
Buffer exchange for protein used in crystallization experiments was carried out by diafiltration (Vivaspin 15 mL column with Hydrosart membrane, 30 KDa MWCO, 3000 xg). The protein solution used for crystallization optimization contained 7 mg/mL CalO2 in 10 mM Tris- HCl (pH 8.0). CalO2 crystals were grown by the hanging drop vapor-diffusion method. The reservoir solution contained 10% (w/v) methyl ether polyethylene glycol 5000, 1.44 M tetramethylammonium chloride, and 100 mM triethanolamine (pH 8.0). The hanging drop consisted of 2 μL of protein solution mixed with 2 μL of reservoir solution. Crystallization trays were stored at 293 K. Under these conditions, CalO2 crystals required two weeks to reach full size (100×100×100 μm). Crystals were subsequently soaked in increasing concentrations of ethylene glycol in mother liquor up to a final concentration of 20% (v/v) and flash-frozen in a stream of liquid nitrogen.
Diffraction data collection and structure determination
Diffraction data were collected at the Advanced Photon Source on General Medicine and Cancer Institutes Collaborative Access Team beamline 23-ID-D. Data were collected with 1° oscillations per frame, an exposure time of 5 s, and 200-fold attenuation of the incident beam at a temperature of 93 K. Reflections were indexed, integrated, and scaled using the HKL2000 package.32 The data were phased by molecular replacement via MOLREP.33,34 A number of P450 homologs (both with and without sidechains) were initially used as MR models without success. Eventually a homology model was generated with Jackal using the sequence of CalO2 and the coordinates of Streptomyces venezuelae PikC (33% identity, PDB ID 2BVJ).35 This resulted in a solution with a slightly improved correlation coefficient and a map that visually corresponded to certain portions of the output structure. Regions of the model that did not fit the electron density were removed and the resulting structure was used as the model in another molecular replacement run. This process was repeated multiple times until an optimal molecular replacement solution was achieved. Phases were further improved using density modification as implemented in DM36 and the structure was finalized through multiple rounds of model building with Coot and refinement with REFMAC.37,38 TLS groups, selected based on the output of the TLSMD web server,39 were incorporated during the final stages of refinement.40 Relevant crystallographic statistics are summarized in Table I. Structure figures were generated in Pymol.41 In an attempt to further understand the improvement in the molecular replacement solution solved with the Jackal calculated homology model, the final structure was aligned with PikC (2BVJ), the homology model calculated by Jackal, and the final truncated homology model. This revealed that there was a negative correlation between the observed quality of the molecular replacement solution and the root mean square deviations of the mainchain atoms between the model used for molecular replacement and the final structure. These were 5.65 Å for PikC (over 1565 atoms), 3.63 Å for the initial homology model (over 1564 atoms), and 2.12 Å for the truncated homology model (over 1140 atoms)
Table I.
Data-Collection and Refinement Statistics.
| Data collection statistics | |
| Space group | C41212 |
| Unit Cell Parameters (Å) | a=71.2 b=71.2 c=204.5 |
| Observed reflections | 480,092 |
| Unique reflections | 9109 |
| Redundan cy | 38.6 (12.4)a |
| Rmergeb (%) | 8.0 (47.5) |
| Mean I/σ | 14.7 (2.1) |
| Completeness (%) | 96.9 (77.0) |
| Refinement Statistics | |
| Resolution Range (Å) | 67.27-2.47 (2.54-2.47) |
| Rcryst c | 0.198 |
| Rfree d | 0.257 |
| Mean B factor (Å2) | 32.6 |
| r.m.s.d. bonds (Å) | 0.009 |
| r.m.s.d. angles (°) | 1.2 |
| Ramachandran plot (%) | |
| Core region | 91.2 |
| Allowed region | 8.8 |
Values in parentheses refer to the highest resolution s hell.
Rmerge = ΣhΣi|Ii(h) - <I(h)>|/ ΣhΣiIi(h), where I i(h) is the intensity of an individual measurement of the reflecti on and <I(h)> is the mean intensity of the reflection.
Rcryst = Σh||Fobs|-|Fcalc||/Σh|Fobs|, where F obs and F calc are the observed and calculated structure-factor amplitudes, resp ectively.
Rfree was calculated as Rcryst using 5.0 % of the randomly selecte d unique reflections that were omitted from structure refinement.
Docking Studies
A homology model for the acyl-carrier protein (ACP) domain of the iterative PKS CalO5 (residues 1181 through 1257) was constructed with Jackal using the ACP coordinates from Protein Data Bank entry 1ACP.35 The ACP homology model coordinates and CalO2 coordinates were subsequently submitted to the ClusPro web server,42,43 and putative complexes were obtained using both DOT and ZDOCK docking software.44,45
RESULTS AND DISCUSSION
Protein characterization and ligand binding
P450s are known to participate in a variety of biosynthetic transformations including general oxidation, oxidative coupling reactions, oxidative degradative reactions, and a plethora of aliphatic/aromatic hydroxylations.46-49 Two P450-encoding genes (calO2 and calE10) reside within the calicheamicin biosynthetic locus.19 The calO2 gene is co-localized within a subcluster containing calO1 (AdoMet-dependent aromatic O-methyltransferase), calO3 (a flavin-dependent halogenase), calO4 (3-oxoacyl-ACP synthase III), calO5 (orsellinic acid synthase, iterative type I PKS) and calO6 (AdoMet-dependent aromatic O-methyltransferase). In contrast, calE10 is located in a distinct location and has recently been characterized as the aminosugar oxidase responsible for the biosynthesis of a novel hydroxylaminosugar nucleotide aryltetrasaccharide precursor en route to CLM (Johnson, H.; Thorson, J. S., unpublished). Given the common role of P450s in aromatic hydroxylation, the localization of calO2 within the set of genes associated with the biosynthesis and modification of the iterative type I PKS product, and the in vitro characterization of CalE10 as a specific aminosugar oxidase, CalO2 has been put forth as the putative orsellinate hydroxylase - prior to, or after, iodination (Fig. 1).
Despite the ability to purify the recombinant holo-CalO2 from S. lividans, attempts to reconstitute the in vitro activity of CalO2 using a variety of electron transport systems (including the successful spinach ferredoxin/spinach ferredoxin reductase system for the related CalE10) were unsuccessful. To assess the interaction of CalO2 with the putative substrates employed for these in vitro assays, a series of ligand-binding studies were subsequently pursued (Fig. 3). From this analysis, very weak perturbation of the heme spectrum was observed with free acids 6 - 8, indicative of weak active-site binding, wherein the iodo-substituted analogs 7 and 8 led to the greatest perturbation. Stronger association was observed in the presence of SNAc variants with iodination again critical for affinity (12 Kd = 0.2 ± 0.6 μM μM > 13 Kd = 120 μM ± 40 μM > 11 Kd = 170 ± 30 μM > 14 Kd = 405 ± 100 μM). This cumulative analysis suggests a substrate thioester conjugate (e.g. ACP or CoA) and the iodine are important for CalO2 recognition and thus, CalO3-catalyzed iodination may occur prior to CalO2-catalyzed hydroxylation as illustrated in Fig. 1.
Structure quality and overall fold
The CalO2 model was refined to a nominal resolution of 2.47 Å. The model included all 397 residues encoded by the calO2 gene, however; there was insufficient electron density to build in the N-terminal His-tag. In addition to one chain of CalO2, the asymmetric unit contained a heme group and 82 water molecules. The final model was refined to an R and Rfree of 19.8% and 25.7%. All of the amino acid residues were in energy allowed regions of a Ramachandran plot. Additional model statistics are listed in Table I. The structure factors as well as the final coordinates have been deposited to the Protein Data Bank under ID 3BUJ. The enzyme core is a four-helix bundle made up of helices D, E, I, and L (Fig. 4). The heme group is localized between helices I and L and sits at the bottom of a deep cavity wherein Cys343 serves as the heme iron axial ligand and resides on the loop directly preceeding helix L. In addition to the core domain, the active site cavity is also defined by the two-helix bundle containing helices B' and B", the loops between helices F and G, as well as the β-sheets β1 and β4. The distance between the heme iron and the surface of the protein is 27.5 Å.
Fig. 4.
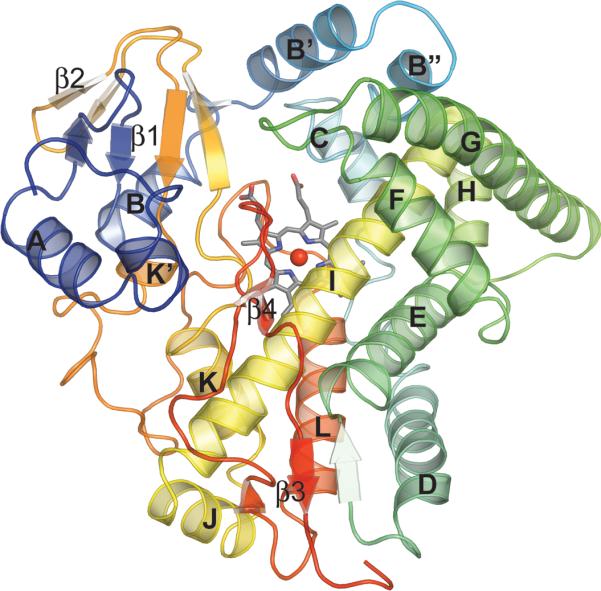
Cartoon representation of CalO2. Color corresponds to protein sequence (blue-N terminus, red-C terminus).
Comparison to other P450 enzymes
The overall fold of CalO2 is very similar to that of other P450 enzymes with the most dramatic alteration occurring between residues 54 through 81 (helices B' and B") of CalO2, a key structural variable region among P450s. In many P450s this region forms a random loop that defines multiple solvent channels to provide substrates active site access.50,51 In contrast, this region in CalO2 forms a two-helix bundle that blocks many of the solvent channels observed in previous P450 structures while maintaining a large central opening for substrate access to the active site. With the exception of the axial ligand (Cys343), only two polar groups (Thr233 and Thr237, 5.4 Å and 5.3 Å, respectively) are within close proximity of the CalO2 heme iron, one of which (Thr237) is highly conserved among P450 enzymes and is believed to form a hydrogen bond with the iron-coordinated dioxygen intermediate.52 In contrast, sequence analysis reveals that Thr233 appears to be relatively unique among P450 enzymes (Fig. 5). The uniqueness of this residue in the CalO2 active site suggests that it may play a role in substrate recognition.
Fig. 5.

Sequence alignment of several P450 enzymes involved in natural product synthesis. Residues colored red are homologous to residues predicted to be involved in substrate recognition in CalO2. Residues in blue are homologous to residues predicted to be involved in binding of ACP.
Analysis of natural product-related P450 complexes - including P450eryF bound to androstendione (PDB ID 1eup),53 P450NOR complexed with 3-pyridinealdehyde adenine dinucleotide (PDB ID 1xqd),54 StaP bound to chromopyrrolic acid (PDB ID 2z3u),55 and PikC complexed with narbomycin (PDB ID 2c7x) and YC-17 (PDB ID 2cd8)56 - reveal a predominance of non-conserved hydrophobic contributions to substrate recognition with few common hydrogen bonding and electrostatic interactions. In addition to the putative role of Thr233 highlighted in previous paragraph, another exception to this general hydrophobicbinding model includes Arg67 in StaP - postulated to coordinate a carboxyl group of chromopyrrolic acid. As illustrated in Fig. 6, the homologous residue (Arg55) in CalO2 could potentially be involved in substrate recognition. CalO2 Arg341 substitutes for a histidine common to many P450s (Fig. 5) and may therefore, also be involved in CalO2-substrate specific interactions. Finally, CalO2 Phe383 is homologous to Phe403 in StaP, which is involved in T-stacking π-π interactions with the aromatic chromopyrrolic acid in StaP. Thus, CalO2 Phe383 may play a similar role in binding the aromatic orsellinate substrate in CalO2.
Fig. 6.

Stereoscopic illustration of predicted binding mode of iodoorsellinic acid in CalO2. The carboxyl group is positioned such that a putative pantetheinyl arm would extend from the active site out to helix B' (blue). In this model, C3 of the substrate analogue is approximately 4 Å from the heme iron. Additional helices are labeled as in Figure 4.
ACP docking and implications for catalysis
As previously described, the lack of successful reconstitution of CalO2 activity in vitro may be due to the requirement of ACP- or CoA-thioester substrate conjugates. Consistent with this, a CalO2 BLAST search revealed the strongest homology to Biol (33% identity), a P450 known to act upon an ACP bound substrate.57 Similar protein-bound substrates have been established for other biosynthetic P450s including NovI (novobiocin biosynthesis), NikQ (nikkomycin biosynthesis), and OxyB (vancomycin biosynthesis).58-61 While an ACP-binding consensus among such biosynthetic P450s has not been determined, docking algorithms have been used to accurately predict residues that may be critical to ACP binding in polyketide synthases.62 In addition, a number of bacterial ACP protein complex structures have been solved by crystallography.63-65 These studies reveal binding to primarily engage helix 2 of the ACP and involve a network of acidic and hydrophobic residues on the ACP interacting with a series of basic and hydrophobic residues on the respective protein. In an effort to assess the feasibility of an interaction between CalO2 and an ACP-bound substrate, docking simulations were subsequently conducted.
The top ten simulated solutions from both ZDOCK and DOT were analyzed. Four of the top eight solutions from ZDOCK placed the ACP domain near the active site of the molecule. Of these, two oriented the conserved ACP serine (which would bear the pantetheine arm conjugated to the substrate) into the active site. Notably, the model complex predicted by both ZDOCK and DOT (Fig. 7) most closely resembles the ACP-protein complex structures described in the literature. In this model, helix 2 of the CalO5 ACP domain interacts with helix B' of CalO2. In contrast to previous structures, the two helices both contain a mixture of positively charged and negatively charged residues (CalO2 helix 4 - Gly57, lle58, Arg59, Arg60, Phe61, Trp62, Thr63, Asp64, Leu65, and Val66; CalO5 ACP domain - Met1214, Asp1215, Ser1216, Val1217, Met1218, Thr1219, Val1220, lle1221, Val1222, Arg1223, Arg1224, and Arg1225). In the docked CalO2-CalO5 ACP model, the parallel orientation of the two interacting helices to maintain correct electrostatic interactions despite the electrostatic heterogeneity of the two helices. Additional regions of interaction, derived from this model, are also highlighted in Fig. 5.
Fig. 7.

Stereoscopic image of the CalO2 - CalO5 ACP domain complex as predicted by DOT and ZDOCK. The primary region of interaction is between helix 2 of ACP (blue) and helix B' of CalO2 (green). The iodoorsellinic acid group is modeled into the active site and a line is drawn between the sidechain oxygen of Ser1216 (homolog of Ser36 in Escherichia coli ACP) and the carboxyl oxygen of the modeled substrate to depict the expected path of the proposed pantetheine group. Additional helices are labeled as in Figure 4.
The docked CalO2-CalO5 ACP model places Ser1216 directly up against the edge of the open cleft on the surface of CalO2. Ser1216 is the homolog of Ser36 in Escherichia coli ACP that binds the pantetheinate group. The distance between the modeled iodoorsellinic acid and Ser1216 is 19 Å. Given that the reach of the pantetheinate arm is approximately 18 Å, this model suggests that the proposed ACP-linked substrate could reach directly into the active site as opposed to being released from the ACP and diffusing the rest of the way into the active site.
CONCLUSION
We have determined the crystal structure of CalO2, a P450 enzyme responsible for the modification of a bacterial iterative type I polyketide product. In addition, we have probed the active site affinity for a number of substrate analogs. The results suggest that the CalO2 reaction follows substrate iodination and may require the presence of a CoA or ACP bound substrate. Structural analysis of CalO2 revealed a two helix bundle lining the active site cavity which docking studies indicate could serve as a binding site in a putative CalO2 CalO5 binding interaction.
ACKNOWLEDGEMENTS
Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. W-31-109-ENG-38. General Medicine and Cancer Institutes Collaborative Access Team has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). We are grateful to the School of Pharmacy Analytical Instrumentation Center for analytical support. Special thanks to Achim Ahlert for his help in producing the overexpression plasmids. This work was also supported in part by National Institutes of Health Grants CA84374 and U19 CA113297. J.S.T is a UW HI Romnes Fellow and H.D.J. was supported in part as a UW NIH Chemical Biology Training Grant Trainee. J.G.M. was supported by an NHGRI training grant to the Genomic Sciences Training Program (5T32HG002760)
REFERENCES
- 1.Thorson JS, Sievers EL, Ahlert J, Shepard E, Whitwam RE, Onwueme KC, Ruppen M. Understanding and exploiting nature's chemical arsenal: the past, present and future of calicheamicin research. Curr Pharm Des. 2000;6(18):1841–1879. doi: 10.2174/1381612003398564. [DOI] [PubMed] [Google Scholar]
- 2.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Antitumor antibiotics: bleomycin, enediynes, and mitomycin. Chem Rev. 2005;105(2):739–758. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 3.Biggins JB, Onwueme KC, Thorson JS. Resistance to enediyne antitumor antibiotics by CalC self-sacrifice. Science. 2003;301(5639):1537–1541. doi: 10.1126/science.1086695. [DOI] [PubMed] [Google Scholar]
- 4.Singh S, Hager MH, Zhang C, Griffith BR, Lee MS, Hallenga K, Markley JL, Thorson JS. Structural insight into the self-sacrifice mechanism of enediyne resistance. ACS Chem Biol. 2006;1(7):451–460. doi: 10.1021/cb6002898. [DOI] [PubMed] [Google Scholar]
- 5.Zein N, Sinha AM, McGahren WJ, Ellestad GA. Calicheamicin γ1I: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science. 1988;240(4856):1198–1201. doi: 10.1126/science.3240341. [DOI] [PubMed] [Google Scholar]
- 6.Zein N, Poncin M, Nilakantan R, Ellestad GA. Calicheamicin γ1I and DNA: Molecular recognition process responsible for site-specificity. Science. 1989;244(4905):697–699. doi: 10.1126/science.2717946. [DOI] [PubMed] [Google Scholar]
- 7.Kumar RA, Ikemoto N, Patel DJ. Solution structure of the calicheamicin γ1I-DNA complex. J Mol Biol. 1997;265(2):187–201. doi: 10.1006/jmbi.1996.0718. [DOI] [PubMed] [Google Scholar]
- 8.Walker S, Murnick J, Kahne D. Structural characterization of a calicheamicin DNA complex by NMR. J Am Chem Soc. 1993;115(18):7954–7961. [Google Scholar]
- 9.Wu AM, Senter PD. Arming antibodies: Prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 10.Damle NK. Tumour-targeted chemotherapy with immunoconjugates of calicheamicin. Expert Opinion on Biological Therapy. 2004;4(9):1445–1452. doi: 10.1517/14712598.4.9.1445. [DOI] [PubMed] [Google Scholar]
- 11.Hamann PR. Monoclonal antibody-drug conjugates. Exp Opin on Therapeut Patents. 2005;15(9):1087–1103. [Google Scholar]
- 12.Zhang CS, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li LJ, Thorson JS. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science. 2006;313(5791):1291–1294. doi: 10.1126/science.1130028. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy DR, Gawron LS, Ju JH, Liu W, Shen B, Beerman TA. Single chemical modifications of the C-1027 enediyne core, a radiomimetic antitumor drug, affect both drug potency and the role of ataxia-telangiectasia mutated in cellular responses to DNA doublestrand breaks. Cancer Res. 2007;67(2):773–781. doi: 10.1158/0008-5472.CAN-06-2893. [DOI] [PubMed] [Google Scholar]
- 14.Shen B, Liu W, Nonaka K. Enediyne natural products: Biosynthesis and prospect towards engineering novel antitumor agents. Curr Med Chem. 2003;10(21):2317–2325. doi: 10.2174/0929867033456701. [DOI] [PubMed] [Google Scholar]
- 15.Jones GB, Fouad FS. Designed enediyne antitumor agents. Curr Pharm Des. 2002;8(27):2415–2440. doi: 10.2174/1381612023392810. [DOI] [PubMed] [Google Scholar]
- 16.LAM KS, Veitch JA, Golik J, Krishnan B, Klohr SE, Volk KJ, Forenza S, Doyle TW. Biosynthesis of esperamicin-a1, an enediyne antitumor antibiotic. J Am Chem Soc. 1993;115(26):12340–12345. [Google Scholar]
- 17.Hensens OD, Giner JL, Goldberg IH. Biosynthesis of Ncs chrom-a, the chromophore of the antitumor antibiotic neocarzinostatin. J Am Chem Soc. 1989;111(9):3295–3299. [Google Scholar]
- 18.Tokiwa Y, Miyoshisaitoh M, Kobayashi H, Sunaga R, Konishi M, Oki T, Iwasaki S. Biosynthesis of dynemicin-a, a 3-ene-1,5-diyne antitumor antibiotic. J Am Chem Soc. 1992;114(11):4107–4110. [Google Scholar]
- 19.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang KX, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. The calicheamicin gene cluster and its iterative type I enediyne PKS. Science. 2002;297(5584):1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Christenson SD, Standage S, Shen B. Biosynthesis of the enediyne antitumor antibiotic C-1027. Science. 2002;297(5584):1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 21.Liu W, Ahlert J, Gao QJ, Wendt-Pienkowski E, Shen B, Thorson JS. Rapid PCR amplification of minimal enediyne polyketide synthase cassettes leads to a predictive familial classification model. Proc. Natl. Acad. Sci. USA. 2003;100(21):11959–11963. doi: 10.1073/pnas.2034291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zazopoulos E, Huang KX, Staffa A, Liu W, Bachmann BO, Nonaka K, Ahlert J, Thorson JS, Shen B, Farnet CM. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat Biotechnol. 2003;21(2):187–190. doi: 10.1038/nbt784. [DOI] [PubMed] [Google Scholar]
- 23.Liu W, Nonaka K, Nie LP, Zhang J, Christenson SD, Bae J, Van Lanen SG, Zazopoulos E, Farnet CM, Yang CF, Shen B. The neocarzinostatin biosynthetic gene cluster from Streptomyces carzinostaticus ATCC 15944 involving two iterative type I polyketide synthases. Chem Biol. 2005;12(3):293–302. doi: 10.1016/j.chembiol.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 24.Van Lanen SG, Oh TJ, Liu W, Wendt-Pienkowski E, Shen B. Characterization of the maduropeptin biosynthetic gene cluster from Actinomadura madurae ATCC 39144 supporting a unifying paradigm for enediyne biosynthesis. J. Am. Chem. Soc. 2007;129(43):13082–13094. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao QJ, Thorson JS. The biosynthetic genes encoding for the production of the dynemicin enediyne core in Micromonospora chersina. Manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr Opin Chem Biol. 2003;7(2):285–295. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- 27.Gaisser S, Trefzer A, Stockert S, Kirschning A, Bechthold A. Cloning of an avilamycin biosynthetic gene cluster from Streptomyces viridochromogenes Tü57. J Bacteriol. 1997;179(20):6271–6278. doi: 10.1128/jb.179.20.6271-6278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sthapit B, Oh TJ, Lamichhane R, Liou K, Lee HC, Kim CG, Sohng JK. Neocarzinostatin naphthoate synthase: an unique iterative type I PKS from neocarzinostatin producer Streptomyces carzinostaticus. FEBS Lett. 2004;566(1-3):201–206. doi: 10.1016/j.febslet.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 29.Jia XY, Tian ZH, Shao L, Qu XD, Zhao QF, Tang J, Tang GL, Liu W. Genetic characterization of the chlorothricin gene cluster as a model for spirotetronate antibiotic biosynthesis. Chem Biol. 2006;13(6):575–585. doi: 10.1016/j.chembiol.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Shao L, Qu XD, Jia XY, Zhao QF, Tian ZH, Wang M, Tang GL, Liu W. Cloning and characterization of a bacterial iterative type I polyketide synthase gene encoding the 6- methylsalicyclic acid synthase. Biochem Biophys Res Commun. 2006;345(1):133–139. doi: 10.1016/j.bbrc.2006.04.069. [DOI] [PubMed] [Google Scholar]
- 31.Zucai S, Huawei C, Walsh CT. Acyl-CoA hydrolysis by the high molecular weight protein 1 subunit of yersiniabactin synthetase: Mutational evidence for a cascade of four acyl-enzyme intermediates during hydrolytic editing. Proc. Natl. Acad. Sci. USA. 2000;97:14188–14193. doi: 10.1073/pnas.260495697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276(A):307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Vagin A, Teplyakov A. MOLREP: An automated program for molecular replacement. J Appl Crystall. 1997;30:1022–1025. [Google Scholar]
- 34.Collaborative Computational Project N The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Xiang JZ. Jackal. 1.5. New York: [Google Scholar]
- 36.Cowtan K. 'dm': An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994;31:34–48. [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 39.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39:109–111. [Google Scholar]
- 40.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 4):439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 41.DeLano WL. The PyMOL User's Manual. Delano Scientific; Palo Alto: 2002. [Google Scholar]
- 42.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20(1):45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 43.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization and Dynamics Calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 44.Mandell JG, Roberts VA, Pique ME, Kotlovyi V, Mitchell JC, Nelson E, Tsigelny I, Ten Eyck LF. Protein docking using continuum electrostatics and geometric fit. Protein Eng. 2001;14(2):105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 45.Chen R, Li L, Weng Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins. 2003;52(1):80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 46.Isin EM, Guengerich FP. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim Biophys Acta. 2007;1770(3):314–329. doi: 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Pylypenko O, Schlichting I. Structural aspects of ligand binding to and electron transfer in bacterial and fungal p450s. Annu Rev Biochem. 2004;73:991–1018. doi: 10.1146/annurev.biochem.73.011303.073711. [DOI] [PubMed] [Google Scholar]
- 48.Lewis DFV, Wiseman A. A selective review of bacterial forms of cytochrome P450 enzymes. Enzyme Microbial Technol. 2005;36(4):377–384. [Google Scholar]
- 49.Urlacher VB, Eiben S. Cytochrome P450 monooxygenases: perspectives for synthetic application. Trends Biotechnol. 2006;24(7):324–330. doi: 10.1016/j.tibtech.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 50.Wade RC, Winn PJ, Schlichting I, Sudarko A survey of active site access channels in cytochromes P450. J Inorg Biochem. 2004;98(7):1175–1182. doi: 10.1016/j.jinorgbio.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 51.Cojocaru V, Winn PJ, Wade RC. The ins and outs of cytochrome P450s. Biochim Biophys Acta. 2007;1770(3):390–401. doi: 10.1016/j.bbagen.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 52.Nagano S, Poulos TL. Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam. Implications for the dioxygen activation mechanism. J Biol Chem. 2005;280(36):31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 53.Cupp-Vickery J, Anderson R, Hatziris Z. Crystal structures of ligand complexes of P450eryF exhibiting homotropic cooperativity. Proc Natl Acad Sci U S A. 2000;97(7):3050–3055. doi: 10.1073/pnas.050406897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oshima R, Fushinobu S, Su F, Zhang L, Takaya N, Shoun H. Structural evidence for direct hydride transfer from NADH to cytochrome P450nor. J Mol Biol. 2004;342(1):207–217. doi: 10.1016/j.jmb.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 55.Makino M, Sugimoto H, Shiro Y, Asamizu S, Onaka H, Nagano S. Crystal structures and catalytic mechanism of cytochrome P450 StaP that produces the indolocarbazole skeleton. Proc Natl Acad Sci U S A. 2007;104(28):11591–11596. doi: 10.1073/pnas.0702946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sherman DH, Li S, Yermalitskaya LV, Kim Y, Smith JA, Waterman MR, Podust LM. The structural basis for substrate anchoring, active site selectivity, and product formation by P450 PikC from Streptomyces venezuelae. J Biol Chem. 2006;281(36):26289–26297. doi: 10.1074/jbc.M605478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stok JE, De Voss J. Expression, purification, and characterization of Biol: a carbon-carbon bond cleaving cytochrome P450 involved in biotin biosynthesis in Bacillus subtilis. Arch Biochem Biophys. 2000;384(2):351–360. doi: 10.1006/abbi.2000.2067. [DOI] [PubMed] [Google Scholar]
- 58.Eustaquio AS, Luft T, Wang ZX, Gust B, Chater KF, Li SM, Heide L. Novobiocin biosynthesis: inactivation of the putative regulatory gene novE and heterologous expression of genes involved in aminocoumarin ring formation. Arch Microbiol. 2003;180(1):25–32. doi: 10.1007/s00203-003-0555-2. [DOI] [PubMed] [Google Scholar]
- 59.Chen H, Hubbard BK, O'Connor SE, Walsh CT. Formation of beta-hydroxy histidine in the biosynthesis of nikkomycin antibiotics. Chem Biol. 2002;9(1):103–112. doi: 10.1016/s1074-5521(02)00090-x. [DOI] [PubMed] [Google Scholar]
- 60.Chen H, Walsh CT. Coumarin formation in novobiocin biosynthesis: beta-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI. Chem Biol. 2001;8(4):301–312. doi: 10.1016/s1074-5521(01)00009-6. [DOI] [PubMed] [Google Scholar]
- 61.Zerbe K, Woithe K, Li DB, Vitali F, Bigler L, Robinson JA. An oxidative phenol coupling reaction catalyzed by oxyB, a cytochrome P450 from the vancomycin-producing microorganism. Angew Chem Int Ed. 2004;43(48):6709–6713. doi: 10.1002/anie.200461278. [DOI] [PubMed] [Google Scholar]
- 62.Zhang YM, Rao MS, Heath RJ, Price AC, Olson AJ, Rock CO, White SW. Identification and analysis of the acyl carrier protein (ACP) docking site on beta-ketoacyl-ACP synthase III. J Biol Chem. 2001;276(11):8231–8238. doi: 10.1074/jbc.M008042200. [DOI] [PubMed] [Google Scholar]
- 63.Parris KD, Lin L, Tam A, Mathew R, Hixon J, Stahl M, Fritz CC, Seehra J, Somers WS. Crystal structures of substrate binding to Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric arrangement of molecules resulting in three active sites. Structure. 2000;8(8):883–895. doi: 10.1016/s0969-2126(00)00178-7. [DOI] [PubMed] [Google Scholar]
- 64.Rafi S, Novichenok P, Kolappan S, Zhang X, Stratton CF, Rawat R, Kisker C, Simmerling C, Tonge PJ. Structure of acyl carrier protein bound to FabI, the FASII enoyl reductase from Escherichia coli. J Biol Chem. 2006;281(51):39285–39293. doi: 10.1074/jbc.M608758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang YM, Wu B, Zheng J, Rock CO. Key residues responsible for acyl carrier protein and beta-ketoacyl-acyl carrier protein reductase (FabG) interaction. J Biol Chem. 2003;278(52):52935–52943. doi: 10.1074/jbc.M309874200. [DOI] [PubMed] [Google Scholar]