

Abstract

4-Trifluoromethylbenzenepropargyl ethers are stable and sterically minimal alcohol protecting groups that are readily cleaved in a single step by exposure to lithium naphthalenide. In conjunction with the 4,6-O-benzylidene protecting group, glycosylation reactions of 2-O-4-trifluoromethylbenzenepropargyl-protected mannosyl donors are extremely β selective.
Introduction
Protecting groups continue to play a central role in modern organicsynthesis,1 and the ability/inability to achieve selective deprotection of one protecting group in the presence of another is often key to the success/failure of a synthetic route. In response to a problem arising from the influence of protecting group size on the stereoselectivity of a glycosylation reaction, we described the application of a propargyl ether as a sterically minimal donor protecting group for β-mannosylation with donor 1.2
While the propargyl ethers were readily introduced and had the anticipated effect on stereoselectivity, they required a two-step deprotection protocol, namely an initial treatment with the base followed by catalytic osmylation of the resulting allenyl ether.2 Subsequently, to facilitate deprotection, the propargyl ether system was modified in such a way as to be cleavable in a single step, orthogonal to the ubiquitous benzyl ethers. This was achieved through the use of the naphthyl propargyl system, which is removed in a single step with DDQ.3 Donors 2 and 3, however, had two limitations: incompatibility with glycosylation reactions owing to cyclization onto the oxacarbenium ion and direct reaction of the electron rich triple bond with glycosidation promoters. Nevertheless, the naphthylpropargyl system was ideal for use at the 3-O-position of donors, as in 4, provided that it was used in conjunction with the sulfoxide glycosylation method and a sacrificial alkene.3 Extending this series we now describe an electron poor arylpropargyl system compatible with the 2-O-position and removable under dissolving metal conditions.
Alkylation of 1,2:5,6-di-O-acetone-d-glucofuranose with sodium hydride and 4-trifluoromethylphenyl propargyl bromide 54 gave the model ether 6, with which different reductive conditions were explored (Scheme 1). Exposure of 6 to samarium iodide, generated under standard conditions,5 resulted in no reaction. When 6 was exposed to samarium iodide generated by mixing samarium and iodine,6 partial loss of an isopropylidene group was observed with no formation of the desired product. Presumably unreacted iodine acted as a Lewis acid resulting in the loss of an isopropylidene group.7 However, sodium in liquid ammonia,8 and lithium naphthalenide9 proved effective in removing the electron-deficient propargyl ether and in affording the required deprotected sugar in good yield.
Scheme 1.

Model Protection & Deprotection of d-Glucofuranose
To examine the effect of the new electron poor protecting group on the stereoselectivity of glycosylation reactions when located at O2, donor 8 was prepared by reaction of glycoside 710 with propargyl bromide 5 (Scheme 2).
Scheme 2.

Preparation of Donor 8
Treatment of 8 with triflic anhydride in the presence of BSP11 and TTBP12 (Figure 2) at −70 °C in CH2Cl2, to give an intermediate glycosyl triflate,11 followed by addition of 1-adamantanol, resulted in the formation of the β-mannoside 9 with impeccable selectivity (Table 1, entry 1). A number of further couplings were then conducted with more standard glycosyl acceptors, leading to the yields collected in Table 1.
Figure 2.

Coupling Reagents
Table 1.
Coupling Reactions of Donor 8















Isolated yields after column chromatography
1H NMR spectroscopy of crude reaction mixtures revealed only β isomer formation in all cases.
Exposure of mannoside 9 to Na in liquid ammonia cleanly gave fully deprotected 1-adamantanyl-β-d-mannopyranoside 16 (Scheme 3).
Scheme 3.

Complete deprotection with sodium in liquid ammonia
Selective deprotection was achieved using lithium naphthalenide in THF by warming a −70 °C reaction mixture to 0 °C over a period of 2–3 h when 17 was obtained in 85% yield. This strategy was extended to glycosides 10 to 15 (Table 2). In almost all cases minor amounts of debenzylated products were obtained as the side products.
Table 2.
Cleavage of 4-Trifluoromethylbenzenepropargyl Ethers
 | ||
|---|---|---|
| Entry | Deprotected Product | Yield a |
| 1 |  |
85% |
| 2 |  |
67% |
| 3 |  |
63% |
| 4 |  |
61% |
| 5 |  |
60% |
| 6 | 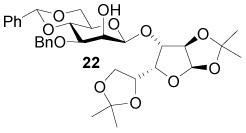 |
59% |
| 7 |  |
55% |
Isolated yields after column chromatography
Conclusion
To conclude, we report the development of the electron poor 4-trifluoromethylbenzenepropargyl ether system as an alcohol protecting group. In conjunction with the BSP glycosylation method, when introduced at the 2-position of 4,6-O-benzylidene-protected mannosyl donors, this system affords extremely β-selective coupling reactions and the possibility of orthogonal cleavage in a single step with lithium naphthalenide.
Experimental Section
3-O-[3-(4-Trifluoromethylphenyl)-prop-2-ynyl]-1,2:5,6-di-O-isopropylidene-α-d-glucofuranose (6)
Sodium hydride (60% in mineral oil, 16 mg, 1.1 eq) was added to a stirred solution of diacetone d-glucose (99 mg, 0.4 mmol, 1 eq) in DMF (8 mL) at 0 °C and allowed to stir for 30 min. This yellow mixture was cooled to 0 °C and treated with propargyl bromide 5 (120 mg, 1.1 eq) in DMF (1 mL). The dark brown reaction mixture was allowed to stir for an hour at room temperature and quenched (at 5 °C–10 °C) with saturated aqueous ammonium chloride (2 mL). The crude product was extracted with methylene chloride (3×3 mL), dried (MgSO4) and concentrated. The residue was purified by column chromatography on silica (5% to 20% Ethyl acetate:Hexane as eluent) to afford 6 (100 mg, 60%). Viscous oil. [α]24d −6.3 (c 1.7, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 1.29 (s, 3H), 1.32 (s, 3H), 1.40 (s, 3H), 1.48 (s, 3H), 4.00 (q, J = 8.5 Hz, 1H), 4.07–4.10 (m, 3H), 4.14 (d, J = 9.1 Hz, 1H), 4.30 (q, J = 6.9 Hz, 1H), 4.96 (s, 2H), 4.66 (d, J = 3.5 Hz, 1H), 5.88 (d, J = 3.5 Hz, 1H), 7.51–7.56 (m, 4H). 13C-NMR (125 MHz, CDCl3): δ 25.3, 26.1, 26.7, 26.8, 58.6, 67.2, 72.5, 76.8, 81.0, 81.5, 82.8, 85.1, 87.2, 105.2, 109.0, 111.9, 125.2, 126.2, 131.9; 19F-NMR (471 MHz, CDCl3): δ −64.47. ESI-HRMS calcd for C22H25F3O6 [M]+ 442.1603, found 442.1593.
Phenyl 4,6-O-benzylidene-2-O-[3-(4-trifluoromethylphenyl)-prop-2-ynyl]-1-thio-α-d-mannopyranoside (8)
Sodium hydride (60% in mineral oil, 0.29 g, 1.6 eq) was added in three portions to a stirred solution of alcohol 7 (2.1 g, 4.7 mmol, 1 eq) in DMF (100 mL) at 0 °C and allowed to stir for 30 min. This yellow mixture was cooled to 0 °C and treated with propargyl bromide 5 (1.33 g, 1.08 eq) in DMF (10 mL). Dark brown reaction mixture was allowed to stir for an hour at room temperature and quenched (at 5 °C–10 °C) with saturated aqueous ammonium chloride (20 mL). The crude product was extracted with methylene chloride (3×30 mL), dried (MgSO4) and concentrated. The residue was purified by column chromatography on silica (5% to 20% Ethyl acetate: Hexane in 5% increments) to afford 2 g (68%) of mannopyranoside 8. Viscous oil. [α]24d +69.4 (c 1.7, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 3.95 (t, J = 10 Hz, 1H), 4.06 (dd, J = 3.2 Hz, 10 Hz, 1H), 4.29–4.39 (m, 4H), 4.68 (q, J = 16.2 Hz, 2H), 4.82 (d, J = 12.5 Hz, 1H), 4.97 (d, J = 12.5 Hz, 1H), 5.70 (s, 1H), 5.77 (s, 1H), 7.26–7.32 (m, 2H), 7.32–7.34 (m, 2H), 7.42–7.47 (m, 10H), 7.55–7.60 (m, 5H); 13C-NMR (125 MHz, CDCl3): δ 59.6, 65.4, 68.6, 73.5, 76.1, 78.6, 79.3, 85.6, 87.5, 87.8, 101.6, 125.31, 125.33, 126.1, 127.7, 127.8, 128.3, 128.5, 129.0, 129.2, 131.5, 132.0, 133.9, 137.6, 138.3; ESI-HRMS calcd for C36H31F3O5SNa [M+Na]+ 655.1742, found 655.1739.
General Procedure for Coupling of Mannosyl Donor 8 with Acceptors
To a stirred solution of donor 8 (0.10 g, 0.16 mmol, 1 eq). BSP (1.5 eq), TTBP (2 eq), and 4Å molecular sieves in 3.2 mL (0.05 M) of dichloromethane at −70 °C under an Ar atmosphere, was added Tf2O (1.1 eq) slowly. The resulting yellow mixture was stirred at this temperature for 30 min, and then a solution of acceptor (2.0 eq) in dichloromethane 2 mL was added. The stirring was continued for another 30 min and the reaction mixture was poured into saturated aqueous NaHCO3 solution. The crude product was extracted with dichloromethane (3×4 mL). The organic layer was separated, dried (MgSO4) and concentrated. The residue was purified by column chromatography (hexane/ethyl acetate) on silica gel to give the corresponding coupled products.
1-Adamantanyl 4,6-O-benzylidene-2-O-[3-(4-trifluoromethylphenyl)-prop-2-ynyl]-β-d-mannopyranoside (9)
Viscous oil. [α]25d −37.5 (c 1, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 1.58–1.67 (m, 6H), 1.78–1.87 (m, 6H), 2.26 (s, 3H), 3.32–3.37 (m, 1H), 3.68 (dd, J = 10 Hz, J = 3.2 Hz, 1H), 3.93 (t, J = 8.1 Hz, 1H), 4.07 (d, J = 2.5 Hz, 1H), 4.16 (t, J = 9.7 Hz, 1H), 4.28 (dd, J = 10 Hz, J = 4.7 Hz, 1H), 4.80–4.92 (m, 5H), 5.61 (s, 1H), 7.23 (m, 4H), 7.38 (m, 5H), 7.53 (m, 5H). 13C-NMR (125 MHz, CDCl3): δ 30.6, 36.2, 42.4, 60.8, 67.2, 68.8, 72.5, 75.5, 76.9, 78.5, 84.8, 88.7, 94.7, 101.4, 125.2, 126.1, 126.8, 127.5, 128.3, 128.4, 128.9, 132.1, 137.7, 138.4. 19F-NMR (471 MHz, CDCl3): δ −64.33. ESI-HRMS calcd for C40H41F3O6Na [M+Na]+ 697.2748, found 697.2749.
Methyl 2,3,6-tri-O-benzyl-4-O-[4,6-O-benzylidene-2-O-[3-(4-trifluoromethylphenyl)-prop-2-ynyl]-3-O-benzyl-β-d-mannopyranosyl]-α-d-glucopyranoside (10)
Viscous oil. [α]14d −13.4 (c 0.5, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 3.02–3.07 (m, 1H), 3.35 (s, 3H), 3.38–3.40 (m, 1H), 3.50–3.56 (m, 2H), 3.60 (d, J = 9 Hz, 1H), 3.67–3.77 (m, 2H), 3.86–3.89 (m, 2H), 3.92–3.96 (m, 1H), 4.03–4.07 (m, 2H), 4.35 (d, J = 12 Hz,1H), 4.45 (s, 1H), 4.63–4.68 (m, 3H), 4.72 (d, J = 7 Hz, 2H), 4.79–4.83 (m, 3H), 5.05 (d, J = 10.5 Hz, 1H), 5.52 (s, 1H), 7.24–7.27 (m, 6H), 7.30–7.37 (m, 8H), 7.40–7.41 (m, 4H), 7.43–7.44 (m, 5H), 7.48 (dd, J = 8 Hz, J = 1.5 Hz, 3H), 7.52 (d, J = 8 Hz, 2H) . 13C-NMR (125 MHz, CDCl3): δ 55.4, 60.6, 67.2, 68.5, 68.6, 69.6, 72.7, 73.6, 75.3, 76.4, 77.7, 78.6, 79.1, 80.2, 88.4, 98.4, 101.3, 101.4, 125.2, 126.1, 127.3, 127.4, 127.6, 127.8, 127.9, 128.1, 128.2, 128.24, 128.3, 128.6, 128.9, 132.0, 137.5, 137.6, 138.3, 138.5, 139.3. 19F-NMR (471 MHz, CDCl3): δ −64.40. ESI-HRMS calcd for C58H57F3O11Na [M+Na]+ 1009.3745, found 1009.3744.
Methyl 2,3,6-tri-O-benzyl-6-O-[4,6-O-benzylidene-2-O-[3-(4-trifluoromethylphenyl)-prop-2-ynyl]-3-O-benzyl-β-d-mannopyranosyl]-α-d-glucopyranoside (11)
Viscous oil. [α]14d −6.0 (c 0.7, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 3.20–3.25 (m, 1H), 3.34 (s, 3H), 3.47–3.50 (m, 3H), 3.54 (dd, J = 9.7 Hz, J = 3.5 Hz, 1H), 3.78 (m, 1H), 3.90 (t, J = 10.5 Hz, 1H), 4.01 (t, J = 9 Hz, 1H), 4.05–4.14 (m, 4H), 4.27 (dd, J = 9.7 Hz, J = 4.5 Hz, 1H), 4.55 (d, J = 3.5 Hz, 1H), 4.59 (d, J = 11.7 Hz, 1H), 4.65 (d, J = 11.7 Hz, 1H), 4.77–4.78 (m, 2H), 4.79–4.83 (m, 4H), 4.84 (d, J = 8.5 Hz, 1H), 4.88 (d, J = 11.5 Hz, 1H), 5.00 (d, J = 11 Hz, 1H), 5.58 (s, 1H), 7.21–7.23 (m, 8H), 7.39–7.33 (m, 15H), 7.43–7.45 (m, 2H), 7.49–7.52 (m, 4H). 13C-NMR (125 MHz, CDCl3): δ 55.1, 60.4, 67.4, 68.3, 68.5, 69.6, 72.5, 73.4, 74.8, 75.0, 76.1, 76.6, 77.4, 78.5, 79.9, 82.2, 85.4, 88.1, 97.9, 101.4, 102.0, 125.3, 126.0, 127.6, 127.7, 127.8, 127.9, 128.1, 128.2, 128.3, 128.4, 128.5, 128.9, 132.1, 137.5, 138.0, 138.3, 138.4, 138.6. 19F-NMR (471 MHz, CDCl3): δ −64.34. ESI-HRMS calcd for C58H57F3O11Na [M+Na]+ 1009.3745, found 1009.3744.
Methyl 4-O-[4,6-O-benzylidene-2-O-[3-(4-trifluoromethylphenyl)-prop-2-ynyl]-3-O-benzyl-β-d-mannopyranosyl]-2,3-O-isopropylidene-α-l-rhamnopyranoside (12)
Viscous oil. [α]16d −49.3 (c 1, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 1.32 (s, 3H), 1.36 (s, 3H), 1.54 (s, 3H), 3.34 (s, 3H), 3.31–3.40 (m, 4H), 3.60–3.62 (m, 1H), 3.66–3.71 (m, 2H), 3.96 (t, J = 10.2 Hz, 1H), 4.08 (d, J = 5.5 Hz, 1H), 4.15–4.18 (m, 2H), 4.22 (d, J = 3.5 Hz, 1H), 4.28 (dd, J = 10.5 Hz, J = 5 Hz, 1H), 4.76–4.79 (m, 2H), 4.81 (s, 1H), 4.85–4.87 (m, 2H), 5.05 (s, 1H), 5.61 (s, 1H), 7.22–7.26 (m, 3H), 7.34–7.41 (m, 5H), 7.49–7.52 (m, 4H), 7.56 (d, J = 8.5Hz, 2H). 13C-NMR (125 MHz, CDCl3): δ 17.7, 26.4, 27.8, 54.9, 60.6, 64.0, 67.6, 68.6, 72.5, 76.0, 76.1, 77.4, 77.9, 78.3, 78.7, 84.9, 88.4, 97.8, 99.9, 101.4, 109.5, 125.2, 125.3, 126.0, 126.7, 127.5, 128.2, 128.3, 128.9, 132.1, 137.5, 138.3. 19F-NMR (471 MHz, CDCl3): δ −64.38. ESI-HRMS calcd for C40H43F3O10Na [M+Na]+ 763.2701, found 763.2672
1,6-Anhydro-4-O-{4,6-O-benzylidene-2-O-[3-(4-trifluoromethylphenyl)-prop-2-ynyl]-3-O-benzyl-β-d-mannopyranosyl}-2,3-O-isopropylidene-β-d-mannopyranose (13)
Viscous oil. [α]15d −68.5 (c 0.9, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 1.30 (s, 3H), 1.53 (s, 3H), 3.34–3.39 (m, 1H), 3.68 (dd, J = 10 Hz, J = 3 Hz, 1H), 3.77 (t, J = 7 Hz, 1H), 3.94 (t, J = 10 Hz, 1H), 3.98 (d, J = 7 Hz, 1H), 4.02 (dd, J = 6 Hz, J = 3 Hz, 1H), 4.09 (s, 1H), 4.20 (t, J = 9.5 Hz, 1H), 4.25 (d, J = 3 Hz, 1H), 4.32 (d, J = 10.5 Hz, J = 5 Hz, 1H), 4.37 (d, J = 6 Hz, 1H), 4.59 (d, J = 6 Hz, 1H), 4.77–4.89 (m, 5H), 5.33 (d, J = 2.5 Hz, 1H), 5.62 (s, 1H), 7.23–7.26 (m, 4H), 7.33 –7.41 (m, 5H), 7.49–7.57 (m, 5H); 13C-NMR (125 MHz, CDCl3): δ 25.9, 26.0, 61.0, 64.3, 67.7, 68.4, 72.1, 72.4, 72.8, 74.9, 75.3, 76.1, 78.4, 85.1, 88.0, 99.5, 99.9, 110.0, 125.2, 125.3, 126.0, 126.6, 127.6, 128.3, 128.4, 129.0, 132.1, 137.3, 138.0; 19F-NMR (471 MHz, CDCl3): δ −64.39; ESI-HRMS calcd for C39H39F3O10Na [M+Na]+ 747.2389, found 747.2373
3-O-[4,6-O-Benzylidene-2-O-[3-(4-trifluoromethylphenyl)-prop-2-ynyl]-3-O-benzyl-β-d-mannopyranosyl]-1,2:5,6-di-O-isopropylidene-α-d-glucofuranose (14)
Viscous oil. [α]23d −18.8 (c 1.2, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 1.25 (s, 3H), 1.37 (s, 3H), 1.44 (s, 3H), 1.49 (s, 3H), 3.33 (m, 1H), 3.68 (d, J = 8 Hz, 1H), 3.92 (t, J = 10.2 Hz, 1H), 4.09 (d, J = 8.5 Hz, 1H), 4.16–4.18 (m, 2H), 4.21–4.22 (m, 2H), 4.31–4.32 (m, 2H), 4.37–4.40 (m, 2H), 4.50 (s, 1H), 4.62 (s, 1H), 4.71–4.79 (m, 3H), 4.90 (d, J = 12.5 Hz, 1H), 5.62 (s, 1H), 5.85 (s, 1H), 7.25–7.27 (m, 3H), 7.35–7.38 (m, 5H), 7.48–7.49 (m, 2H), 7.55–7.56 (m, 4H). 13C-NMR (125 MHz, CDCl3): δ 25.6, 26.2, 26.5, 26.7, 60.6, 66.0, 67.7, 68.4, 73.0, 73.5, 75.8, 77.8, 78.6, 80.3, 80.6, 82.6, 85.3, 87.9, 99.5, 101.5, 104.9, 108.5, 111.9, 125.3, 126.0, 126.5, 127.6, 127.8, 128.3, 128.4, 129.0, 132.1, 137.4, 138.1. 19F-NMR (471 MHz, CDCl3): δ −64.47. ESI-HRMS calcd for C42H45F3O11Na [M+Na]+ 805.2795, found 805.2806.
6-O-[4,6-O-Benzylidene-2-O-[3-(4-trifluoromethylphenyl)-prop-2-ynyl]-3-O-benzyl-β-d-mannopyranosyl]-1,2:3,4-di-O-isopropylidene-α-d-galactopyranose (15)
Viscous oil. [α]15d −76.2 (c 1.1, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 1.32 (s, 6H), 1.4 (s, 3H), 1.50 (s, 3H), 3.33–3.35 (m, 1H), 3.63–3.69 (m, 2H), 3.92 (t, J = 10.2 Hz, 1H), 4.05 (dd, J = 8 Hz, J = 2 Hz, 1H), 4.13–4.17 (m, 2H), 4.2 (dd, J = 8 Hz, J = 1.5 Hz, 1H), 4.28 (d, J = 3 Hz, 1H), 4.30–4.34 (m, 2H), 4.60 (dd, J = 8 Hz, J = 2.5 Hz, 1H), 4.63 (s, 1H), 4.77 (d, J = 6 Hz, 1H), 4.8 (d, J = 2H, 1H), 4.83 (s, 1H), 4.86 (d, J = 3.5 Hz, 1H), 7.55 (d, J = 5 Hz, 1H), 5.6 (s, 1H), 7.18–7.22 (m, 4H), 7.30–7.33 (m, 2H), 7.36–7.40 (m, 3H), 7.48–7.51 (m, 2H), 7.53–7.56 (m, 3H). 13C-NMR (125 MHz, CDCl3): δ 24.4, 25.1, 26.0, 60.8, 67.4, 68.1, 68.5, 70.1, 70.7, 71.5, 72.6, 75.8, 78.5, 84.8, 88.5, 96.3, 101.5, 102.5, 108.8, 109.6, 125.2, 126.0, 126.8, 127.5, 127.6, 128.2, 128.3, 128.9, 132.1, 132.2, 137.5, 138.2. 19F-NMR (471 MHz, CDCl3): δ −64.38. ESI-HRMS calcd for C42H45F3O11Na [M+Na]+ 805.2806, found 805.2787.
Global deprotection with Sodium in liquid ammonia: 1-Adamantyl-β-d-mannopyranoside (16)
Mannopyranoside 9 (0.1 g, 0.12 mmol, 1 eq) was dissolved in a mixture of liquid ammonia (2 mL), THF (4 mL) and tertiary butanol (2 mL). To this solution at −70 °C was added sodium (0.03 g, 10 eq) in 4 pieces and the reaction mixture was warmed to −40 °C and the intense blue solution was stirred for 1 h. After the reaction completion (TLC 10% CH3OH:CHCl3), the reaction mixture at −20 °C was slowly quenched with saturated aqueous ammonium chloride solution (4 mL). The product was extracted with dichloromethane (3×4 mL). The organic layer was dried (MgSO4) and concentrated to afford a crude product which was purified on a silica chromatography (2%, 4%, 5%, 8%, 10% CHCl3:CH3OH) to afford 27 mg (72%) of 16. White solid: M.pt.: 218–220 °C: [α]11d −27.4 (c 0.7, CH3OH); 1H-NMR (400 MHz, CD3OD): δ 1.63–1.70 (m, 6H), 1.79–1.92 (m, 6H), 2.13 (m, 3H), 3.16–3.20 (m, 1H), 3.45 (dd, J = 8.8 Hz, J = 3.2 Hz, 1H), 3.65–3.69 (m, 2H), 3.82 (dd, J = 11.6 Hz, J = 2 Hz, 1H), 4.82 (d, 1H). 13C-NMR (125 MHz, CD3OD): δ 31.0, 36.2, 42.3, 61.8, 67.3, 72.9, 74.3, 74.8, 76.6, 93.4. ESI-HRMS calcd for C16H26NaO6 [M+Na]+ 337.1627, found 337.1613.
General procedure for selective reduction. 1-Adamantyl 3-O-benzyl-4,6-O-benzylidene-β-d-mannopyranoside (17)
Lithium naphthalenide in THF (0.25 M) was generated by dissolving Li (3–5 mg) in a solution of naphthalene (64 mg, 0.5 mmol) in THF (2 mL) at room temperature and allowing the reaction mixture to stir under Ar for 2 h. This dark green solution of lithium naphthalide (10 eq) was dropwise added to a solution of sugar 9 (34 mg, 0.05 mmol, 1 eq) in THF (1 mL) kept at −78 °C. The resultant reaction mixture was allowed to warm up to 0 °C over a period of 3 h and then quenched with a saturated solution of aqueous ammonium chloride (2.5 mL). The crude product was extracted with methylene chloride (3×4 mL). The organic layer was separated, dried (MgSO4) and concentrated. The residue was purified by column chromatography on silica (3%, 5%, 10%, 15%, 20%, 25% Ethyl acetate:Hexane) to afford 17 (21 mg, 85%). Viscous oil. [α]11d −8.7 (c 0.6, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 1.59–1.66 (m, 6H), 1.77–1.86 (m, 6H), 2.16 (m, 3H), 2.64 (s, 1H), 3.31–3.35 (m, 1H), 3.66 (dd, J = 9.8 Hz, J = 3 Hz, 1H), 3.88 (t, J = 9.8 Hz, 1H), 3.98 (d, J = 3 Hz, 1H), 4.15 (t, J = 9.8 Hz, 1H), 4.27 (dd, J = 9.8 Hz, J = 5 Hz, 1H), 4.78–4.86 (m, 3H), 7.26–7.32 (m, 4H), 7.33–7.40 (m, 4H), 7.49–7.50 (m, 2H). 13C-NMR (125 MHz, CDCl3): δ 30.7, 36.2, 42.4, 66.7, 68.8, 71.6, 72.3, 75.8, 78.3, 93.3, 126.1, 127.7, 127.9, 128.2, 128.9, 137.6, 138.2; ESI-HRMS calcd for C30H36O6Na [M+Na]+ 515.2410, found 515.2413.
Compounds 18 to 23
The spectral data for these compounds are consistent with the literature data.13
Supplementary Material
Copies of 1H and 13C NMR spectra of all compounds. This material is available free of charge via the internet at http://pubs.acs.org.
Figure 1.

Various propargyl ether protecting groups
Acknowledgement
We thank the NIH (GM 57335) for support of this work.
References
- 1.(a) Greene TW, Wuts PGM. Protective groups in Organic Synthesis. New York: Wiley and Sons; 2007. [Google Scholar]; (b) Kocienski PJ. Protecting Groups. Stuttgart: Thieme; 1994. [Google Scholar]
- 2.Crich D, Jayalath P. Org. Lett. 2005;7:2277. doi: 10.1021/ol050680g. [DOI] [PubMed] [Google Scholar]
- 3.(a) Crich D, Wu B. Org. Lett. 2006;8:4879. doi: 10.1021/ol061938l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crich D, Wu B, Jayalth P. J. Org. Chem. 2007;72:6806. doi: 10.1021/jo071009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.This compound was prepared using a two-step literature procedure starting from commercially available 1-bromo-4-trifluoromethylbenzene. See Wrobel J, Li Z, Dietrich A, McCabel M, Mihan B, Sredy J, Sullivan D. J. Med. Chem. 1998;41:1084. doi: 10.1021/jm9706168..
- 5.(a) Girard P, Namy JL, Kagan HB. J. Am. Chem. Soc. 1980;102:2693. [Google Scholar]; (b) Molander GA, Mckie JA. J. Org. Chem. 1992;57:3132. [Google Scholar]
- 6.Li J, Xu H, Zhang Y. Tetrahedron Lett. 2005;46:1931. [Google Scholar]
- 7.Szarek WA, Zamojski A, Tiwari K, Ison E. Tetrahedron Lett. 1986;27:3827. [Google Scholar]
- 8.Iserloh U, Dudkin V, Wang Z-G, Danishefsky SJ. Tetrahedron Lett. 2002;43:7027. [Google Scholar]
- 9.Liu H-J, Yip J, Shia K-S. Tetrahedron Lett. 1997;38:2253. [Google Scholar]
- 10.Crich D, Li W, Li H. J. Am .Chem .Soc. 2004;126:15081. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- 11.Crich D, Smith M. J. Am. Chem. Soc. 2001;123:9015. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 12.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001;2:323. [Google Scholar]
- 13.Crich D, Jayalath P, Hutton TK. J. Org. Chem. 2006;71:3064. doi: 10.1021/jo0526789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of 1H and 13C NMR spectra of all compounds. This material is available free of charge via the internet at http://pubs.acs.org.