

Abstract

Two series of neoglucosyl donors are prepared based on connection of the allylic disulfide motif to the anomeric center via either a simple O-glycosyl linkage or N-glycosyl amide unit. Conjugation of both sets of donors to cysteine in peptides is demonstrated through classical disulfide exchange followed by the phosphine-mediated desulfurative allylic rearrangement resulting in neoglycopeptides characterized by a simple thioether spacer. The conjugation reaction functions in the absence of protecting groups on both the neoglycosyl donor and peptide in aqueous media at room temperature.
Introduction
We have introduced the desulfurative rearrangement of allylic disulfides (Scheme 1) as a facile means of electrophile-free, permanent functionalization of thiols suitable for the modification of cysteine and other thiols.1 Herein we report on the extension of this chemistry to the ligation of carbohydrates to cysteine in peptides and proteins.
Scheme 1.

Ligation via the Desulfurative Allylic Disulfide Rearrangement
The ability to prepare glycosylated peptides and proteins is critical to the pursuit and advancement of glycomics and glycoscience.2 The generation of native linkages between oligosaccharides and peptides typically requires the assembly of preglycosylated peptide building blocks into larger peptides, as exemplified by recent groundbreaking syntheses employing the method of native chemical ligation,3 whereas the synthesis of neoglycoconjugates in principle allows the conjugation of oligosaccharides to fully assembled peptides and even proteins.4 Among the many elegant methods devised,1b,5 the disulfide ligation, as applied to peptides and proteins by the Boons and Davis groups,6 stands out for the mildness of conditions and broad functional group tolerance. The impermanence of the disulfide ligation, due to the ease of reduction and/or scrambling processes, has promoted the search for methods to convert the disulfide to a simple thioether which have so far been marred by a loss of stereochemical integrity of the cysteine residue involved in the linkage.7 The application of the desulfurative allylic rearrangement that we describe here combines the broad functional group compatibility of the disulfide linkage with the formation of permanent thioether type bond, and the maintenance of stereochemical integrity of the peptide chain.
Results and Discussion
In our approach to neoglycoconjugate synthesis we envisaged the use of a short spacer, resulting from the ligation reaction itself, to link the carbohydrate to the peptide. We preferred to accommodate the inherent requirement of the desulfurative allylic disulfide rearrangement for an allylic sulfide through the use of an appropriate tether, rather than attempt incorporation of this functionality into the saccharide itself to limit the number of chemical manipulations prior to the ligation reaction. To this end, in a first generation approach, a series of glycosyl bromides were coupled in β-selective reactions to cis-butene-1,4-diol (Scheme 2). Subsequent functionalization of the residual alcohol as a thionocarbonate ester was followed by heating in toluene at reflux to provoke [3,3]-sigmatropic rearrangement8 and the formation of allylic thiolcarbonates as mixtures of stereoisomers at the newly generated stereogenic center. Selective hydrazinolysis of the thiolcarbonate gave the corresponding allylic thiols which were not isolated but immediately converted to the benzothiazolyl disulfides in high yield (Scheme 2).
Scheme 2.

First Generation Approach to Precursor Synthesis
Exactly analogous chemistry with a sialosyl chloride gave an α-sialyl glycoside carrying the allylic disulfide moiety at the anomeric position (Scheme 3).
Scheme 3.

First Generation Approach Applied to a Sialic Acid Derivative
A more efficient second generation protocol involved application of the allylic thionocarbonate rearrangement to the mono-(tert-butyldimethylsilyl) ether of cis-butene1,4-diol, and desilylation followed by glycosylation by the trichloroacetimidate method9 to intercept the first generation method at the stage of the glycosylated thiolcarbonates 4a–c (Scheme 4).10
Scheme 4.

Second Generation Approach
The ideal approach (third generation) involved direct glycosylation of the known3 hydroxylated allylic disulfide 14, accessible from 12 by cleavage of the thiolcarbonate and sulfenylation in the usual manner, with glycosyl donors 13a–c. Unfortunately, while this approach was successful (Scheme 5), it provided the desired products in only moderate yield and resulted in complications in a subsequent step (vide infra).
Scheme 5.

Third Generation Approach
With four protected neoglycosyl donors in hand a series of couplings were attempted to model cysteine-containing peptides. These reactions were conducted at room temperature by mixing the peptide and the neoglycosyl donor in acetonitrile/methanol (1:1) as solvent and, after the sulfenyl transfer stage was complete, adding triphenylphosphine to provoke the key rearrangement and render the ligation complete. Initial reactions were conducted with protected peptides, before compatibility with free glutathione was employed in an aqueous medium. In all cases the ligation proceeded efficiently, with the products being obtained in a highly trans-selective manner with respect to the linker (Table 1).
Table 1.
Neoglycoconjugate Formation with Protected Donors.
| Neoglycosyl Donor | Peptides | Solvent | Neoglycopeptide (Yield) |
|---|---|---|---|
 |
 |
MeCN/ MeOH (1:1) |
 |
 |
15 | MeCN/ MeOH (1:1) |
 |
 |
15 | MeCN/ MeOH (1:1) |
 |
| 5a | 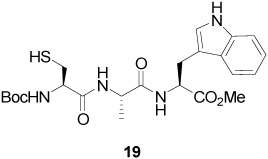 |
MeCN/ MeOH (1:1) |
 |
| 5b | 19 | MeCN/ MeOH (1:1) |
 |
| 5a | 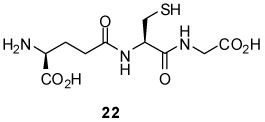 |
MeCN/ Tris buffer pH 8.0 (1:1) |
 |
 |
22 | MeCN/ Tris buffer pH 8.0 (1:1) |
 |
Deprotection of the neoglycoconjugate donors encountered several problems owing to the sensitivity of both disulfides11 and allylic thiols8c to basic conditions. Thus, Zemplen type cleavage of disulfides 5a–c and 10 gave complex reaction mixtures from which only low yields of the desired products could be isolated. Optimal conditions were eventually found involving a mild saponification at the level of the thiolcarbonates 4b, c and 9, with concomitant removal of the thiolcarbonate and direct sulfenylation of the released thiol without isolation (Scheme 6). The optimum synthesis of the free neoglycosyl donors is therefore the combination of the second generation approach to the protected donor (Scheme 3), followed by saponification and sulfenylation (Scheme 6).
Scheme 6.

Preparation of Unprotected Neoglycosyl Donors
Conjugation of the fully deprotected neoglycosyl donors 25 and 26 to the model peptide glutathione was achieved without issue in pH 8.0 phosphate buffer by simply mixing to achieve sulfenyl transfer and adding triphenylphosphine to bring about the sigmatropic rearrangement (Table 2). Interestingly, the yields in the protecting group free reactions conducted in phosphate buffer (Table 2) are routinely higher than those conducted with protected neoglycosyl donors in organic solvent mixtures (Table 1), which we attribute to the established facilitation of the desulfurative allylic disulfide rearrangement in polar solvents1c at the expense of the background phosphine-mediated disulfide cleavage reaction.
Table 2.
Protecting Group-Free Neoglycoconjugate Preparation in Phosphate Buffer
| Neoglycosyl Donor | Peptide | Neoglycopeptide (Yield) |
|---|---|---|
 |
22 | 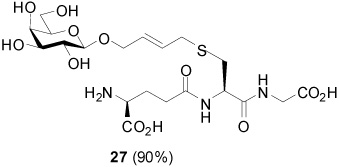 |
 |
22 | 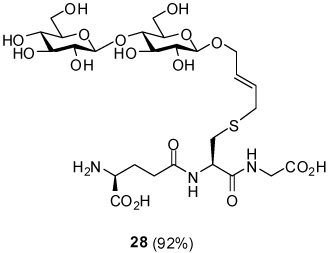 |
 |
22 | 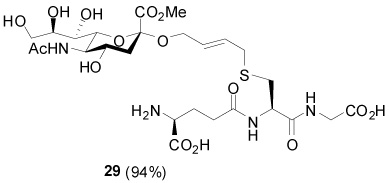 |
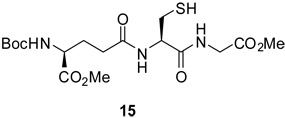
A second allylic disulfide linker was prepared as set out in Scheme 7 with a view to mimicking the N-glycosyl amides linkage. Thus, aldol 31 was subjected to the thio-Mitsunobu reaction12 to give the thioacetate 32. Saponification and sulfenylation then afforded disulfide 33 which was converted to the free acid 34 by exposure to trifluoroacetic acid.
Scheme 7.

Preparation of an Alternative Linker
Standard peptide coupling reactions then enabled acid 34 to be affixed to glycosylamines in the complete absence of protecting groups (Scheme 8). Although the yields for this coupling process are only moderate at the present time, it possesses the very obvious advantages of convergency and the direct provision of neoglycosyl donors ready for conjugation without the need for further protecting group manipulation.
Scheme 8.

Preparation of Amide-Based Neoglycosyl Donors
Neoglycoconjugate synthesis with donors 36 was conducted in aqueous buffer and proceeded in excellent yield (Table 3).
Table 3.
Amide-Based Neoglycopeptide Synthesis
| Neoglycosyl Donor | Peptide | Neoglycopeptide (Yield) |
|---|---|---|
 |
 |
 |
 |
22 | 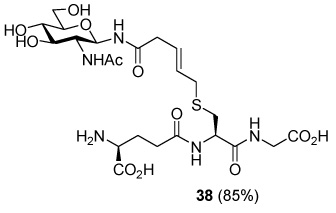 |
 |
 |
 |
Experimental Section
General
All reagents were purchased from commercial sources and used as received, unless otherwise indicated, and all reaction solvents were anhydrous. All reactions were conducted under an atmosphere of dry nitrogen. Organic extracts were dried over sodium sulfate, and concentrated under aspirator vacuum at room temperature.
General Procedure for the Reaction of Glycosyl Bromides with cis-But-2-ene-1,4-diol (2a-c, 7).13
cis-But-2-ene-1,4-diol (10 mL, 120 mmol), Ag2CO3 (750 mg, 2.7 mmol), CaSO4 (2 g) and an iodine crystal were stirred with the exclusion of moisture and light for 0.5 h at room temperature before a glycosyl bromide (1a–c) (1.9 mmol) or the sialyl chloride (6) was added. The reaction mixture was stirred at room temperature for 24 h, then diluted with dichloromethane (25 mL), filtered through Celite and the filtrate washed with water. The solvent was removed by evaporation and the mixture was purified by chromatography over silica gel to afford the glycosides (2a–c and 7).
(Z)-4-Hydroxybut-2-enyl Tetra-O-acetyl-β-d-glucopyranoside (2a)
White solid, eluted from silica gel with hexane/ethyl acetate (2:1) in 84% yield. M.p. 97 °C; [α]D25 −15.3° (c, 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 5.82 (m, 1H), 5.60 (m, 1H), 5.18 (t, J = 9.5 Hz, 1H), 5.06 (t, J = 9.5 Hz, 1H), 4.97 (dd, J = 8.0, J = 9.5 Hz, 1H), 4.55 (d, J = 8.0 Hz, 1H), 4.34 (dd, J = 6.0, J = 12.5 Hz, 1H), 4.24 (m, 2H), 4.17 (d, J = 6.5 Hz, 2H), 4.14 (dd, J = 2.0, J = 10.0 Hz, 1H), 3.69 (m, 1H), 2.08 (s, 3H), 2.03 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.8, 170.3, 169.5, 133.4, 126.7, 99.3, 72.8, 71.8, 71.2, 68.4, 64.3, 62.0, 58.5, 20.8, 20.7, 20.6; Anal. Calcd for C18H26O11 C, 51.33; H, 6.28; N, 2.49; Found, C, 51.22; H, 6.01; N, 2.44.
(Z)-4-Hydroxybut-2-enyl Tetra-O-acetyl-β-d-galactopyranoside (2b)
Syrup, eluted from silica gel with hexane/ethyl acetate (2:1) in 83% yield. [α]D15 −9.6° (c, 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 5.75 (m, 1H), 5.52 (m, 1H), 5.30 (d, J = 3.5 Hz, 1H), 5.11 (dd, J = 8.0, J = 10.5 Hz, 1H), 4.93 (dd, J = 3.5, J = 10.5 Hz, 1H), 4.45 (d, J = 8.0 Hz, 1H), 4.28 (dd, J = 7.5, J = 12.5 Hz, 1H), 4.20 (dd, J = 7.5, J = 12.5 Hz, 1H), 4.11-4.02 (m, 1H), 3.86 (t, J = 6.5 Hz, 1H), 2.07 (s, 3H), 1.98 (s, 6H), 1.89 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.5, 170.3, 170.1, 169.6, 133.5, 126.4, 99.6, 70.9, 70.6, 68.7, 67.1, 64.2, 61.4, 58.3, 20.7, 20.64, 20.5; ESIHRMS Calcd. for C18H26O11Na [M+Na]+ 441.1368, found 441.1361.
(Z)-4-Hydroxybut-2-enyl Hepta-O-acetyl-β-d-cellobioside (2c)
White solid, eluted from silica gel with hexane/ethyl acetate (1:3) in 81% yield; M.p. 171 °C; [α]D15 −21.8° (c, 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 5.83 (m, 1H), 5.60 (m, 1H), 5.15 (m, 2H), 5.05 (t, J = 9.5 Hz, 1H), 5.90 (m, 2H), 4.55-4.50 (m, 3H), 4.35(dd, J = 4.5, J = 12.5 Hz, 1H), 4.31(dd, J = 6.0, J = 13.0 Hz, 1H), 4.23(dd, J = 7.5, J = 12.5 Hz, 1H), 4.17 (d, J = 6.5 Hz, 2H), 4.09-4.03 (m, 2H), 3.76 (t, J = 9.5 Hz, 1H), 3.64 (m, 1H), 3.59 (m, 1H), 2.13 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H), 2.003 (s, 3H), 1.998 (s, 3H), 1.97 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.6, 170.4, 170.2, 169.8, 169.7, 169.3, 169.1, 133.5, 126.6, 100.7, 99.1, 76.4, 72.9, 72.7, 72.4, 71.9, 71.6, 71.5, 67.7, 64.3, 61.8, 61.5, 58.4, 20.9, 20.70, 20.66, 20.5; ESIHRMS Calcd. for C30H42O19Na [M+Na]+ 729.2218, found 729.2191.
(Z)-4-Hydroxybut-2-enyl (Methyl 4,7,8,9-tetra-O-acetyl-5-acetylamino-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate) (7)
White solid, eluted from silica gel with ethyl acetate in 58% yield. M.p. 130.1 °C; [α]D24 −11.2° (c, 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 5.74 (m, 1H), 5.57 (m, 1H), 5.44 (m, 1H), 5.30 (dd, J = 0.5, J = 8.5 Hz, 1H), 5.25 (dd, J = 0.5, J = 9.5 Hz, 1H), 4.83 (m, 1H), 4.34 (m, 1H), 4.20 (m, 1H), 4.12 (dd, J = 6.5, J = 13.5 Hz, 1H), 4.05 (m, 4H), 3.78 (s, 3H), 2.59 (dd, J = 4.5, J = 12.5 Hz, 1H), 2.16 (s, 3H), 2.14 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 1.87 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 171.04, 170.96, 170.5, 170.3, 168.4 (C-1, JC-1,H-3ax = 5.0 Hz), 131.8, 127.5, 98.4, 72.4, 69.0, 68.4, 67.3, 62.7, 61.1, 58.3, 52.8, 49.4, 38.1, 23.2, 21.2, 20.9, 20.8; Anal. Calcd for C24H35NO14 C, 51.33; H, 6.28; N, 2.49; Found, C, 51.22; H, 6.01; N, 2.44.
General Procedure for the Reaction of Hydroxybutenyl Glycosides with Phenyl Thionochloroformate (3a-c, 8)
A solution of phenyl chlorothionocarbonate (158 µL, 1.14 mmol) in dichloromethane (3 mL) was added to a solution of the alcohols 2a–c (0.38 mmol), pyridine (184 µL, 2.28 mmol) and DMAP (6 mg) in dichloromethane (2 mL), and the resulting yellow solution was stirred at room temperature for 5 h. The reaction mixture was poured into H2O (10 mL), and extracted with dichloromethane (3×10 mL). The combined organic phases were dried, filtered, evaporated, and purified by chromatography over silica gel.
(Z)-4-(Phenyloxythionocarbonyloxy)-2-butenyl Tetra-O-acetyl-β-d-glucopyranoside (3a)
Yellow syrup, eluted from silica gel with hexane/ethyl acetate (1:1) in 96% yield. [α]D21 −9.0° (c, 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.42 (t, J = 8.0 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.09 (t, J = 8.0 Hz, 2H), 5.87 (m, 1H), 5.80 (m, 1H), 5.19 (t, J = 9.5 Hz, 1H), 5.09 (m, 3H), 5.00 (dd, J = 8.0, J = 9.5 Hz, 1H), 4.56 (d, J = 8.0 Hz, 1H), 4.38 (dq, J = 5.5, J = 13.5 Hz, 2H), 4.25 (dd, J = 4.5, J = 12.0 Hz, 1H), 4.15 (dd, J = 2.0, J = 12.0 Hz, 1H), 3.69 (m, 1H), 2.07 (s, 3H), 2.05 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 194.9, 170.7, 170.3, 169.41, 169.39, 153.4, 130.6, 129.6, 126.7, 126.1, 121.9, 99.5, 72.8, 71.9, 71.2, 69.2, 68.3, 64.6, 61.9, 20.79, 20.75, 20.6; ESIHRMS Calcd. for C25H30O12SNa [M+Na]+ 577.1356, found 577.1338.
(Z)-4-(Phenyloxythionocarbonyloxy)-2-butenyl Tetra-O-acetyl-β-d-galactopyranoside (3b)
Yellow syrup, eluted from silica gel with hexane/ethyl acetate (1:1) in 90% yield. [α]D15 −3.5° (c, 1.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.38 (m, 2H), 7.26 (dt, J = 1.0, J = 7.5 Hz, 1H), 7.07 (m, 2H), 5.81 (m, 2H), 5.34 (dd, J = 0.5, J = 3.5 Hz, 1H), 5.19 (dd, J = 8.0, J = 10.5 Hz, 1H), 5.07 (m, 2H), 4.97 (dd, J = 3.5, J = 10.5 Hz, 1H), 4.50 (d, J = 8.0 Hz, 1H), 4.39 (dd, J = 5.5, J = 12.5 Hz, 1H), 4.32 (dd, J = 6.5, J = 12.5 Hz, 1H), 4.12(d, J = 7.0 Hz, 1H), 3.88 (dt, J = 1.5, J = 6.5 Hz, 1H), 2.10 (s, 3H), 2.02 (s, 3H), 1.98 (s, 3H), 1.93 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 194.9, 170.3, 170.2, 170.1, 169.4, 153.4, 130.7, 129.6, 126.7, 125.9, 121.9, 100.0, 70.9, 70.8, 69.2, 68.7, 67.1, 64.6, 61.3, 20.8, 20.7, 20.6; ESIHRMS Calcd. for C25H30O12SNa [M+Na]+ 577.1356, found 577.1344.
(Z)-4-(Phenyloxythionocarbonyloxy)-2-butenyl Hepta-O-acetyl-β-d-cellobioside (3c)
Yellow syrup, eluted from silica gel with hexane/ethyl acetate (1:1) in 96% yield, [α]D15 −18.1° (c, 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.42 (t, J = 8.0 Hz, 2H), 7.30 (m, 1H), 7.09 (m 2H), 5.87 (m, 1H), 5.79 (m, 1H), 5.19-5.03 (m, 5H), 4.91 (m, 2H), 4.54-4.48 (m, 3H), 4.40-4.31(m, 3H), 3.78 (t, J = 9.5 Hz, 1H), 3.64 (m, 1H), 3.58 (m, 1H), 2.13 (s, 3H), 2.08 (s, 3H), 2.04 (s, 3H), 2.01 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H), 1.97 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 195.0, 170.5, 170.33, 170.26, 169.8, 169.7, 169.3, 169.1, 153.4, 130.6, 129.6, 126.7, 126.1, 121.9, 100.8, 99.3, 76.4, 72.9, 72.8, 72.5, 72.0, 71.6, 71.4, 69.2, 67.8, 64.6, 61.8, 61.5, 21.0, 20.8, 20.7, 20.6; ESIHRMS Calcd. for C37H46O20SNa [M+Na]+ 865.2201, found 865.2173.
(Z)-4-(Phenyloxythionocarbonyloxy)-2-butenyl (Methyl 4,7,8,9-tetra-O-acetyl-5-acetylamino-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate) (8)
Yellow syrup, eluted from silica gel with dichloromethane/methanol (10:1) in 99% yield. [α]D24 −12.6° (c, 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.41(d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 1H), 7.09 (d, J = 8.0 Hz, 2H), 5.79 (m, 1H), 5.43 (m, 1H), 5.32 (dd, J = 2.0, J = 8.5 Hz, 1H), 5.24 (d, J = 9.0 Hz, 1H), 5.14 (dd, J = 1.5, J = 6.5 Hz, 2H), 4.85 (m, 1H), 4.41(dd, J = 5.0, J = 13.5 Hz, 1H), 4.29 (dd, J = 2.5, J = 12.5 Hz, 1H), 4.06 (m, 4H), 3.79 (s, 3H), 2.60 (dd, J = 4.5, J = 13.0 Hz, 2H), 2.16 (s, 3H), 2.12 (s, 3H), 2.023 (s, 3H), 2.017 (s, 3H), 1.87 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 194.9, 171.0, 170.7, 170.24, 170.18, 170.1, 168.2, 153.5, 130.3, 129.6, 126.6, 125.5, 121.9, 98.5, 72.5, 69.8, 69.0, 68.3, 67.2, 61.0, 52.9, 49.4, 38.0, 23.2, 21.2, 20.9, 20.8; ESIHRMS Calcd. for C31H39NO15SNa [M+Na]+ 720.1933, found 720.1928.
General Procedure for the [3,3]-Sigmatropic Rearrangement Reaction of Allylic Thionocarbonates (4a–c, 9)
A solution of 3a–c or 8 in toluene was heated at reflux for 8h–12h. Evaporation of the solvent afforded 4a–c or 9 as yellow syrups in quantitative yield.
2-(Phenyloxycarbonylthioxy)-3-butenyl Tetra-O-acetyl-β-d-glucopyranoside (4a)
An approximately 1:1 mixture of diastereomers with 1H NMR (500 MHz, CDCl3) δ: 7.38 (m, 2×2H), 7.24 (t, J = 7.5 Hz, 2×1H), 7.15 (m, 2×2H), 5.90 (m, 2×1H), 5.39 (dd, J = 3.5, J = 17.0 Hz, 2×1H), 5.22 (m, 2×2H), 5.08 (dt, J = 3.5, J = 10.0 Hz, 2×1H), 5.02 (dt, J = 2.0, J = 8.0 Hz, 2×1H), 4.58 (d, J = 8.0 Hz, 1H), 4.56 (d, J = 8.0 Hz, 1H), 4.16 (m, 2×4H), 3.79 (m, 2×1H), 3.70 (m, 2×1H), 2.07 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 2.018 (s, 3H), 2.017 (s, 3H), 2.003 (s, 3H), 2.000 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.7, 170.3, 169.4, 169.3, 169.0, 168.96, 168.92, 168.31, 168.29, 151.1, 133.6, 129.6, 126.3, 121.3, 119.0, 118.9, 101.3, 100.5, 72.7, 72.6, 71.9, 71.8, 71.04, 71.98, 70.5, 68.4, 68.3, 61.9, 61.8, 48.5, 47.9, 20.8, 20.6 ; ESIHRMS Calcd. for C25H30O12SNa [M+Na]+ 577.1356, found 577.1339.
2-(Phenyloxycarbonylthioxy)-3-butenyl Tetra-O-acetyl-β-d-galactopyranoside (4b)
An approximately 1:1 mixture of diastereomers with 1H NMR (500 MHz, CDCl3) δ: 7.38 (m, 2×2H), 7.25 (t, J = 7.5 Hz, 2×2H), 7.15 (m, 2×2H), 5.92 (m, 2×1H), 5.39 (m, 2×2H), 5.24 (m, 2×2H), 5.02 (m, 2×1H), 4.53 (d, J = 8.5 Hz, 1H), 4.51 (d, J = 8.5 Hz, 1H), 4.21-4.10 (m, 2×4H), 3.91 (m, 2×1H), 3.79(m, 2×1H), 2.513 (s, 3H), 2.148 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 1.99 (s, 3H), 1.98 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.4, 170.3, 170.2, 169.5, 169.4, 168.9, 151.1, 133.8, 133.4, 129.6, 126.3, 121.3, 118.9, 118.8, 101.8, 101.1, 71.7, 70.83, 70.76, 70.5, 68.6, 68.5, 67.0, 61.3, 48.6, 48.0, 20.88, 20.83, 20.7, 20.6; ESIHRMS Calcd. for C25H30O12SNa [M+Na]+ 577.1356, found 577.1331.
2-(Phenyloxycarbonylthioxy)-3-butenyl Hepta-O-acetyl-β-d-cellobioside (4c)
An approximately 1:1 mixture of diastereomers with 1H NMR (500 MHz, CDCl3) δ: 7.37 (t, J = 8.0 Hz, 2×2H), 7.24 (t, J = 7.5 Hz, 2×1H), 7.14 (m 2×2H), 5.87 (m, 2×1H), 5.36 (d, J = 17.5 Hz, 1H), 5.35 (d, J = 17.5 Hz, 1H), 5.24-5.11 (m, 2×3H), 5.05 (m, 2×1H), 4.91 (m, 2×2H), 4.54-4.49 (m, 2×3H), 4.35 (dd, J = 4.5, J = 12.5 Hz, 2×1H), 4.21-4.02 (m, 2×4H), 3.76 (m, 2×2H), 3.65 (m, 2×1H), 3.59 (m, 2×1H), 2.10 (s, 3H), 2.09 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H), 2.024 (s, 3H), 2.015 (s, 2×3H), 2.01 (s, 2×3H), 2.00 (s, 2×3H), 1.97 (s, 2×3H); 13C NMR (125 MHz, CDCl3) δ: 170.5, 170.3, 170.2, 169.8, 169.6, 169.5, 169.3, 169.1, 168.92, 168.87, 151.1, 133.7, 133.4, 129.6, 129.3, 121.3, 118.9, 118.8, 101.2, 100.8, 100.3, 72.9, 72.8, 72.34, 72.29, 72.0, 71.7, 71.6, 71.3, 71.2, 70.4, 67.8, 61.74, 61.7, 61.6, 48.4, 47.9, 20.9, 20.74, 20.72, 20.68, 20.6; ESIHRMS Calcd. for C37H46O20SNa [M+Na]+ 865.2201, found 865.2164.
2-(Phenyloxycarbonylthioxy)-3-butenyl (Methyl 4,7,8,9-tetra-O-acetyl-5-acetylamino-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate) (9)
An approximately 1:1 mixture of diastereomers with 1H NMR (500 MHz, CDCl3) δ: 7.36 (t, J = 8.0 Hz, 2×2H), 7.22 (m, 2×1H), 7.15 (d, J = 8.0 Hz, 2×2H), 5.92 (m, 2×1H), 5.39 (m, 2×2H), 5.32 (m, 2×1H), 5.21 (m, 2×1H), 4.86 (m, 2×1H), 4.27 (t, J = 12.5 Hz, 2×1H), 4.15-4.01 (m, 2×6H), 3.80 (s, 3H), 3.78 (s, 3H), 3.58 (m, 2×1H), 2.61 (m, 2×1H), 2.12 (s, 2×3H), 2.11 (s, 2×3H), 2.03 (s, 2×3H), 2.01 (s, 2×3H), 1.864 (s, 3H), 1.861 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 171.2, 170.9, 170.45, 170.37, 170.2, 169.0, 168.4, 168.3, 151.4, 134.4, 134.0, 129.7, 129.3, 128.5, 126.4, 121.6, 118.7, 118.6, 98.9, 98.8, 72.8, 69.3, 68.7, 67.5, 66.5, 66.4, 62.6, 62.5, 53.11, 53.07, 49.7, 48.8, 48.5, 38.1, 23.5, 21.3, 21.10, 21.06, 21.0; ESIHRMS Calcd. for C31H39NO15SNa [M+Na]+ 720.1933, found 720.1936.
General Procedure for the Conversion of Allylic Thiocarbonates to Allylic Benzothiazolyl Disulfides
The thiocarbonate (0.48 mmol) and NH2NH2•H2O (36 µL, 0.72 mmol) were dissolved with stirring in DMF (3 mL), followed after 2 min, by the dropwise addition of glacial acetic acid (41 µL, 0.72 mmol) at room temperature over 0.5 h. The resulting solution was added dropwise to a well stirred suspension of 2, 2’-dithiobis(benzothiazole) (242 mg, 0.72 mmol) in dichloromethane (3 mL) over 15 min, followed by stirring at room temperature until completion (~3 h). The solvent was evaporated and the residue was taken up in dichloromethane, and washed with water. The combined organic phases were dried, filtered, evaporated, and purified by chromatography over silica gel.
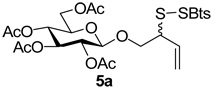
2-(Benzothiazol-2-yldisulfanyl)-3-enyl Tetra-O-acetyl-β-d-glucopyranoside (5a)
Yellow foam, eluted from silica gel as an approximately 1:1 mixture of diastereomers with hexane/ethyl acetate (2:1) in 76% yield. 1H NMR (500 MHz, CDCl3) δ: 7.84 (m, 2×2H), 7.44 (t, J = 7.5 Hz, 2×1H), 7.35 (t, J = 7.5 Hz, 2×1H), 5.84 (m, 1H), 5.75 (m, 1H), 5.33−5.18 (m, 2×3H), 5.10−5.01 (m, 2×2H), 4.55 (d, J = 8.0 Hz, 1H), 4.54 (d, J = 8.0 Hz, 1H), 4.26-4.17 (m, 2×2H), 4.12 (dt, J = 2.0, J = 12.0 Hz, 2×1H), 3.85-3.75 (m, 2×2H), 3.68 (m, 2×1H), 2.08 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.024 (s, 3H), 2.021 (s, 3H), 2.009 (s, 3H), 2.008 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 172.7, 172.4, 170.1, 170.3, 169.41, 169.35, 155.0, 135.9, 132.6, 132.3, 126.4, 124.7, 122.23, 122.18, 121.2, 120.7, 120.5, 101.1, 100.8, 72.7, 72.6, 72.0, 71.0, 70.3, 70.1, 68.3, 54.2, 20.8, 20.6; ESIHRMS Calcd. for C25H29NO10S3Na [M+Na]+ 622.0846, found 622.0851.
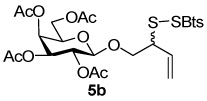
2-(Benzothiazol-2-yldisulfanyl)-3-enyl Tetra-O-acetyl-β-d-galactopyranoside (5b)
Yellow foam, eluted from silica gel as an approximately 1:1 mixture of diastereomers with hexane/ethyl acetate (2:1) in 48% yield. 1H NMR (500 MHz, CDCl3) δ: 7.86 (m, 2×1H), 7.82 (d, J = 8.0 Hz, 2×1H), 7.44 (m, 2×1H), 7.33 (m, 2×1H), 5.85 (m, 1H), 5.76 (m, 1H), 5.39 (2s, 2×1H), 5.34-5.22 (m, 2×3H), 5.01 (m, 2×1H), 4.51 (d, J = 8.0 Hz, 1H), 4.50 (d, J = 8.0 Hz, 1H), 4.22 (m, 2×1H), 4.19-4.09 (m, 2×2H), 3.89 (m, 2×1H), 3.85 (m, 2×1H), 3.78 (d, J = 10.0 Hz, 1H), 3.77 (d, J = 10.0 Hz, 1H), 2.514 (s, 3H), 2.148 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 1.99 (s, 2×3H); 13C NMR (125 MHz, CDCl3) δ: 172.7, 172.3, 170.4, 170.3, 170.2, 169.5, 169.4, 155.0, 135.9, 132.7, 132.3, 126.38, 126.35, 124.8, 124.7, 122.24, 122.18, 121.2, 120.7, 120.5, 101.6, 101.3, 70.84, 70.79, 70.7, 70.2, 70.1, 68.5, 66.9, 61.2, 54.2, 20.98, 20.95, 20.7, 20.6; ESIHRMS Calcd. for C25H29NO10S3Na [M+Na]+ 622.0846, found 622.0836.
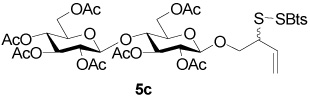
2-(Benzothiazol-2-yldisulfanyl)-3-enyl Hepta-O-acetyl-β-d-cellobioside (5c)
Yellow foam, eluted from silica gel as an approximately 1:1 mixture of diastereomers with hexane/ethyl acetate (1:1) in 50% yield. 1H NMR (500 MHz, CDCl3) δ: 7.84 (m, 2×2H), 7.43 (t, J = 7.5 Hz, 2×1H), 7.34 (t, J = 7.5 Hz, 2×1H), 5.83-5.70 (m, 2×1H), 5.31−5.22 (m, 2×2H), 5.18-5.11 (m, 2×2H), 5.06 (m, 2×1H), 4.93 (m, 2×2H), 4.53−4.47 (m, 2×3H), 4.38 (m, 1H), 4.35 (m, 1H), 4.13 (m, 2×1H), 4.10−4.02 (m, 2×2H), 3.85-3.73 (m, 2×3H), 3.65 (m, 2×1H), 3.55 (m, 2×1H), 2.17 (s, 3H), 2.10 (s, 3H), 2.082 (s, 3H), 2.079 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 2.023 (s, 3H), 2.020 (s, 3H), 2.015 (s, 2×3H), 2.00 (s, 2×3H), 1.98 (s, 2×3H); 13C NMR (125 MHz, CDCl3) δ: 172.7, 172.4, 170.5, 170.29, 170.26, 169.8, 169.61, 169.56, 169.3, 169.1, 155.0, 135.9, 132.7, 132.4, 126.3, 124.7, 122.21, 122.18, 121.2, 120.6, 120.5, 100.8, 100.6, 72.9, 72.8, 72.33, 72.26, 72.0, 71.6, 71.3, 70.2, 70.1, 67.8, 61.7, 61.6, 54.24, 54.21, 20.88, 20.83, 20.80, 20.7, 20.6; ESIHRMS Calcd. for C37H46O18NS3 [M+H]+ 888.1877, found 888.1834.
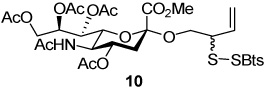
2-(Benzothiazol-2-yldisulfanyl)-3-enyl (Methyl 4,7,8,9-tetra-O-acetyl-5-acetylamino-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate) (10)
Yellow foam, eluted from silica gel as an approximately 1:1 mixture of diastereomers with dichloromethane/methanol (10:1) in 60% yield. 1H NMR (500 MHz, CDCl3) δ: 7.85(dd, J = 3.0, J = 8.0 Hz, 2×1H), 7.81 (d, J = 8.0 Hz, 2×1H), 7.42 (t, J = 8.0 Hz, 2×1H), 7.33 (t, J = 8.0 Hz, 2×1H), 5.83 (m, 2×1H), 5.40 (m, 2×1H), 5.31 (m, 2×2H), 5.24 (m, 2×1H), 5.19 (m, 2×1H), 4.89 (m, 2×1H), 4.28(m, 2×1H), 4.10 (m, 2×4H), 3.79 (s, 3H), 3.76 (s, 3H), 3.68 (m, 1H), 3.59 (m, 1H), 2.61 (m, 2×1H), 2.12 (m, 2×6H), 2.03 (m, 2×6H), 1.98 (t, J = 12.50 Hz, 2×1H), 1.88 (s, 2×3H); 13C NMR (125 MHz, CDCl3) δ: 171.0, 170.6, 170.3, 170.1, 168.1, 155.1, 135.9, 132.8, 132.6, 126.2, 124.6, 122.1, 121.1, 120.3, 120.2, 98.7, 98.5, 69.0, 68.5, 68.4, 67.2, 65.8, 68.4, 67.2, 65.8, 65.5, 62.4, 54.8, 54.7, 52.9, 49.4, 37.9, 23.2, 21.1, 20.9, 20.8; ESIHRMS Calcd. for C31H38N2O19S3Na [M+Na]+ 765.1429, found 765.1423.
2-(Phenoxycarbonylthioxy)-3-butenol (12)
A solution of phenyl chlorothionocarbonate (5 g, 29 mmol) in dichloromethane (3 mL) was added to a solution of (E)-4-(tert-butyldimethylsiloxy)but-2-en-1-ol14 (1.95 g, 9.65 mmol), pyridine (4.6 mL, 58 mmol), and 4-(dimethylamino)pyridine (236 mg) in dichloromethane (48 mL). The yellow solution was stirred for 5 h then was poured into H2O, and extracted with dichloromethane. The combined organic phases were dried, filtered, evaporated. The residue was taken up in toluene (200 mL) and heated at reflux for 4 h at room temperature. Evaporation of the solvent afforded a yellow syrup which was dissolved in dichloromethane/methanol (9:1), and p-TsOH•H2O (1.87 g, 9.65 mmol) was added in one portion. On completion, the reaction mixture was poured into H2O, and extracted with dichloromethane. The combined organic phases were dried, filtered, evaporated and purified by chromatography over silica gel, eluting with with hexane/ethyl acetate (2:1), to give the title compound as a yellow liquid in (1.2 g, 68%). 1H NMR (500 MHz, CDCl3) δ: 7.38 (t, J = 8.0 Hz, 2×2H), 7.25 (t, J = 7.5 Hz, 2×1H), 7.16 (d, J = 8.0 Hz, 2×2H), 5.92 (m, 2×1H), 5.44 (d, J = 17.0 Hz, 2×1H), 5.32 (d, J = 10.0 Hz, 2×1H), 4.14 (m, 2×1H), 3.89 (m, 2×2H), 1.92 (t, J = 6.0 Hz, 2×1H); 13C NMR (125 MHz, CDCl3) δ: 169.3, 151.3, 133.7, 129.8, 126.5, 121.5, 119.7, 64.6, 51.3; ESIHRMS Calcd. for C11H12O3SNa [M+Na]+ 247.0405, found 247.0430.
General Procedure for the Glycosylation of 2-(Phenoxycarbonylthioxy)-3-butenol with Glycosyl Trichloroacetimidates
The trichloroacetimidate (0.2 mmol), alcohol 12 (0.4 mmol) and activated 4Å powdered molecular sieves were mixed in dichloromethane (2 mL) and stirred at room temperature for 0.5 h before TMSOTf (0.06mmol) was added at 0 °C. The reaction mixture was allowed to warm to room temperature, and stirred for 2–4 h, when TLC showed the donor to hae been consumed. 4-(dimethylamino)pyridine (0.4 mmol) and Ac2O (0.4 mmol) were added at 0 °C and stirring continued for 4 h at room temperature before saturated aqueous NaHCO3 was added at 0 °C, and the reaction mixture was filtered, and washed with brine. The organic layer was dried and concentrated under reduced pressure and the glycosides were isolated by silica gel column chromatography (hexane/ethyl acetate from 95/5 to 2/1).
General Procedure for the Glycosylation of 2-(Benzothiazol-2-yldisulfanyl)-but-3-enol with Glycosyl Trichloroacetimidates
The trichloroacetimidate (0.2 mmol), 2-(benzothiazol-2-yldisulfanyl)-but-3-en-1-ol1c (0.4 mmol), and activated 4 Å powdered molecular sieves were mixed in dichloromethane (2 mL) and stirred at room temperature for 0.5 h before Zn(OTf)2 (74 mg, 0.2 mmol) was added at 0 °C. The reaction mixture was allowed to warm to room temperature and was stirred for 8–12 h until TLC indicated consumption of the donor.15 The reaction mixture was quenched with saturated aqueous NaHCO3 at 0 °C, filtered, and washed with brine. The organic layer was dried and concentrated under reduced pressure, and the glycosides were isolated by silica gel column chromatography (hexane/ethyl acetate from 95/5 to 2/1).
General Procedure for the Ligation of Protected Neoglycosyl Donors with Protected Peptides
A solution of cysteine containing peptide (0.05 mmol) and neoglycosyl donor (0.075 mmol) in methanol/acetonitrile (0.5 mL/0.5 mL) was stirred at room temperature until TLC indicated completion, after which triphenylphosphine (39 mg, 0.15 mmol) was added and the reaction mixture was stirred at room temperature for 10–24 h. The solution was evaporated and the mixture purified by chromatography eluting with dichloromethane/methanol (100/1).
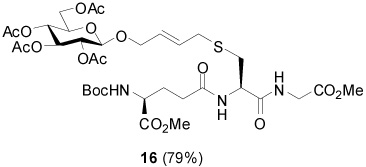
S-[4-(Tetra-O-acetyl-β-d-glucopyranosyloxy)-2E-butenyl] N-(t-Butyloxycarbonyl)-γ-l-Glu-(α-OMe)-l-Cys-Gly-OMe (16)
79% yield. [α]D23 −40.8° (c, 0.2, MeOH); 1H NMR (500 MHz, CDCl3) δ: 5.75 (m, 2H), 5.27 (t, J = 9.5 Hz, 1H), 5.04 (t, J = 10.0 Hz, 1H), 4.92 (m, 1H), 4.76 (d, J = 8.0 Hz, 1H), 4.73 (m, 1H), 4.30 (m, 2H), 4.16 (m, 3H), 3.98 (d, J = 5.5 Hz, 2H), 3.88 (m, 1H), 3.73 (s, 6H), 3.40 (m, 2H), 3.21 (dd, J = 5.0 Hz, J = 14.0 Hz, 1H), 2.91 (m, 1H), 2.40 (t, J = 7.5 Hz, 2H), 2.14 (m, 1H), 2.08 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H), 1.98 (s, 3H), 1.93 (m, 1H), 1.45 (s, 9H); 13C NMR (125 MHz, CDCl3) δ: 172.94, 172.90, 171.2, 170.8, 170.6, 170.0, 169.54, 169.46, 155.8, 129.7, 128.8, 99.5, 80.3, 72.8, 71.7, 71.4, 69.1, 68.4, 62.0, 60.4, 52.6, 52.5, 52.44, 52.36, 41.3, 33.6, 32.5, 32.2, 28.9, 28.7, 28.3, 27.8, 21.1, 20.8, 20.7; ESIHRMS Calcd. for C35H53N3O18SNa [M+Na]+ 858.2943, found 858.2936.
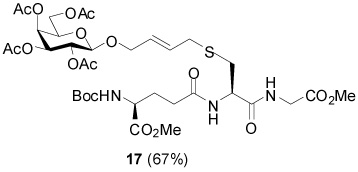
S-[4-(Tetra-O-acetyl-β-d-galactopyranosyloxy)-2E-butenyl] N-(t-Butyloxycarbonyl)-γ-l-Glu-(α-OMe)-l-Cys-Gly-OMe (17)
67% yield, [α]D23 −36.6° (c, 0.1, MeOH); 1H NMR (500 MHz, CD3OD) δ: 5.70 (m, 2H), 5.37 (m, 1H), 5.20 (dd, J = 3.5 Hz, J = 11.0 Hz, 1H), 5.09 (m, 1H), 4.74 (d, J = 7.5 Hz, 1H), 4.54 (m, 1H), 4.29 (m, 1H), 4.14 (m, 5H), 3.722 (s, 3H), 3.716 (s, 3H), 3.31 (m, 2H), 2.98 (dd, J = 5.5 Hz, J = 14.0 Hz, 1H), 2.69 (m, 1H), 2.39 (t, J = 7.5 Hz, 2H), 2.14 (s, 3H), 2.13 (m, 1H), 2.06 (s, 3H), 2.04 (s, 3H), 1.94 (s, 3H), 1.93 (m, 1H), 1.44 (s, 9H); 13C NMR (125 MHz, CD3OD) δ: 173.5, 173.2, 171.9, 170.8, 170.7, 170.2, 170.13, 170.10, 170.0, 156.7, 129.6, 128.6, 99.2, 79.3, 71.0, 70.4, 69.1, 68.3, 67.6, 61.3, 53.1, 52.4, 51.4, 51.3, 40.5, 32.6, 31.8, 31.5, 27.3, 27.0, 19.4, 19.3, 19.1; ESIHRMS Calcd. for C35H53N3O18SNa [M+Na]+ 858.2943, found 858.2940.
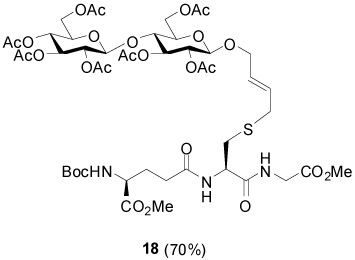
S-[4-(Hepta-O-acetyl-β-d-cellobiosyloxy)-2E-butenyl] N-(t-Butyloxycarbonyl)-γ-l-Glu-(α-OMe)-l-Cys-Gly-OMe (18)
70% yield, [α]D21 −15.4° (c, 0.7, CHCl3); 1H NMR (500 MHz, CD3OD) δ: 5.69-5.61 (2H, m), 5.24-5.16 (m, 2H), 5.00 (t, J = 9.6 Hz, 1H), 5.09 (m, 1H), 4.80 (t, J = 8.4 Hz, 2H), 4.70 (dd, J = 2.4, J = 8.0 Hz, 1H), 4.61 (dd, J = 4.0, J = 8.0 Hz, 1H), 4.53 (m, 2H), 4.39 (m, 1H), 4.26 (m, 1H), 4.14 (m, 3H), 4.04 (dd, J = 2.0, J = 12.4 Hz, 1H), 3.97 (s, 2H), 3.89-3.81 (m, 2H), 3.78 (m, 1H), 3.72 (s, 3H), 3.71 (s, 3H), 3.18 (m, 2H), 2.96 (m, 1H), 2.67 (m, 1H), 2.37 (t, J = 7.4 Hz, 2H), 2.12 (m, 4H), 2.05 (s, 3H), 2.02 (s, 3H), 2.01 (s, 3H), 1.98 (s, 3H), 1.93 (m, 4H), 1.43 (s, 9H); 13C NMR (125 MHz, CD3OD) δ: 173.7, 173.3, 171.8, 171.7, 171.2, 171.0, 170.7, 170.4, 170.1, 170.0, 169.8, 156.5, 133.5, 126.1, 100.7, 99.0, 79.5, 76.9, 73.3, 73.1, 72.8, 71.9, 71.8, 71.7, 68.1, 64.1, 62.4, 61.6, 57.5, 55.74, 55.70, 53.3, 51.6, 51.5, 40.8, 40.7, 31.7, 27.6, 27.2, 25.8, 19.8, 19.6, 19.53, 19.5, 19.40, 19.38; ESIHRMS Calcd. for C47H69N3O26SNa [M+Na]+ 1146.3788, found 1146.3786.
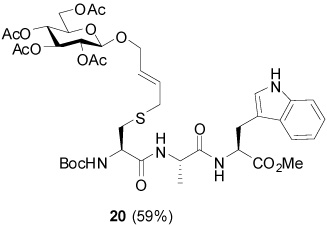
S-[4-(Tetra-O-acetyl-β-d-glucopyranosyloxy)-2E-butenyl] N-(t-Butyloxycarbonyl)-l-Cys-l-Ala-l-Trp-OMe (20)
59% Yield, [α]D23 −19.9° (c, 0.9, MeOH); 1H NMR (500 MHz, CD3OD) δ: 7.50 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.09 (m, 2H), 7.02 (m, 1H), 5.63 (m, 2H), 5.25 (t, J = 10.0 Hz, 1H), 4.99 (t, J = 10.0 Hz, 1H), 4.73 (m, 1H), 4.64 (d, J = 8.0 Hz, 1H), 4.42 (m, 1H), 4.24 (m, 2H), 4.17 (m, 1H), 4.09 (m, 3H), 3.76 (m, 1H), 3.64 (s, 3H), 3.27 (m, 1H), 3.22 (m, 1H), 3.10 (d, J = 2.5 Hz, 1H), 2.81 (m, 1H), 2.04 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H), 1.95 (s, 3H), 1.44 (s, 9H), 1.32 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ: 173.0, 172.2, 171.6, 171.0, 170.3, 169.9, 169.8, 156.3, 136.6, 129.3, 128.5, 127.4, 123.3, 121.1, 118.5, 117.8, 111.0, 109.1, 99.2, 79.5, 72.9, 71.5, 71.3, 68.6, 68.4, 61.7, 60.2, 53.8, 53.4, 51.3, 48.9, 32.7, 32.4, 27.3, 27.1, 19.5, 19.4, 19.3, 19.2, 16.8, 13.1; ESIHRMS Calcd. for C41H57N4O16S [M+H]+ 893.3485, found 893.3488.
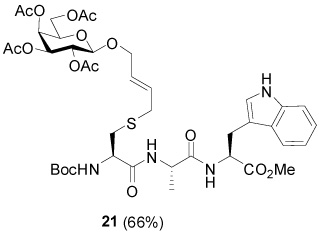
S-[4-(Tetra-O-acetyl-β-d-galactopyranosyloxy)-2E-butenyl] N-(t-Butyloxycarbonyl)-l-Cys-l-Ala-l-Trp-OMe (21)
66% yield, [α]D21 −10.5° (c, 1.1, MeOH); 1H NMR (500 MHz, CDCl3) δ: 8.61 (br. s, 1H), 7.49 (t, J = 8.0 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.16 (t, J = 7.5 Hz, 1H), 7.09 (t, J = 7.5 Hz, 1H), 7.00 (s, 1H), 6.83 (d, J = 7.5 Hz, 1H), 6.79 (d, J = 7.0 Hz, 1H), 5.63 (m, 2H), 5.38 (d, J = 3.0 Hz, 1H), 5.32 (d, J = 7.0 Hz, 1H), 5.23 (m, 1H), 5.13 (dd, J = 3.5, J = 10.5 Hz, 1H), 4.88 (m, 1H), 4.61 (d, J = 7.5 Hz, 1H), 4.50 (t, J = 7.2 Hz, 1H), 4.33 (d, J = 11.5 Hz, 1H), 4.20 (m, 2H), 4.11 (m, 2H), 4.01 (m, 2H), 3.69 (s, 3H), 3.42 (dd, J = 5.0, J = 15.0 Hz, 1H), 3.28 (dd, J = 5.5, J = 14.5 Hz, 1H), 3.07 (d, J = 4.5 Hz, 2H), 2.75 (d, J = 6.0 Hz, 2H), 2.11 (s, 3H), 2.06 (s, 3H), 2.01 (s, 3H), 1.95 (s, 3H), 1.47 (s, 9H), 1.34 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 172.1, 171.5, 170.8, 170.5, 170.0, 136.3, 129.8, 129.1, 127.7, 123.6, 122.3, 119.7, 118.6, 111.6, 109.7, 99.9, 80.7, 71.1, 70.7, 69.3, 69.1, 67.4, 61.6, 53.0, 52.7, 49.1, 33.8, 33.2, 29.9, 28.5, 27.6, 21.1, 20.88, 20.85, 20.8, 18.1; ESIHRMS Calcd. for C41H57N4O16S [M+H]+ 893.3485, found 893.3480.
General Procedure for the Ligation of Protected Neoglycosyl Donors with Glutathione
A solution of glutathione (0.05 mmol) and neoglycosyl donor (0.075 mmol) in Tris buffer (pH 8.0)/acetonitrile (0.5 mL/0.5 mL) was stirred at room temperature until completion. Triphenylphosphine (39 mg, 0.15 mmol) dissolved in tetrahydrofuran (0.5 mL) was added and the reaction mixture was stirred at room temperature for 10–24 h. The solution was evaporated and the residue purified by reverse phase HPLC using a gradient of 50% A to 100% A developed over 50 min (A, 0.1%TFA/CH3CN; B, 0.1% TFA/H2O; column: Varian Microsorb C18 250 × 21.4 mm; flow rate: 8mL/min; UV detection: 215nm).
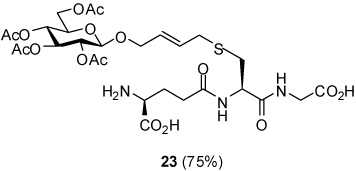
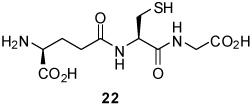
S-[4-(Tetra-O-acetyl-β-d-glucopyranosyloxy)-2E-butenyl] γ-l-Glu-l-Cys-Gly (23)
75% yield, [α]D24 −13.3° (c, 0.6, MeOH); 1H NMR (500 MHz, D2O) δ: 5.60 (m, 2H), 5.24 (t, J = 9.5 Hz, 1H), 4.98 (t, J = 9.5 Hz, 1H), 4.84 (t, J = 8.5 Hz, 1H), 4.76 (d, J = 8.5 Hz, 1H), 4.42 (m, 1H), 4.23 (m, 2H), 4.10 (m, 2H), 3.91 (m, 3H), 3.85 (t, J = 6.5 Hz, 1H), 3.10 (d, J = 6.0 Hz, 2H), 2.87 (dd, J = 5.4, J = 14.0 Hz, 1H), 2.68 (m, 1H), 2.44 (m, 2H), 2.08 (m, 2H), 1.99 (s, 3H), 1.98 (s, 3H), 1.95 (s, 3H), 1.91 (s, 3H); 13C NMR (125 MHz, D2O) δ: 174.3, 173.7, 173.1, 172.85, 172.77, 172.66, 171.9, 130.6, 128.2, 98.8, 73.1, 71.5, 71.1, 69.5, 68.3, 61.8, 52.8, 52.5, 41.0, 32.6, 31.4, 30.9, 25.5, 20.14, 20.11, 20.03, 19.99; ESIHRMS Calcd. for C28H41N3O16SNa [M+Na]+ 730.2105, found 730.2102.
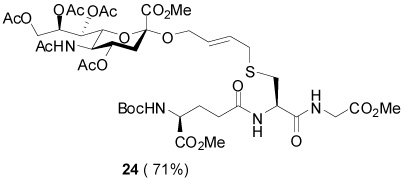
S-[4-(Methyl 4,7,8,9-tetra-O-acetyl-5-acetylamino-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate)-2E-butenyl] γ-l-Glu-l-Cys-Gly (24)
71% yield, [α]D24 −9.2° (c, 0.6, MeOH/H2O, 1:1); 1H NMR (500 MHz, D2O) δ: 5.64 (m, 2H), 5.29 (m, 2H), 4.80 (m, 2H), 4.45 (m, 1H), 4.26 (m, 1H), 4.15 (m, 3H), 3.99 (t, J = 6.5 Hz, 1H), 3.95 (m, 1H), 3.92 (s, 2H), 3.83 (t, J = 10.5 Hz, 1H), 3.77 (s, 3H), 3.13 (d, J = 6.5 Hz, 2H), 2.89 (dd, J = 5.5, J = 14.0 Hz, 2H), 2.71 (m, 1H), 2.63 (dd, J = 5.0, J = 13.0 Hz, 1H), 2.50 (m, 2H), 2.16 (m, 2H), 2.11 (s, 3H), 2.06 (s, 3H), 1.99 (s, 3H), 1.94 (s, 3H), 1.82 (s, 3H); 13C NMR (125 MHz, D2O) δ: 175.1, 174.5, 173.9, 173.7, 173.6, 173.2, 173.1, 172.6, 169.0, 132.7, 126.5, 98.8, 71.8, 69.4, 68.5, 67.5, 62.4, 57.4, 55.8, 53.9, 53.6, 49.0, 41.6, 37.2, 26.1, 25.5, 22.0, 20.7, 20.4, 20.3; ESIHRMS Calcd. for C34H50N4O19SNa [M+Na]+ 873.2688, found 873.2699.
General Procedure for Preparation of Deprotected Neoglycosyl Donors from Acetylated Allylic Thiocarbonates
The peracetyl glycosyl thiolcarbonate (0.2 mmol) was dissolved in MeOH (1 mL) at 0 °C with stirring and 1M KOH in MeOH (1.2 mmol-2.1 mmol, 1.5 eq per acetate group) was added dropwise. After 10–15 min, the pH of the reaction mixture was adjusted 7 by careful addition of Amberlyst-15. The reaction mixture was then filtered and the filtrate was added dropwise over 15 min to a well stirred solution of 2,2′-dipyridyl disulfide (68 mg, 0.3 mmol) in MeOH (1 mL) at room temperature, and the resulting solution stirred until completion (TLC, ~3 h). The solvent was removed in vacuo and the residue taken up in H2O and washed with ether several times. Evaporation of the aqueous phase followed by chromatographic purification gave the products.
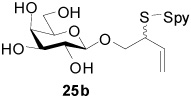
2-(2-Pyridyldisulfanyl)-3-enyl β-d-Galactopyranoside (25b)
White foam, eluted from silica gel as an approximately 1:1 mixture of diastereomers with dichloromethane/methanol (20:1) in 92% yield, 1H NMR (500 MHz, CD3OD) δ: 8.36 (m, 2×1H), 7.94 (m, 2×1H), 7.81(m, 2×1H), 7.20 (m, 2×1H), 5.86 (m, 1H), 5.81 (m, 1H), 5.23 (m, 2×1H), 5.14 (d, J = 10.5 Hz, 2×1H), 4.24 (m, 2×1H), 4.13 (m, 2×1H), 3.80 (m, 2×3H), 3.72 (m, 2×2H), 3.55 (2×1H, m), 3.47(m, 2×2H); 13C NMR (125 MHz, CD3OD) δ: 160.6, 148.8, 138.0, 134.24, 134.20, 121.0, 120.4, 118.4, 118.2, 104.04, 103.96, 75.6, 73.8, 71.3, 70.1, 69.1, 61.3, 61.2, 54.3; ESIHRMS Calcd. for C15H21NO6S2Na [M+Na]+ 398.0708, found 398.0706.
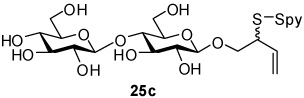
2-(2-Pyridyldisulfanyl)-3-enyl β-d-Cellobioside (25c)
White foam, eluted from silica gel as an approximately 1:1 mixture of diastereomers with dichloromethane/methanol (15:1) in 92% yield, 1H NMR (500 MHz, CD3OD) δ: 8.53 (m, 2×1H), 8.13(m, 2×2H), 7.50 (m, 2×1H), 5.86 (m, 2×1H), 5.28 (m, 2×1H), 5.18 (m, 2×1H), 4.41 (d, J = 7.5 Hz, 2×1H), 4.32 (dd, J = 8.0, J = 17.0 Hz, 2×1H), 4.16 (m, 1H), 4.09 (m, 1H), 3.90 (m, 2×4H), 3.80 (m, 2×2H), 3.66 (m, 2×1H), 3.52 (m, 2×2H), 3.36 (m, 2×3H), 3.25 (m, 2×2H); 13C NMR (125 MHz, CD3OD) δ: 160.6, 148.8, 138.0, 134.2, 134.1, 121.1, 120.4, 118.5, 118.3, 103.4, 103.2, 79.5, 76.9, 76.7, 75.4, 75.2, 73.7, 73.6, 70.2, 70.1, 61.3, 60.7, 54.2, 54.1; ESIHRMS Calcd. for C21H31NO11S2Na [M+Na]+ 560.1236, found 560.1255.
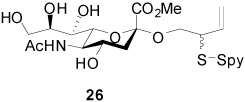
2-(2-Pyridyldisulfanyl)-3-enyl (Methyl 5-acetylamino-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranosylonate (26)
White foam, eluted from silica gel as an approximately 1:1 mixture of diastereomers with dichloromethane/methanol (20:1) in 78% yield, 1H NMR (500 MHz, CD3OD) δ: 8.37 (m, 2×1H), 7.87 (m, 2×1H), 7.81 (m, 2×1H), 7.21 (m, 2×1H), 5.77 (m, 2×1H), 5.20 (m, 2×2H), 4.05 (m, 2×1H), 3.82 (m, 2×2H), 3.80 (s, 3H), 3.78 (s, 3H), 3.64 (m, 2×4H), 3.54 (m, 2×1H), 3.49 (m, 2×1H), 2.66 (m, 2×1H), 1.994 (s, 3H), 1.991 (s, 3H), 1.71(m, 2×1H); 13C NMR (125 MHz, CD3OD) δ: 174.0, 169.47, 169.49, 160.5, 160.4, 148.97, 148.89, 138.0, 133.91, 133.86, 121.2, 121.1, 120.24, 120.21, 118.7, 118.5, 99.05, 99.00, 73.9, 71.2, 71.1, 68.98, 68.9, 67.3, 64.9, 64.87, 63.6, 63.5, 54.2, 53.9, 52.6, 52.3, 40.3, 40.28, 21.5; ESIHRMS Calcd. for C21H30N2O9S2Na [M+Na]+ 541.1290, found 541.1305.
General Procedure for the Ligation of Unprotected Neoglycosyl Donors with Glutathione
A solution of glutathione (21.7 mg, 0.07 mmol) and neoglycosyl donor (0.11 mmol) in phosphate buffer (pH 8.0)/acetonitrile (0.5 mL/0.5mL) was stirred at room temperature until completion (12–24 h). Triphenylphosphine (27.5 mg, 0.11 mmol) dissolved in THF (0.1 mL) was added and the reaction mixture was stirred at room temperature for 10–24 h. The solution was evaporated and the mixture purified by reverse phase HPLC using a gradient of 100% B to 50% B developed over 50 min (A, 0.1%TFA/CH3CN; B, 0.1% TFA/H2O; column: Varian Microsorb C18 250 × 21.4 mm; flow rate: 8mL/min; UV detection: 215nm).
S-[4-(β-d-Galactopyranosyloxy)-2E-butenyl] γ-l-Glu-l-Cys-Gly (27)
90% yield, [α]D23 −18.9° (c, 1.0, MeOH); 1H NMR (400 MHz, D2O) δ: 5.77 (m, 2H), 4.52 (m, 1H), 4.38 (d, J = 8.4 Hz, 1H), 4.35 (dd, J = 3.2, J = 12.0 Hz, 1H), 4.20 (m, 1H), 4.08 (t, J = 6.4 Hz, 1H), 3.99 (s, 2H), 3.88 (d, J = 3.2 Hz, 1H), 3.73 (m, 2H), 3.61 (m, 2H), 3.48 (m, 1H), 3.20 (d, J = 5.6 Hz, 2H), 2.96 (m, 1H), 2.79 (m, 1H), 2.56 (m, 2H), 2.22 (m, 2H); 13C NMR (100 MHz, D2O) δ: 174.5, 173.13, 173.07, 171.7, 130.7, 130.0, 102.0, 75.4, 73.1, 71.0, 69.7, 68.91, 61.2, 53.2, 52.4, 41.4, 33.0, 31.8, 31.1, 25.7; ESIHRMS Calcd. for C20H31N3O12S [M−H]− 538.1707, found 538.1689.
S-[4-(β-d-Cellobiosyloxy)-2E-butenyl] γ-l-Glu-l-Cys-Gly (28)
92% yield, [α]D23 −0.4° (c, 1.7, MeOH); 1H NMR (400 MHz, CD3OD) δ: 5.76 (m, 2H), 4.57 (m, 1H), 4.40 (d, J = 8.0 Hz, 1H), 4.35 (m, 2H), 4.16 (m, 1H), 4.05 (m, 1H), 3.94 (s, 2H), 3.86 (m, 2H), 3.66 (m, 1H), 3.54 (m, 2H), 3.39 (m, 2H), 3.31 (m, 3H), 3.25 (m, 2H), 3.19 (d, J = 5.6 Hz, 2H), 2.95 (m, 1H), 2.71 (m, 1H), 2.57 (m, 2H), 2.20 (m, 2H); 13C NMR (100 MHz, CD3OD) δ: 173.2, 171.9, 171.5, 170.3, 129.6, 129.3, 103.5, 101.9, 79.6, 76.9, 76.7, 75.3, 75.2, 73.7, 73.6, 70.2, 68.9, 61.2, 60.6, 53.0, 52.3, 48.7, 40.6, 33.1, 32.2, 31.1, 25.9; ESIHRMS Calcd. for C26H42N3O17S [M-H]− 700.2235, found 700.2229.
S-[4-(Methyl (5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-non-2-ulopyranoside)onate)-2E-butenyl] γ-l-Glu-l-Cys-Gly (29)
94% yield, [α]D23 −13.8° (c, 1.4, MeOH); 1H NMR (400 MHz, D2O) δ: 5.77 (m, 2H), 4.51 (m, 1H), 4.27 (m, 1H), 4.06 (m, 2H), 3.99 (s, 2H), 3.83 (m, 7H), 3.71 (m, 1H), 3.62 (m, 1H), 3.52 (d, J = 9.2 Hz, 1H), 3.18 (d, J = 5.6 Hz, 1H), 2.94 (m, 1H), 2.77 (m, 1H), 2.67 (m, 1H), 2.56 (m, 2H), 2.22 (m, 2H), 2.00 (s, 3H), 1.78 (t, J = 12.0 Hz, 1H); 13C NMR (100 MHz, D2O) δ: 175.3, 174.5, 173.1, 173.0, 171.7, 170.3, 130.5, 128.7, 99.1, 73.2, 70.9, 68.5, 67.4, 65.0, 63.4, 53.7, 53.2, 52.4, 52.0, 41.3, 39.6, 33.0, 31.8, 31.1, 25.7, 22.3; ESIHRMS Calcd. for C26H42N4O15SNa [M+Na]+ 705.2265, found 705.2265.
t-Butyl 3-Acetylthio-4-pentenoate (32)
Diisopropyl azodicarboxylate (5.1 mL, 26 mmol) was added to a stirred solution of triphenylphosphine (6.85g, 26 mmol) in tetrahydrofuran (46 mL) at 0 °C over 0.5 h, resulting in the formation of a white precipitate. A solution of alcohol 3116 (3.0 g, 17.4 mmol) and thiolacetic acid (1.87 mL, 26 mmol) in tetrahydrofuran (20 mL) was then added dropwise and the mixture was stirred for 1 h at 0°C and at room temperature for 13 h resulting in a clear yellow solution. The reaction mixture was diluted with ethyl acetate and washed with water. The organic layer was dried, filtered, evaporated and purified by chromatography (eluting with hexane/ethyl acetate, 10:1) to give 32 (3.06g, 76%) as yellow oil; 1H NMR (500 MHz, CDCl3) δ: 5.86 (m, 2×1H), 5.26 (d, J = 17.0 Hz, 2×1H), 5.10 (d, J = 10.5 Hz, 2×1H), 4.42 (q, J = 7.5 Hz, 2×1H), 2.63 (m, 2×2H), 2.31 (s, 2×3H), 1.43(s, 2×9H); 13C NMR (125 MHz, CDCl3) δ: 195.4, 169.8, 136.6, 116.7, 81.4, 42.5, 40.2, 30.8, 28.3; ESIHRMS Calcd. for C11H18O3SNa [M+Na]+ 253.0874, found 253.0895.
t-Butyl 3-[(2-Pyridinyl)disulfanyl]-4-pentenoate (33)
Lithium hydroxide monohydrate (30 mg, 0.71 mmol) was added in one portion to a stirred solution of thiolacetate 32 (163 mg, 0.71 mmol) in methanol (3.5 mL) at 0 °C. After 0.5 h the pH of the solution was adjusted to 7 with 3M HCl, and the resulting solution was added dropwise to a stirred solution of 2,2′-dipyridyl disulfide (239 mg, 1.06 mmol) in dichloromethane (3 mL) over 15 min. After stirring for 3 h, the solvent was removed. The residue was dissolved in ethyl acetate and washed with water. The organic layer was dried, filtered, evaporated and purified by chromatography on silica gel17 (eluting with hexane/ethyl acetate, 10:1) to afford 33 (144 mg, 68%) as yellow oil. 1H NMR (500 MHz, CDCl3) δ: 8.44 (dd, J = 0.5, J = 4.5 Hz, 2×1H), 7.72 (d, J = 7.5 Hz, 2×1H), 7.63 (dt, J = 2.0, J = 7.5 Hz, 2×1H), 7.08 (dt, J = 1.0, J = 7.5 Hz, 2×1H), 5.76 (m, 2×1H), 5.18 (d, J = 17.0 Hz, 2×1H), 5.10 (d, J = 10.0 Hz, 2×1H), 3.86 (q, J = 8.5 Hz, 2×1H), 2.77 (dd, J = 6.5, J = 15.5 Hz, 2×1H), 2.59 (dd, J = 8.5, J = 16.0 Hz, 2×1H), 1.43(s, 2×9H); 13C NMR (125 MHz, CDCl3) δ: 170.0, 160.6, 149.6, 137.1, 135.6, 120.9, 120.0, 118.3, 81.5, 50.0, 39.8, 28.3; ESIHRMS Calcd. for C14H19NO2S2Na [M+Na]+ 320.0755, found 320.0766.
3-[(2-Pyridinyl)disulfanyl]-4-pentenoic Acid (34)
Trifluoroacetic acid (1.36 mL, 17.5 mmol) was added dropwise over 20 min into a stirred solution of ester 33 (208 mg, 0.7 mmol) in dichloromethane (1.4 mL) at 0 °C. The reaction mixture was stirred for 4–5 h at room temperature, then concentrated, taken up in toluene, and concentrated again to afford 34 (169 mg, 100%) as yellow syrup; 1H NMR (400 MHz, CDCl3) δ: 12.34 (br. s, 2×1H), 8.59 (d, J = 4.8 Hz, 2×1H), 7.87 (m, 2×2H), 7.35 (t, J = 6.0 Hz, 2×1H), 5.80 (m, 2×1H), 5.20 (m, 2×2H), 3.93 (q, J = 7.2 Hz, 2×1H), 2.82(m, 2×2H); 13C NMR (100 MHz, CDCl3) δ: 159.0, 147.1, 140.3, 134.7, 122.7, 122.4, 119.2, 50.0, 38.4, 29.9; ESIHRMS Calcd. for C10H11NO2S2Na [M+Na]+ 264.0129, found 264.0147.
General Procedure for Formation of N-Glycosyl 3-[(2-Pyridinyl)disulfanyl]-4-pentenamides18
A solution of β-d-glucopyranosyl azide or 2-acetamido-2-deoxy-β-d-glucopyranosyl azide (1.18 mmol) in n-methylpyrrolidone (4 mL) was stirred with 10% Pd/C under H2 (1 atm) at room temperature until TLC indicated completion (~5 h). After filtration the reaction mixture was added dropwise over 0.5 h to a stirred, premixed solution of O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) (231 mg, 0.59 mmol) and diisopropylethylamine (202 µL, 1.18 mmol) and acid 34 (142 mg, 0.59 mmol) in DMF (1.7 mL) at 0 °C. The reaction mixture was stirred at room temperature overnight, then concentrated and the residue taken up in toluene and concentrated again to give yellow oil, which was lyophilized to remove N-methylpyrrolidone and then subjected to flash chromatography to afford the pure N-glycosyl amide.
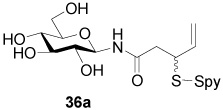
N-(β-d-Glucopyranosyl) 3-[(2-Pyridinyl)disulfanyl]-4-pentenamide (36a)
White foam, eluted from silica gel with dichloromethane/methanol (3:1) as an approximately 1/1 mixture of diastereomers in 46% yield. 1H NMR (400 MHz, D2O) δ: 8.34 (d, J = 4.8 Hz, 2×1H), 7.79 (m, 2×2H), 7.25 (m, 2×1H), 5.73 (m, 2×1H), 5.13 (d, J = 17.2 Hz, 2×1H), 5.04 (d, J = 9.6 Hz, 2×1H), 3.87 (m, 2×2H), 3.69 (m, 2×1H), 3.50 (m, 2×2H), 3.35 (m, 2×2H), 2.77 (m, 2×2H); 13C NMR (100 MHz, D2O) δ: 174.4, 159.0, 149.1, 138.8, 135.1, 122.2, 121.8, 118.6, 118.5, 79.6, 77.9, 76.8, 72.0, 69.5, 60.8, 49.93, 49.86, 39.5, 39.4; ESIHRMS Calcd. for C16H22N2O6S2Na [M+Na]+ 425.0817, found 428.0824.
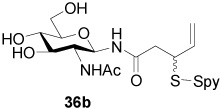
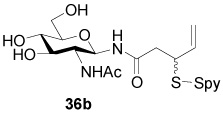
N-(2-Acetamido-2-deoxy-β-d-Glucopyranosyl) 3-[(2-Pyridinyl)disulfanyl]-4-pentenamide (36b)
White foam, eluted from silica gel with dichloromethane/methanol (3:1) as an approximately 1/1 mixture of diastereomers in 43% yield. 1H NMR (400 MHz, D2O) δ: 8.37 (m, 2×1H), 7.83 (m, 2×2H), 7.28 (m, 2×1H), 5.72 (m, 2×1H), 5.07 (m, 2×3H), 3.88 (m, 2×2H), 3.77 (m, 2×2H), 3.60 (m, 2×1H), 3.35 (m, 2×2H), 2.74 (m, 2×2H), 1.98 (s, 3H), 1.97 (s, 3H); 13C NMR (100 MHz, CD3OD) δ: 173.1, 173.0, 171.5, 160.24, 160.15, 149.0, 148.9, 137.91, 137.87, 135.6, 135.5, 121.2, 121.1, 120.20, 120.15, 117.5, 117.3, 79.1, 79.0, 78.62, 78.59, 75.20, 75.16, 70.61, 70.58, 61.5, 54.9, 50.2, 50.0, 39.7, 39.6, 21.9, 21.8; ESIHRMS Calcd. for C18H25N3O6S2Na [M+Na]+ 466.1082, found 466.1100.
General Procedure for Ligation of N-Glycosyl 3-[(2-Pyridinyl)disulfanyl]-4-pentenamides with Peptides
A solution of cysteine containing peptide (0.05 mmol) and the N-glycosyl amide (0.075 mmol) in phosphate buffer (pH 8.0)/acetonitrile (0.5 mL/0.5 mL) was stirred at room temperature until completion (12–24 h). Triphenylphosphine (26 mg, 0.1 mmol) dissolved in THF (0.1 mL) then was added and the reaction mixture was stirred at room temperature for 10–24 h. The solution was evaporated and the residue purified by reverse phase HPLC using a gradient of 100% B to 50% B developed over 70 min (A, 0.1%TFA/CH3CN; B, 0.1% TFA/H2O; column: Varian Microsorb C18 250 × 21.4 mm; flow rate: 8mL/min; UV detection: 215nm).
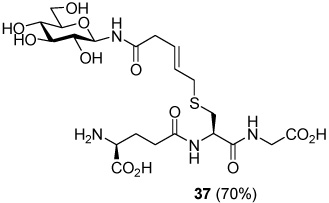
S-[N-(β-d-Glucopyranosyl)-5-carboxamido-2E-butenyl] γ-l-Glu-l-Cys-Gly (37)
70% yield, [α]D23 −18.5° (c, 0.6, MeOH/H2O 1:1); 1H NMR (400 MHz, D2O) δ: 5.66 (m, 2H), 4.94 (d, J = 8.8 Hz, 1H), 4.53 (m, 1H), 4.04 (t, J = 6.4 Hz, 1H), 4.00 (s, 2H), 3.86 (m, 1H), 3.70 (dd, J = 4.8, J = 12.0 Hz, 1H), 3.51 (m, 1H), 3.59 (t, J = 8.4 Hz, 1H), 3.38 (m, 2H), 3.27 (m, 2H), 3.20 (d, J = 6.4 Hz, 2H), 3.11 (d, J = 4.8 Hz, 1H), 2.97 (dd, J = 5.4, J = 14.2 Hz, 1H), 2.80 (m, 1H), 2.57 (m, 2H), 2.22 (m, 2H); 13C NMR (100 MHz, D2O) δ: 176.1, 174.6, 173.2, 173.1, 172.2, 130.6, 126.1, 79.6, 77.9, 76.8, 72.0, 69.5, 60.8, 53.2, 52.8, 47.8, 41.4, 39.2, 33.2, 31.6, 31.2, 25.9; ESIHRMS Calcd. for C21H35N4O12S [M+H]+ 567.1972, found 567.1967.
S-[N-(2-Acetamido-2-deoxy-β-d-glucopyranosyl)-5-carboxamido-2E-butenyl] γ-l-Glu-l-Cys-Gly (38)
85% yield, [α]D23 −3.4° (c, 0.7, MeOH/H2O 1:1); 1H NMR (400 MHz, D2O) δ: 5.52 (m, 2H), 5.03 (d, J = 9.6 Hz, 1H), 4.52 (m, 1H), 4.04 (t, J = 6.4 Hz, 1H), 4.00 (s, 2H), 3.86 (dd, J = 1.6, J = 12.0 Hz, 1H), 3.80 (t, J = 10.0 Hz, 1H), 3.73 (dd, J = 4.8, J = 12.0 Hz, 1H), 3.59 (t, J = 8.4 Hz, 1H), 3.48 (m, 2H), 3.19 (d, J = 5.6 Hz, 2H), 3.03 (d, J = 4.8 Hz, 2H), 3.00 (dd, J = 5.6, J = 14.0 Hz, 1H), 2.78 (dd, J = 8.8, J = 14.4 Hz, 1H), 2.57 (m, 2H), 2.22 (m, 2H), 1.98 (s, 3H); 13C NMR (100 MHz, D2O) δ: 175.5, 175.0, 174.6, 173.2, 173.1, 130.7, 126.0, 78.8, 77.9, 74.4, 69.8, 60.8, 54.6, 53.2, 52.7, 41.4, 39.3, 33.3, 31.8, 31.2, 25.8, 22.3; ESIHRMS Calcd. for C23H37N5O12SNa [M+Na]+ 630.2057, found 630.2072.
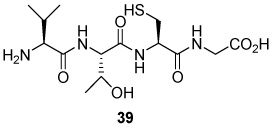
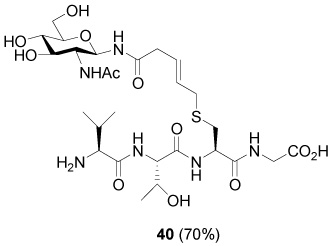
S-[N-(2-Acetamido-2-deoxy-β-d-glucopyranosyl)-5-carboxamido-2E-butenyl] l-Val-l-Thr-l-Cys-Gly (40)
70% yield, [α]D23 −8.9° (c, 0.2, MeOH/H2O 1:1); 1H NMR (500 MHz, D2O) δ: 5.50 (m, 2H), 4.92 (d, J = 10.0 Hz, 1H), 4.45 (m, 1H), 4.32 (d, J = 6.5 Hz, 1H), 4.03 (m, 1H), 3.88 (s, 2H), 3.80 (d, J = 5.5 Hz, 1H), 3.75 (m, 1H), 3.69 (t, J = 10.0 Hz, 1H), 3.62 (dd, J = 4.5, J = 12.0 Hz, 1H), 3.48 (t, J = 9.2 Hz, 1H), 3.37 (m, 2H), 3.07 (d, J = 6.0 Hz, 2H), 2.92 (d, J = 4.5 Hz, 2H), 2.86 (d, J = 5.0 Hz, J = 14.0 Hz, 1H), 2.69 (m, 1H), 2.12 (m, 1H), 1.87 (s, 3H), 1.10 (d, J = 6.0 Hz, 3H), 0.90 (t, J = 7.0 Hz, 6H); 13C NMR (125 MHz, D2O) δ: 175.3, 174.9, 173.1, 172.3, 171.2, 169.7, 130.6, 125.8, 78.7, 77.8, 74.3, 69.7, 67.3, 60.7, 59.3, 58.6, 54.5, 52.9, 50.6, 41.4, 39.2, 30.5, 24.0, 22.2, 19.0, 17.9, 17.0; ESIHRMS Calcd. for C27H47N6O12S [M+H]+ 679.2973, found 679.2997.
Supplementary Material
Copies of 1H and 13C-MR data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgement
We thank the NIH (GM62160) for partial support of this work and Dr. Franck Brebion for help and advice in the initial stages of the project.
References
- 1.a) Crich D, Krishnamurthy V, Hutton TK. J. Am. Chem. Soc. 2006;128:2544–2545. doi: 10.1021/ja057521c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Crich D, Zou Y, Brebion F. J. Org. Chem. 2006;71:9172–9177. doi: 10.1021/jo061439y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Crich D, Krishnamurthy V, Brebion F, Karatholuvhu M, Subramanian V, Hutton TK. J. Am. Chem. Soc. 2007;129:10282–10294. doi: 10.1021/ja072969u. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Li Z, Wang C, Fu Y, Guo Q-X, Liu L. J. Org. Chem. 2008;73:0000–0000. doi: 10.1021/jo800747g. [DOI] [PubMed] [Google Scholar]
- 2.a) Paulson JC, Blixt O, Collins BE. Nature Chem. Biol. 2006;2:238–248. doi: 10.1038/nchembio785. [DOI] [PubMed] [Google Scholar]; b) Dwek RA, Butters TD. Chem. Rev. 2002;102:283–284. [Google Scholar]; c) Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J, editors. Essentials of Glycobiology. Cold Spring Harbor: Cold Spring Harbor Press; 1999. [PubMed] [Google Scholar]; e) Buskas T, Ingale S, Boons G-J. Glycobiology. 2006;16:113R–136R. doi: 10.1093/glycob/cwj125. [DOI] [PubMed] [Google Scholar]; f) Werz DB, Seeberger PH. Chem. Biol. 2007;2:661–691. doi: 10.1021/cb700178s. [DOI] [PubMed] [Google Scholar]; g) Ernst B, Hart GW, Sinaÿ P, editors. Carbohydrates in Chemistry and Biology. Weinheim: Wiley-VCH; 2000. [Google Scholar]; h) Fraser-Reid B, Kuniaki T, Thiem J, editors. Glycoscience: Chemistry and Chemical Biology. Berlin: Springer-Verlag; 2001. [Google Scholar]; i) Fukuda M, Hindsgaul O, editors. Molecular and Cellular Glycobiology. Oxford: Oxford University Press; 2000. [Google Scholar]; j) Demchenko AV, editor. Frontiers in Modern Carbohydrate Chemistry. Washington, DC: American Chemical Society; 2007. [Google Scholar]; k) Davis BG. Chem. Rev. 2002;102:579–601. doi: 10.1021/cr0004310. [DOI] [PubMed] [Google Scholar]; l) Murrey HE, Hsieh-Wilson LC. Chem. Rev. 2008;108:1708–1731. doi: 10.1021/cr078215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Yamamoto N, Tanabe Y, Okamoto R, Dawson PE, Kajihara Y. J. Am. Chem. Soc. 2008;130:501–510. doi: 10.1021/ja072543f. [DOI] [PubMed] [Google Scholar]; b) Krauss IJ, Joyce JG, Finnefrock AC, Song HC, Dudkin VY, Geng X, Warren DJ, Chastain M, Shiver JW, Danishefsky SJ. J. Am. Chem. Soc. 2007;129:11042–11044. doi: 10.1021/ja074804r. [DOI] [PubMed] [Google Scholar]; c) Haase C, Seitz O. Top. Curr. Chem. 2007;267:1–36. [Google Scholar]
- 4.a) Stowell CP, Lee YC. Adv. Carbohydr. Chem. Biochem. 1980;37:225–281. doi: 10.1016/s0065-2318(08)60022-0. [DOI] [PubMed] [Google Scholar]; b) Lee YC, Lee RT, editors. Neoglycoconjugates:Preparation and Applications. San Diego: Academic Press; 1994. [Google Scholar]; c) Roy R. In: Carbohydrate Chemistry. Boons G-J, editor. London: Blackie Academic and Professional; 1998. pp. 243–321. [Google Scholar]; d) Specker D, Wittman V. Top. Curr. Chem. 2007;267:65–108. [Google Scholar]
- 5.a) Zhu XM, Pachamuthu K, Schmidt RR. Org. Lett. 2004;6:1083–1085. doi: 10.1021/ol036186z. [DOI] [PubMed] [Google Scholar]; b) Pachamuthu K, Schmidt RR. Chem. Rev. 2006;106:160–187. doi: 10.1021/cr040660c. [DOI] [PubMed] [Google Scholar]; c) Doores KJ, Gamblin DP, Davis BG. Chem. Eur. J. 2006;12:656–665. doi: 10.1002/chem.200500557. [DOI] [PubMed] [Google Scholar]; d) Peri F, Nicotra F. Chem Comm. 2004:623–627. doi: 10.1039/b308907j. [DOI] [PubMed] [Google Scholar]; e) Ohnishi Y, Ichikawa M, Ichikawa Y. Biorg. Med. Chem. Lett. 2000;10:1289–1291. doi: 10.1016/s0960-894x(00)00223-7. [DOI] [PubMed] [Google Scholar]; f) Jobron L, Hummel G. Org. Lett. 2000;2:2265–2267. doi: 10.1021/ol006019o. [DOI] [PubMed] [Google Scholar]; g) Cohen SB, Halcomb RL. Org. Lett. 2001;3:405–407. doi: 10.1021/ol006908b. [DOI] [PubMed] [Google Scholar]; h) Knapp S, Myers DS. J. Org. Chem. 2002;67:2995–2999. doi: 10.1021/jo0110909. [DOI] [PubMed] [Google Scholar]; i) Zhu XM, Schmidt RR. Chem. Eur. J. 2004;10:875–887. doi: 10.1002/chem.200305163. [DOI] [PubMed] [Google Scholar]; j) Thayer DA, Yu HN, Galan MC, Wong C-H. Angew. Chem. Int. Ed. 2005;44:4596–4599. doi: 10.1002/anie.200500090. [DOI] [PubMed] [Google Scholar]; k) Yang Y-Y, Ficht S, Brik A, Wong C-H. J. Am. Chem. Soc. 2007;129:7690–7701. doi: 10.1021/ja0708971. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Ficht S, Payne RJ, Brik A, Wong C-H. Angew. Chem. Int. Ed. 2007;46:5975–5979. doi: 10.1002/anie.200700546. [DOI] [PubMed] [Google Scholar]; m) Ichikawa Y, Matsukawa Y, Isobe M. J. Am. Chem. Soc. 2006;128:3934–3938. doi: 10.1021/ja056253f. [DOI] [PubMed] [Google Scholar]; n) Ladmiral V, Mantovani G, Clarkson GJ, Cauet S, Irwin JL, Haddleton DM. J. Am. Chem. Soc. 2006;128:4823–4830. doi: 10.1021/ja058364k. [DOI] [PubMed] [Google Scholar]; o) Hotha S, Kashyap S. J. Org. Chem. 2006;71:364–367. doi: 10.1021/jo051731q. [DOI] [PubMed] [Google Scholar]; p) Ichikawa Y, Ohara F, Kotsuki H, Nakano K. Org. Lett. 2006;8:5009–5012. doi: 10.1021/ol0616788. [DOI] [PubMed] [Google Scholar]; q) Wittrock S, Becker T, Kunz H. Angew. Chem. Int. Ed. 2007;46:5226–5230. doi: 10.1002/anie.200700964. [DOI] [PubMed] [Google Scholar]; r) Ni J, Song H, Wang Y, Stamatos NM, Wang L-X. Bioconjugate Chem. 2006;17:493–500. doi: 10.1021/bc0502816. [DOI] [PubMed] [Google Scholar]; s) Kolomiets E, Johansson EMV, Renaudet O, Darbre T, Reymound J-L. Org. Lett. 2007;8:14656–11468. doi: 10.1021/ol070119d. [DOI] [PubMed] [Google Scholar]; t) Samantaray S, Marathe U, Dasgupta S, Nandicoori VK, Roy RP. J. Am. Chem. Soc. 2008;130:2132–2133. doi: 10.1021/ja077358g. [DOI] [PubMed] [Google Scholar]
- 6.a) Macindoe WM, van Oijen AH, Boons G-J. Chem. Commun. 1998:847–848. [Google Scholar]; b) Gamblin DP, Garnier SJ, Ward NJ, Oldham AJ, Fairbanks AJ, Davis BG. Org. Biomol. Chem. 2003;1:3642–3644. doi: 10.1039/b306990g. [DOI] [PubMed] [Google Scholar]; c) Gamblin DP, Garnier P, van Kasteren S, Oldham NJ, Fairbanks AJ, Davis BG. Angew. Chem. Int. Ed. 2004;43:828–833. doi: 10.1002/anie.200352975. [DOI] [PubMed] [Google Scholar]; d) van Kasteren SI, Kramer HB, Jensen HH, Campbell SJ, Kirkpatrick J, Oldham NJ, Anthony DC, Davis BG. Nature. 2007;446:1105–1109. doi: 10.1038/nature05757. [DOI] [PubMed] [Google Scholar]; e) Bernardes GJL, Chalker JM, Errey JC, Davis BG. J. Am. Chem. Soc. 2008;130:5052–5053. doi: 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- 7.Bernardes GJL, Grayson EJ, Thompson S, Chalker JM, Errey JC, El Oualid F, Claridge TDW, Davis BG. Angew. Chem. Int. Ed. 2008;47:2244–2247. doi: 10.1002/anie.200704381. [DOI] [PubMed] [Google Scholar]
- 8.a) Garmaise Dl, Uchiyama A, McKay AF. J. Org. Chem. 1962;27:4509–4512. [Google Scholar]; b) Ferrier RJ, Vethaviyasar N. J. Chem. Soc., Chem. Commun. 1970:1385–1387. [Google Scholar]; c) Hackler RE, Balko TW. J. Org. Chem. 1973;38:2106–2110. doi: 10.1021/jo00951a038. [DOI] [PubMed] [Google Scholar]; d) Nakai T, Ari-Izumi A. Tetrahedron Lett. 1976;17:2335–2338. [Google Scholar]; e) Harano K, Ohizumi N, Hisano T. Tetrahedron Lett. 1985;26:4203–4206. [Google Scholar]; f) Harano K, Taguchi T. Chem. Pharm. Bull. 1975;23:467–472. [Google Scholar]; g) Nakai T, Mimura T, Ari-Izumi A. Tetrahedron Lett. 1977:2425–2428. [Google Scholar]; h) Ueno Y, Sano H, Okawara M. Tetrahedron Lett. 1980;21:1767–1770. [Google Scholar]; i) Eto M, Tajiri O, Nakagawa H, Harano K. Tetrahedron. 1998;54:8009–8014. [Google Scholar]; j) Overman LE, Roberts SW, Sneddon HF. Org. Lett. 2008;8:1485–1488. doi: 10.1021/ol8002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Schmidt RR. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. New York: Dekker; 1997. pp. 283–312. [Google Scholar]; b) Schmidt RR, Jung K-H. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Weinheim: Wiley-VCH; 2000. pp. 5–59. [Google Scholar]
- 10.The glycosylation reaction was accompanied by partial cleavage of the O-2 acetate, which was detrimental to the yield of the product. Therefore, an acetylation step was included in the work-up protocol to reinstall any missing acetates.
- 11.a) Danehy JP, Elia VJ. J. Org. Chem. 1972;37:369–373. [Google Scholar]; b) Happer DAR, Mitchell JW, Wright GJ. Aust. J. Chem. 1973;26:121–134. [Google Scholar]; c) Ichimura A, Nosco DL, Deutsch E. J. Am. Chem. Soc. 1983;105:844–850. [Google Scholar]
- 12.Volante RP. Tetrahedron Lett. 1981;22:3119–3122. [Google Scholar]
- 13.Rodriguez EB, Scally GD, Stick RV. Aust. J. Chem. 1990;43:1391–1405. [Google Scholar]
- 14.Sodeoka M, Yamada H, Shibasaki M. J. Am. Chem. Soc. 1990;112:4906–4911. [Google Scholar]
- 15.When TLC indicated orthoester formation TMSOTf (0.1 eq) was added before the reaction was quenched.
- 16.Zibuck R, Streiber JM. J. Org. Chem. 1989;54:4717–4719. [Google Scholar]
- 17.This compound showed a tendency to undergo allylic rearrangement during silica gel chromatography when moisture was not fully excluded.1c
- 18.Wen S, Guo Z. Org. Lett. 2001;3:3773–3776. doi: 10.1021/ol0101988. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of 1H and 13C-MR data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.