

Abstract
Oxidoreductases of the cytochrome bc1/b6f family transfer electrons from a liposoluble quinol to a soluble acceptor protein and contribute to the formation of a transmembrane electrochemical potential. The crystal structure of cyt b6f has revealed the presence in the Qi site of an atypical c-type heme, heme ci. Surprisingly, the protein does not provide any axial ligand to the iron of this heme, and its surrounding structure suggests it can be accessed by exogenous ligand. In this work we describe a mutagenesis approach aimed at characterizing the ci heme and its interaction with the Qi site environment. We engineered a mutant of Chlamydomonas reinhardtii in which Phe40 from subunit IV was substituted by a tyrosine. This results in a dramatic slowing down of the reoxidation of the b hemes under single flash excitation, suggesting hindered accessibility of the heme to its quinone substrate. This modified accessibility likely originates from the ligation of the heme iron by the phenol(ate) side chain introduced by the mutation. Indeed, it also results in a marked downshift of the ci heme midpoint potential (from +100 mV to −200 mV at pH 7). Yet the overall turnover rate of the mutant cytochrome b6f complex under continuous illumination was found similar to the wild type one, both in vitro and in vivo. We propose that, in the mutant, a change in the ligation state of the heme upon its reduction could act as a redox switch that would control the accessibility of the substrate to the heme and trigger the catalysis.
The cytochrome b6f complex of oxygenic photosynthesis is the integral membrane protein, the quinol:plastocyanin oxido-reductase activity of which allows the linear electron flow between the two photosystems (PSI and PSII).4 The turnover of the cytochrome b6f complex depends on the steady state of its redox partners, the liposoluble plastoquinol (PQH2 reduced and protonated plastoquinone PQ) formed by the PSII, and the hydrosoluble plastocyanin oxidized by the PSI. In the Qo site, exposed to the lumenal side, the quinol is oxidized, and this oxidation is coupled to the release of two protons into the lumen. The two electrons provided by the quinol are transferred along two bifurcated pathways, the high and low potential chains. The high potential chain involves two lumenal redox partners, the membrane-anchored flexible Rieske [2Fe-2S] cluster and the cytochrome f, which ultimately interacts with the soluble plastocyanin. In the low potential chain, electrons are transferred to the stroma via the low and high potential b hemes (bL and bH) of the transmembrane b6 subunit. Two turnovers of the cyt b6f complex lead to the reduction of the low potential chain, thereby allowing the reduction of a quinone molecule in the stromal Qi pocket. This mechanism, which recycles reducing equivalents, is referred to as the “Q cycle,” initially described by Mitchell (1) and modified later by Crofts et al. and others (2, 3).
Although this quinol:cytochrome oxidoreductase activity is involved in both the respiratory and photosynthetic electron transfer chains, recent x-ray data (4–6) have evidenced major structural differences between the b6f complex and its mitochondrial counterpart the bc1 complex (for reviews see Refs. 7–10). Indeed an additional heme localized in close contact with heme bH stands as another putative electron carrier as proposed earlier by Lavergne (11). Since it was brought to light by the x-ray studies, knowledge of the basic properties of this heme, named ci in reference to the Qi site (5) or cn in reference to its proximity to the negatively charged side of the membrane (4), has significantly improved. The proteins involved in the assembly machinery of the heme have been identified in Chlamydomonas reinhardtii and Arabidopsis thaliana (12, 13). Consistent with the structure, according to which the only axial ligand could be a water molecule interacting with the proponiate chain of the bH heme, the spectroscopic properties of this heme are those of a high spin heme (14, 15). Evidences for a high spin heme covalently bound to the cytochrome b subunit were also found in Heliobacterium modesticaldum and Heliobacillus mobilis (16).
In the b6f complex from the oxygenic photosynthetic chain, EPR (15) and structural data (17) have shown that NQNO (2-n-nony l-4-hydroxyquinoline N-oxide), an inhibitor of the Qi pocket (18, 19), can act as an axial ligand to ci. This ligation is accompanied by a significant change in the redox properties of ci, because, in the presence of NQNO, at least two titrations waves were observed (13, 14), one with a midpoint potential (Em) similar to that observed in the absence of NQNO and the other with a midpoint potential downshifted by ∼250 mV. This, together with the widespread range of redox potential found for heme ci (11, 14, 15, 20), points to a structural plasticity of the ci ligand network.
This plasticity may arise from the unusual coordination properties of the heme ci. As a matter of fact, the x-ray models of the complex from C. reinhardtii and Mastigocladus laminosus evidenced a water or hydroxyl molecule as a fifth ligand. The sixth position of coordination is directed toward the Qi pocket and appears as free. Nevertheless, the side chain of Phe40 of subunit IV protrudes above the heme plane, leaving little space for any axial ligand to the heme ci. Besides, modeling a quinone analogue in the electron density found in the Qi pocket of structures obtained in presence of Tridecyl- Stigmatellin or NQNO implies a steric clash with the native position of the Phe40 aromatic ring.5 The Phe40 residue of subunit IV therefore stands as a key residue for the plasticity of the site, making it an ideal mutagenesis target when attempting to alter the possible interactions between ci and the quinone or quinol (4, 5) (Fig. 1). Here we present the consequences of the substitution of Phe40 by a tyrosine on the properties and function of the ci heme.
FIGURE 1.

A view of the Qi site from the dimer interface. The coordinates are from the Protein Data Bank 1Q90 model. The Van der Waal's surface of the peptide chains was drawn with Pymol. Phe40 is in van der Waal's contact with the plane of the ci, heme.
EXPERIMENTAL PROCEDURES
Strains, Media, and Growth Conditions
The H6F5 strain (mt+), expressing a cytochrome f version histidine tagged at its C-terminal end (21), and the ΔpetD (mt+) deletion strain (22) were used as recipient strains in chloroplast transformation experiments. H6F5, ΔpetD, and mutant strains were grown on Tris-acetate-phosphate medium (pH 7.2) at 25 °C under dim light (5–6 μE·m−2·s−1) (23). The H6F5 strain was used as a wild type reference in all experiments.
Plasmids, Oligonucleotides, and Mutagenesis
The pWQH6 plasmid, carrying the entire petD sequence, together with the C-terminal end of the sequence coding for histidine-tagged cytochrome f was obtained by ligation of the 0.55-kb EcoRV-AccI fragment recovered from plasmid pFWH6 (5) into the corresponding sites of plasmid pdWQ (21).
The mutated version of the petD gene was created by PCR-mediated site-directed mutagenesis. Plasmid pdΔHI.I (22), carrying the entire petD coding sequence was used as a template in PCRs performed with ArrowTM Taq DNA polymerase, employed according to the manufacturer's instructions, using the primers: DirF40Y (5′-GGGTGGCCAAACGATTTATTATACATGTACCCTGTTGTTATTTTAGGTACATTT-3′) and RevQi (5′-GGGTGGCCAAGCAGGTTCAC CGTAAGTGTT-3′), where nucleotides written in bold differ from the WT sequence. The PCR product was digested with MscI (a single restriction site underlined in the sequence of the primers) and religated onto itself to yield plasmid pdΔHF40Y. This plasmid was digested with HindIII and NcoI, and the resulting 833-bp fragment was ligated into the corresponding sites of plasmid pWQH6 to yield plasmid pdF40Y. Plasmid pdK-F40Y was constructed by introducing the 1.9-kb SmaI-EcoRV aadA cassette (24), in the same orientation as the petD gene, into the EcoRV site of plasmid pdF40Y.
Chloroplast Transformation in C. reinhardtii
The ΔpetD (22) and H6F5 (21) strains were transformed by tungsten particle bombardment according to Boynton et al. (25). When using plasmid pdF40Y to bombard the non-photosynthetic ΔpetD strain, transformed clones were selected on minimum medium at 60 μE·m−2·s−1. When using plasmid pdK-F40Y to bombard the H6F5 strain, transformants were selected on Tris-acetate-phosphate medium supplemented with spectinomycin (100 μg·ml−1). Transformed cells were subcloned several times on selective medium until they reach homoplasmy, assessed by restriction fragment length polymorphism analysis of specific PCR fragments (26).
Preparative and Analytical Techniques
Cells grown to a density of 4 × 106 ml−1 were broken in “bead beater” (Biospec-Products) according to the manufacturer's instructions. The membrane fraction was collected by centrifugation and resuspended in 10 mm Tricine, pH 8, at a chlorophyll concentration of 3 mg·ml−1. For SDS-PAGE, the membrane proteins were resuspended in 100 mm dithiothreitol and 100 mm Na2CO3 and solubilized by 2% SDS at 100 °C for 1 min. Polypeptides were separated on a 12–18% polyacrylamide gel containing 8 m urea. Immunoblotting was performed as described in Pierre et al. (27).
Cytochrome b6f complexes were isolated as described in Ref. 5 and titrated accordingly to Alric et al. (14). In vitro electron transfer activity was measured as described in Ref. 27. Cytochrome b6f complexes were analyzed by size exclusion chromatography in 20 mm Tris-HCl, pH 8.0, 250 mm NaCl, 0.2 mm C12M onto an exclusion Superdex 200 HR Amersham Biosciences column (28).
Fluorescence Induction Measurements
The phenotype of b6f mutants was characterized by their fluorescence induction kinetics. The measurements were performed at room temperature on a home-built fluorimeter, with continuous illumination at 520 nm and fluorescence detection in the far-red region (29–31).
Spectroscopy
Time-resolved light-induced absorbance changes in whole cells of C. reinhardtii were monitored with a pulsed differential LED spectrophotometer JTS 10 Bio-Logic, whereas redox-induced and CO-induced absorbance changes in purified b6f preparations were measured with a xenon flash lamp spectrophotometer, as previously described respectively by Joliot et al. (32) and in Alric et al. (14). To characterize the kinetics of CO binding after laser flash photolysis, the purified b6f complex (5 μm, pH 8) was thoroughly degassed, then reduced by addition of dithionite, and then equilibrated with ∼1 atm of CO.
Cytochrome b redox changes were measured in the presence of NQNO to obtain full inhibition of cytochrome b6 oxidation. NQNO was synthesized in the laboratory according to the procedure described in Ref. 33. FOS-choline-14 (n-tetradecylphosphocholine) was purchased from Anatrace.
RESULTS
Construction of C. reinhardtii Mutants Expressing a Mutated petD Gene
We investigated by site-directed mutagenesis the importance of the accessibility to the heme ci by replacing the Phe40 of subunit IV, in van der Waal's contact with the plane of the ci, heme, by a tyrosine. Our first attempt to recover phototrophic clones following transformation of the ΔpetD strain with plasmid pdF40Y proved unsuccessful. As an alternative strategy, we used the histidine-tagged H6F5 strain as a recipient strain for transformation with the pd-KF40Y plasmid and recovered transformed clones on spectinomycin supplemented medium (24). Five independent dK-F40Y clones, hereafter referred to as F40Y mutants, were brought to homoplasmy with respect to the petD mutation. Because transformation of the ΔpetD strain by the mutated copy of the petD gene failed to rescue phototrophic growth, we expected the recovered homoplasmic transformants to display fluorescence induction kinetics typical of mutants lacking cytochrome b6f activity (30, 31, 34). Surprisingly, they still displayed unimpaired fluorescence induction kinetics (Fig. 2) (26). They grew as the wild type on minimum medium and did not show any photo-sensitivity under high light (200 μE·m−2·s−1), neither in photoautotrophic nor in mixotrophic conditions (data not shown). On this basis, we expected the electron transfer chain to be active and assessed its function by time-resolved absorption spectroscopy.
FIGURE 2.
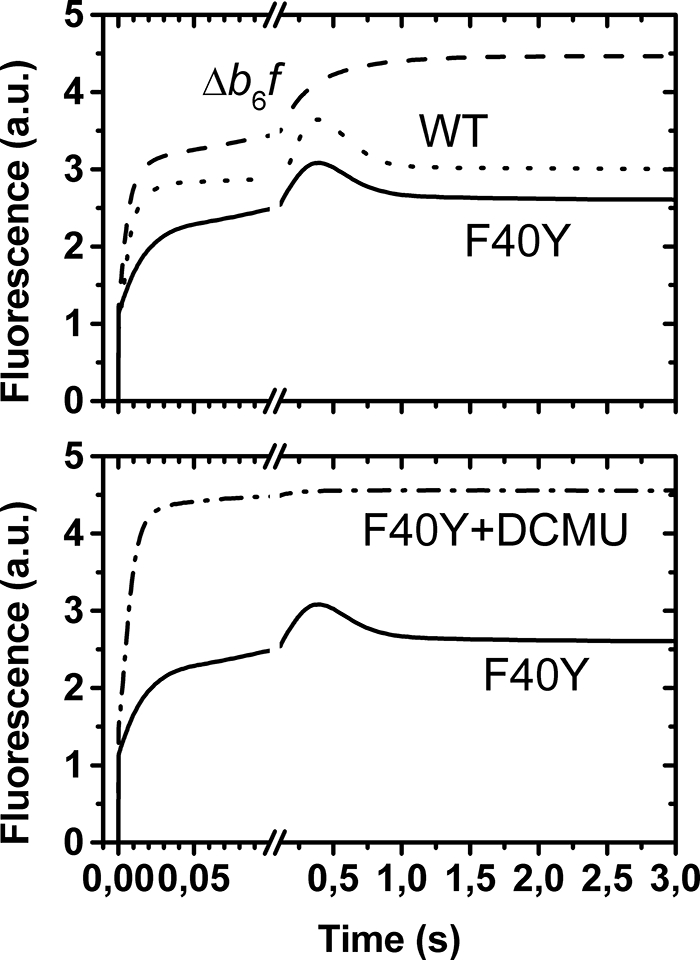
Fluorescence induction curves under aerobic conditions. The top panel displays the light-induced fluorescence changes in the WT (dotted curve), in a mutant lacking the b6f complex (dashed curve), and in the F40Y mutant (solid curve). In the latter, at variance with the b6f lacking mutant, the light-induced fluorescence does not reach the Fmax level, as illustrated in the bottom panel, which displays the fluorescence changes observed in the F40Y mutant in the absence (solid curve) and presence (dashed-dotted curve) of 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU, 10 μm).
Flash-induced Kinetics
Fig. 3A shows the slow electrogenic phase associated with the turnover of the b6f complex in WT and mutant strains. At 100 μs the absorption changes result from the charge separation event at the level of PSI; the subsequent electrogenic phase that develops with a ∼2-ms half-time witnesses the cytochrome b6f turnover (35, 36). In the F40Y mutant, the amplitude of this phase was dramatically decreased.
FIGURE 3.

Flash-induced kinetics of electron transfer in the low potential chain of WT and F40Y cyt b6f complexes. A, the flash-induced absorption changes at 520 nm, which reflect the transient changes in the transmembrane electric field, in the WT (squares) and in the F40Y mutant (circles). B, the flash-induced absorption changes at 563 nm, which reflects the redox changes of the b hemes in the WT (squares) and in the F40Y mutant (circles) in the absence (solid symbols) and presence (open symbols) of NQNO (50 μm).
We then measured the kinetics of the absorption changes associated with the redox changes of the b hemes, at 563 nm (Fig. 3B). In the WT, in the absence of inhibitors, a transient reduction (upward signal) is followed by a pronounced oxidation phase (downward signal). The negative signal observed 1 s after a flash shows that the overall reaction results in the net oxidation of a b heme. Thus, in these experimental conditions, the bH and bL hemes were respectively reduced and oxidized in the dark, so that the injection of one electron into the low potential chain allowed the reduction of a quinone at the Qi site and the associated oxidation of the two b hemes. The addition of NQNO (50 μm) slows down this latter reaction and, accordingly, allows the observation of the full extent of the b heme reduction phase (37). In the mutant, in the absence of NQNO, we observed a pronounced reduction of b heme with a half-time of ∼2 ms similar to the one observed in the WT in the presence of NQNO, followed by a very slow oxidation phase. The addition of NQNO had no further effect on this oxidation phase. This result, together with the decreased amplitude of the electrogenic phase, indicates that in the mutant, under the conditions of the experiments, the bH heme was likely reduced in the dark, whereas the bL heme was oxidized and that quinol oxidation at the Qo site operated normally. The dramatic slow down of the oxidation of the b hemes indicates that quinone reduction at the Qi site is strongly affected, consistent with the quasi absence of electrogenic phase. The F40Y mutation thus seems to cause a strong inhibition of the Qi site. Yet contrasting results were obtained when studying the turnover of the b6f complex under continuous illumination.
Continuous Light Measurements
As shown above, the fluorescence induction curves of F40Y cells are virtually indistinguishable from that commonly obtained with WT. Fig. 2 shows typical measurements carried out under aerobic conditions in the presence or absence of 3-(3,4-dichlorophenyl)-1,1-dimethylurea. Surprisingly, in the F40Y mutant, the steady state fluorescence level, reached after a few seconds of illumination, was similar to the WT one. Thus, despite the apparent loss of Qi site activity, as observed under single turnover flash experiments, the electron flows through the cytochrome b6f complex are similar in both strains.
It is of note however that the fluorescence measurements were performed under aerobic conditions (oxidized PQ pool), whereas the flash-induced kinetics were obtained under anaerobic conditions (reduced PQH2 pool), leaving open the possibility of a different occupancy of the Qi site by its substrate in the two experiments.
We thus measured the overall activity of the photosynthetic chain during illumination under saturating light intensity, in aerobic as well as anaerobic conditions. In such conditions, the limiting step of the photosynthetic electron flow bears on the cytochrome b6f complex. Electrochromic changes of carotenoids, measured at 520 nm, were used as a probe of the transmembrane potential (38). As shown in Fig. 4 the time course of the absorption changes encompasses a very fast rise (with an initial slope of 1000 charges translocated per second), because of the charge separation at the level of the photosynthetic chain (PSII-PQ-b6f-plastocyanin-PSI), followed by a decay likely reflecting the activation of CF0-CF1 ATP synthase. After ∼1 s of illumination, the membrane potential reaches a quasi-steady state regime where the generation of the transmembrane electric field by the activity of the photosynthetic chain nearly compensates its dissipation by the ATP synthase. When the light is switched off, the photochemical process is instantaneously stopped, and the absorption changes decay as the transmembrane potential is rapidly consumed by the CF0-CF1 ATP synthase. The difference between the slopes of the signal before and after the light is switched off is indicative of the photochemical activity (38, 39). We first validated this approach by the study of a b6f lacking mutant (Fig. 4B). Switching off the illumination did not induce any variation of the slope of the electrochromic band shift after 1 s of illumination. In the WT and in the F40Y mutant (Fig. 4, A and C), under aerobic and anaerobic conditions, a clear change in the slope was observed when the light was switched off, showing that, under illumination, the consumption of the transmembrane potential by the ATP synthase is compensated by its photo-induced formation. In the WT, after normalization of the change in slope by the signal induced by a single turnover flash (which yields the absorption changes resulting from the transfer of 2 electrons across the membrane), we estimated the photochemical activity to ∼100 e−·s−1 (aerobic) and 40 e−·s−1 (anaerobic). In the mutant, these rates were estimated to 70 and 40 charges/s, respectively.
FIGURE 4.

Light-induced changes of the transmembrane electric field under continuous illumination. The left, middle, and right panels, respectively show the data obtained with the WT, with a mutant lacking b6f and with the F40Y mutant. The light was switched on and off as indicated by upward and downward arrows, respectively. In each column, the top and bottom panels show the results obtained in aerobic and anaerobic conditions, respectively. Flash-induced kinetics (closed symbols) are also shown under the same conditions to provide a calibration of the light intensity. The amplitude corresponding to one electron transferred by photosystem I (phase a) is depicted by a vertical bar.
At this stage we are thus confronted to an apparent paradox. On the one hand, the quinone reduction activity of the Qi site is strongly impaired as observed in the flash experiments. On the other hand, the overall activity of the mutated b6f complex only slightly differs from the WT one under continuous illumination, suggesting that the functional constraints underlying the blockage of the Qi site function under single flash conditions are relieved in a continuous illumination regime. To obtain a more complete picture of the interaction between the Tyr side chain and the ci heme, we studied the physico-chemical properties of the ci heme in purified b6f complex from the mutant.
Biochemical Characterization of the F40Y Mutant
The mutant accumulated the major subunits of the cytochrome b6f complex at approximately wild type levels (Fig. 5A). In particular the content in subunit IV, which harbors the mutations, was unaltered. To further identify the ci heme associated with cyt b6, the gels were stained with 3,3′,5,5′ tetramethylbenzidine, a specific staining for hemes (40). The peroxidase activity associated with cyt b6 proved to be SDS-resistant consistent with the covalent binding of a heme to this subunit, strongly supporting the presence of the ci heme. The oligomerization state of the cytochrome b6f complex, as measured by size exclusion chromatography, was not affected by the mutation (Fig. 5B).
FIGURE 5.

Biochemical characterization of cytochrome b6f complex mutants. A, immunoblot and specific heme staining (for cyt f and cyt b6) of membrane protein extracts. The membranes were probed with specific antibodies against cytochrome f, cytochrome b6, and subunit IV. Loads for each sample correspond to 20 μg of chlorophyll. B, size exclusion chromatography of WT and dF40Y mutant b6f complex, analyzed on a 10 × 300-mm Superdex 200 HR column. The absorbance of the eluate was monitored at 280 (black line) and 420 (cyan line) nm.
The b6f complex purified from the mutant was then tested for its ability to mediate the transfer of electrons between plastoquinol and oxidized plastocyanin in vitro. Its activity (∼350 e−·f−1·s−1) was found similar to that of the wild type complex (∼400 e−·f−1·s−1). This is satisfyingly consistent with the conclusion drawn from the in vivo activity, described above, that the turnover rate of the mutated complex hardly differs from the WT one.
Redox and Spectral Characteristics of Heme ci
The substitution of the Phe40 by a Tyr has no effect on the midpoint potentials of the bL and bH hemes but strongly modifies the Em of heme ci (Fig. 6B), which was downshifted from +100 mV in the WT to −200 mV in the mutant (at pH 7.0). As for the WT (Fig. 6A), the midpoint potential was pH-dependent with a slope of −60 mV/pH unit (Fig. 6B).
FIGURE 6.

pH dependence of the midpoint potentials of cofactors in the low potential chain of WT (A) and F40Y (B) cyt b6f complexes: bL (solid circles), bH (solid triangles), and ci (solid squares). The open symbols in B show the midpoint potentials of the three hemes in the presence of NQNO (50 μm). In both the absence and presence of NQNO the midpoint potential of ci depends upon pH with a slope of 60 mV per pH unit.
The F40Y mutation also affected the (oxidized − reduced) spectrum of the ci heme (Fig. 7A), which displays a shoulder around 440 nm and an overshoot at 380 nm. These features were even more pronounced after the addition of NQNO (Fig. 7B) and likely reflect changes in the heme environment. In addition to this spectroscopic change, the addition of NQNO also induced a moderate downshift of the midpoint potential of the ci heme in the mutant (Fig. 6B, open squares). However, the amplitude of this downshift was much smaller than that previously reported in the WT, and the corresponding titration curve was satisfyingly fit with a n = 1 Nernst curve, whereas in the WT the titration curve was split into at least two waves (14, 15). Yet the strong modifications of the midpoint potential of ci and the spectroscopic changes of its (oxidized − reduced) spectrum point to a significant change in the environment of the heme in the F40Y mutant, which can still accommodate quinones/analogues.
FIGURE 7.

Spectral characteristics of the ci heme in WT (solid curve) and F40Y (squares) cyt b6f complexes. A, (oxidized − reduced) spectra of the ci heme obtained as the difference between the absorption spectra measured at 240 and 90 mV for the WT b6f complex (at pH 7.0) and −250 and −400 mV for the F40Y b6f complexes (at pH 9.0). B shows the corresponding spectra for WT and F40Y b6f complexes in the presence of NQNO (50 μm).
Carbon Monoxide Binding
Carbon monoxide can bind to the reduced state of most high spin hemes, including ci, as witnessed by the blue shift of its Soret band (14, 19). The reduced F40Y complex was able to bind CO, because incubation in ∼100% CO resulted in the appearance of a pronounced shoulder at ∼410 nm. To assess the hindrance resulting from the F40Y mutation more directly, we measured the kinetics of CO rebinding after its photodissociation by a laser flash (Fig. 8). As regards to the WT, we note that the kinetics of CO rebinding presented here are significantly slower than those previously reported in (14). This likely reflects changes in the structural integrity of the b6f complex in the previously reported experiments, because we could mimic (not shown) similarly fast rebinding kinetics after the addition of FOS-choline 14, a detergent known to induce the progressive dissociation of the complex.6 As shown in Fig. 8, the rebinding of CO was only 2-fold slower in the F40Y mutant than in the WT.
FIGURE 8.

Kinetics of CO recombination after photolysis, followed at 411 nm, for the WT (solid squares) and the F40Y mutant (open circles). The kinetics were normalized to their initial amplitude. For qualitative comparison purposes we fitted the data with a stretched exponential (exp (−(t/τ)n)) (gray lines), and this fit yielded: for the WT, τ = 25 ms and n = 0.5; for the F40Y, τ = 52 ms and n = 0.6.
DISCUSSION
Structural Modification of the Near Environment of ci in the F40Y Mutant
We designed the F40Y mutant with the aim to provide a proteic axial ligand to the heme ci. Data from the redox titration suggest that this goal was achieved. Indeed, a coordination link between the phenol(ate) side chain and the heme iron would account for the strong downshift in midpoint potential of heme ci; the phenol(ate) side chain, a strong electron donor group should stabilize the ferric form of the heme and thus lower its midpoint potential, as observed in tyrosine mutants of heme oxygenase and myoglobin (41, 42). Moreover, previous reports showed that NQNO could act as an axial ligand to the WT heme ci (15) and that this inhibitor induces a strong downshift in redox potential (14). In this respect, the consequences of the F40Y mutation are similar to the addition of NQNO. However, whereas upon the addition of NQNO, the titration curve was split into two n = 1 waves, the redox titration of ci in the mutant was satisfyingly fitted by a single n = 1 Nernst curve, both in the presence and absence of NQNO. On a redox standpoint, the ligation state of ci would thus be more homogenous in the mutant than in the WT in the presence of NQNO. The pronounced shoulder at ∼440 nm, which is enhanced upon addition of NQNO in the (reduced − oxidized) spectrum of ci (Fig. 7), could, however, be taken as an indication of static disorder in the near environment of the heme. Yet a split Soret band of high spin c type cytochrome has been previously assigned to a non-degenerate Soret electronic transition, resulting from electronic inequivalency in the x and y directions of the heme (43, 44), without implying any heterogeneity of the sample. Irrespective of the precise origin of these spectroscopic modifications, the addition of NQNO slightly modifies the spectroscopic as well as the redox properties of ci, showing that the Qi site can accommodate this (semi)quinone analogue, which can interact with ci.
The Functional Consequences of the F40Y Mutation
According to the above proposal that the tyrosine side chain likely acts as a ligand to the ci heme, the accessibility to the heme is expected to be significantly hindered, and this should have significant functional consequences. We found that the reoxidation of the b hemes was dramatically slowed down when Phe40 was substituted by Tyr (Fig. 3B). The decreased amplitude of the slow electrochromic phase (Fig. 3A) shows that, under our experimental conditions, bH is reduced in the dark so that the cytochrome b reduction phase can be ascribed to the reduction of the bL heme. Yet although the low potential chain was normally fed in reducing equivalents in the mutant, as shown by the WT-like reduction rate of the b hemes, the reoxidation of the b hemes occurred in the hundreds of ms time range, i.e. more than 50-fold more slowly than in the WT. This is consistent with the above hypothesis according to which the ci heme is ligated by the Tyr side chain in the mutant and thereby prevents the access of the quinone to its reduction site. Further, it shows that, provided a quinone is present in the site, the direct electron transfer from bH to the quinone cannot occur with a reasonable rate. At first sight, these proposals are contradicted by the puzzling finding that under continuous illumination the overall activity of the b6f complex is far less decreased (Fig. 4). Interestingly, inhibiting the wild type Qi site with NQNO efficiently slows down the reoxidation of the b hemes after a single turnover flash but does not prevent the linear electron flow under steady state illumination (45).
As a possible way to reconcile these apparently contradicting findings, one could consider that the Qo site carries out its role in the quinol-plastocyanin oxidase pathway even though the Qi site is blocked. Such a hypothesis could be in line with the fact that the bc1 complex can also sustain electron transfer between quinol and oxidized cytochrome c when the Qi site is blocked by antimycin (46). However, the rate sustained in the presence of antimycin is only 2% of that obtained in its absence (47, 48). Given the large structural homology between the bc1 and b6f complex regarding the Qo site (which should be qualified however by the different sensitivities to inhibitors such as myxothiazol), blocking the Qi site in the b6f complex should as severely impair the steady state rate of the Qo site turnover, yielding overall turnover rates far lower than those observed here under continuous illumination.
More plausibly, the structural hindrance that accounts for the slowing down of this rate under flash experiments may be, at least partially, relieved under steady state conditions. In other terms, the quinone reduction might be triggered by the illumination regime. What could be the molecular bases of such a switch? As discussed above, the strong downshift of the midpoint potential of ci resulting from the Tyr for Phe substitution implies that the Tyr side chain provides stabilization energy to the oxidized state of the heme. On thermodynamic grounds, this means that the interaction between the heme and the phenol(ate) side chain is weakened upon reduction of the heme. This change in the ligation bond strength upon reduction of ci would readily account for our observations. Indeed, considering the respective midpoint potential of the three hemes in the low potential chain, the F40Y mutation should favor the oxidized state of ci even under anaerobic conditions (when bH is reduced but bL oxidized). Being ligated by the phenol(ate) the midpoint potential of ci is more negative than that of bL; the injection of one electron into the low potential chain results in the reduction of bL and hardly affects either the redox state of ci or its ligation state. Thus, the accessibility of the ci heme to a quinone is hindered, and quinone reduction cannot take place with a reasonable rate. Under continuous illumination, however, the cytochrome b6f complex is submitted to a stronger reducing pressure, which may eventually lead to the reduction of ci, thereby decreasing the ligation strength. This will allow a quinone to displace the Tyr side chain, access the ci heme, and then be reduced. Moreover, assuming that the redox potential of the non-ligated ci is indeed more positive than that of the ligated heme, the reducing pressure continuously undergone by the b6f mutant complex under illumination should significantly increase the steady state concentration of the reduced and weakly ligated heme with respect to the dark-adapted case. Reduction of ci thus weakens the gate for quinone reduction at the Qi site, and the electron flux that ensues would keep the reducing pressure high enough to maintain the gate open.
The binding kinetics of CO to the reduced ci supports this model. Indeed, the moderate decrease of the CO binding rate in the mutant with respect to the WT (2-fold) suggests that the Tyr side chain only weakly hinders the accessibility of CO to the reduced iron. The possibility that the CO replaces the water molecule on the other face of the heme appears less likely because of sterical constraints.
In myoglobin or in the cd1 nitrite reductase from Pseudomonas aeruginosa, mutating the distal histidine resulted in a 7–10-fold increase of the CO rebinding rate (49, 50). The magnitude of these kinetic changes provides a reasonable estimate for the sensitivity of the CO binding rate to moderate modifications of the accessibility to the heme. In the cd1 nitrite reductase from P. aeruginosa the targeted His does not directly interact with the heme iron but rather provides steric hindrance (50). In hemoglobin, the distal His also controls the access to the heme via a weak H-bond (51). Thus, if the Tyr40 remained a strong axial ligand to the reduced ci in the mutant b6f complex, it would likely result in a pronounced decrease of the CO rebinding rate. The finding that this is not the case strongly backs up the proposal that, when reduced, ci is not or weakly liganded by the Tyr side chain and that it can be more easily displaced than when oxidized.
Weakening the ligation strength upon reduction to the ferrous state has already been documented; in mutated heme oxygenase or myoglobin, where the natural His heme ligand was substituted for a Tyr (41, 52), it has been shown that the tyrosinate ligand dissociates upon reduction. Moreover, in the case of the d1 heme of the cd1 nitrite reductase (53), the change in the ligation state undergone by the heme upon its reduction to the ferrous state was proposed to act as a redox switch that controls the accessibility of the substrate to the heme and triggers the catalysis.
Conclusion
We propose that a redox switch is at work to allow the quinone reductase activity in the F40Y mutant. Likewise, the hypothesis that the interaction between the (semi)quinone and ci is controlled by the redox state of the heme in the WT b6f complex is attractive.
In the WT b6f complex, modeling a quinone in the Qi site results in a steric clash with the native position of the Phe40 aromatic ring,5 which has to move to provide access to the sixth ligand position. As a consequence, energy must be provided to allow the interaction between ci and the (semi)quinone. In analogy with the mechanism proposed here, according to which the ligation state of ci is linked to its redox state, this “interaction energy” could be provided by the redox state of the heme.
In addition, such a redox-dependent interaction between the quinone and ci would make the midpoint potentials of both ci and the quinone/semiquinone dependent on their interaction. If the quinone interacts more strongly with the ci heme in its oxidized state than in its reduced one, then the oxidized ci should stabilize the quinone, making the semiquinone more difficult to form. In brief, a stronger reducing redox potential would be required to inject an electron in the interacting quinone-ci ensemble than in the quinone or ci heme alone. In addition, the injection of a single electron in this ensemble would weaken the interaction between the two partners, raising their midpoint potential and facilitating the injection of a second electron. Extrapolating to the WT the present evidence that, in the mutant, the ligation state of ci depends on its redox state and vice versa thus provides a mechanistic insight into the proposal that ci would force the quasi-concerted reduction of the quinone into a quinol at the expense of the two b hemes (15, 54).
Acknowledgments
We are grateful to Frauke Baymann, Yves Choquet, and Daniel Picot for critical reading of the manuscript and fruitful discussions. Thanks are also due to F. Giusti for kindly providing the NQNO and to C. Lebreton for help with biochemical preparations.
D. Picot, personal communication.
Y. Pierre, personal communication.
- PSI
- photosystem I
- PSII
- photosystem II
- bH
- high potential heme b
- bL
- low potential heme b
- NQNO
- 2-n-nonyl-4-hydroxyquinoline N-oxide
- PQ
- plastoquinone
- cyt
- cytochrome
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- WT
- wild type.
REFERENCES
- 1.Mitchell P. (1975) FEBS Lett. 56, 1–6 [DOI] [PubMed] [Google Scholar]
- 2.Crofts A. R., Meinhardt S. W. (1982) Biochem. Soc. Trans. 10, 201–203 [DOI] [PubMed] [Google Scholar]
- 3.Osyczka A., Moser C. C., Dutton P. L. (2005) Trends Biochem. Sci. 30, 176–182 [DOI] [PubMed] [Google Scholar]
- 4.Kurisu G., Zhang H., Smith J. L., Cramer W. A. (2003) Science 302, 1009–1014 [DOI] [PubMed] [Google Scholar]
- 5.Stroebel D., Choquet Y., Popot J. L., Picot D. (2003) Nature 426, 413–418 [DOI] [PubMed] [Google Scholar]
- 6.Baniulis D., Yamashita E., Whitelegge J. P., Zatsman A. I., Hendrich M. P., Hasan S. S., Ryan C. M., Cramer W. A. (2009) J. Biol. Chem. 284, 9861–9869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berry E. A., Guergova-Kuras M., Huang L. S., Crofts A. R. (2000) Annu. Rev. Biochem. 69, 1005–1075 [DOI] [PubMed] [Google Scholar]
- 8.Cramer W. A., Yan J., Zhang H., Kurisu G., Smith J. L. (2005) Photosynth. Res. 85, 133–143 [DOI] [PubMed] [Google Scholar]
- 9.Darrouzet E., Cooley J. W., Daldal F. (2004) Photosynth. Res. 79, 25–44 [DOI] [PubMed] [Google Scholar]
- 10.Crofts A. R. (2004) Photosynth. Res. 80, 223–243 [DOI] [PubMed] [Google Scholar]
- 11.Lavergne J. (1983) Biochim. Biophys. Acta 725, 25–33 [Google Scholar]
- 12.Kuras R., Saint-Marcoux D., Wollman F. A., de Vitry C. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 9906–9910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lezhneva L., Kuras R., Ephritikhine G., de Vitry C. (2008) J. Biol. Chem. 283, 24608–24616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alric J., Pierre Y., Picot D., Lavergne J., Rappaport F. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 15860–15865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baymann F., Giusti F., Picot D., Nitschke W. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ducluzeau A. L., Chenu E., Capowiez L., Baymann F. (2008) Biochim. Biophys. Acta 1777, 1140–1146 [DOI] [PubMed] [Google Scholar]
- 17.Yamashita E., Zhang H., Cramer W. A. (2007) J. Mol. Biol. 370, 39–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musser S. M., Stowell M. H., Lee H. K., Rumbley J. N., Chan S. I. (1997) Biochemistry 36, 894–902 [DOI] [PubMed] [Google Scholar]
- 19.Zhang H., Primak A., Cape J., Bowman M. K., Kramer D. M., Cramer W. A. (2004) Biochemistry 43, 16329–16336 [DOI] [PubMed] [Google Scholar]
- 20.Joliot P., Joliot A. (1988) Biochim. Biophys. Acta 933, 319–333 [Google Scholar]
- 21.Choquet Y., Zito F., Wostrikoff K., Wollman F. A. (2003) Plant Cell 15, 1443–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuras R., Wollman F. A. (1994) EMBO J. 13, 1019–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris E. H. (1989) The Chlamydomonas Source Book: A Comprehensive Guide to Biology and Laboratory Use, Academic Press, San Diego: [DOI] [PubMed] [Google Scholar]
- 24.Goldschmidt-Clermont M. (1991) Nucleic Acids Res. 19, 4083–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boynton J. E., Gillham N. W., Harris E. H., Hosler J. P., Johnson A. M., Jones A. R., Randolph-Anderson B. L., Robertson D., Klein T. M., Shark K. B. (1988) Science 240, 1534–1538 [DOI] [PubMed] [Google Scholar]
- 26.de Lacroix de Lavalette A., Barbagallo R. P., Zito F. (2008) C. R. Biol. 331, 510–517 [DOI] [PubMed] [Google Scholar]
- 27.Pierre Y., Breyton C., Kramer D., Popot J. L. (1995) J. Biol. Chem. 49, 29342–29349 [DOI] [PubMed] [Google Scholar]
- 28.Huang D., Everly R. M., Cheng R. H., Heymann J. B., Schägger H., Sled V., Ohnishi T., Baker T. S., Cramer W. A. (1994) Biochemistry 33, 4401–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennoun P., Delepelaire P. (1982) in Methods in Chloroplast Molecular Biology ( Edelman M., Chua N. H., Hallick R. B. eds) pp. 25–38, Elsevier Biomedical Press [Google Scholar]
- 30.Zito F., Kuras R., Choquet Y., Kössel H., Wollman F. A. (1997) Plant Mol. Biol. 33, 79–86 [DOI] [PubMed] [Google Scholar]
- 31.Lemaire C., Girard-Bascou J., Wollman F. A. (1987) in Progress in Photosynthesis Research ( Biggins J. ed) Vol. IV, pp. 655–658, Martinus Nijhoff Publishers [Google Scholar]
- 32.Joliot P., Beal D., Frilley B. (1980) J. Chim. Phys. 77, 209–216 [Google Scholar]
- 33.Cornforth J. W., James A. T. (1956) Biochem. J. 63, 124–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuras R., de Vitry C., Choquet Y., Girard-Bascou J., Culler D., Büschlen S., Merchant S., Wollman F. A. (1997) J. Biol. Chem. 272, 32427–32435 [DOI] [PubMed] [Google Scholar]
- 35.Joliot P., Delosme R. (1974) Biochim. Biophys. Acta 357, 267–284 [DOI] [PubMed] [Google Scholar]
- 36.Junge W., Witt H. T. (1968) Z. Naturforsch. B. 23, 244–254 [DOI] [PubMed] [Google Scholar]
- 37.Selak M. A., Whitmarsh J. (1984) Photochem. Photobiol. 39, 485–489 [Google Scholar]
- 38.Joliot P., Joliot A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 10209–10214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sacksteder C. A., Kramer D. M. (2000) Photosynth. Res. 66, 145–158 [DOI] [PubMed] [Google Scholar]
- 40.de Vitry C., Desbois A., Redeker V., Zito F., Wollman F. A. (2004) Biochemistry 43, 3956–3968 [DOI] [PubMed] [Google Scholar]
- 41.Liu Y., Moënne-Loccoz P., Hildebrand D. P., Wilks A., Loehr T. M., Mauk A. G., Ortiz de Montellano P. R. (1999) Biochemistry 38, 3733–3743 [DOI] [PubMed] [Google Scholar]
- 42.Hildebrand D. P., Burk D. L., Maurus R., Ferrer J. C., Brayer G. D., Mauk A. G. (1995) Biochemistry 34, 1997–2005 [DOI] [PubMed] [Google Scholar]
- 43.Andrew C. R., Kemper L. J., Busche T. L., Tiwari A. M., Kecskes M. C., Stafford J. M., Croft L. C., Lu S., Moënne-Loccoz P., Huston W., Moir J. W., Eady R. R. (2005) Biochemistry 44, 8664–8672 [DOI] [PubMed] [Google Scholar]
- 44.Strekas T. C., Spiro T. G. (1974) Biochim. Biophys. Acta 351, 237–245 [DOI] [PubMed] [Google Scholar]
- 45.Rich P. R., Madgwick S. A., Moss D. A. (1991) Biochim. Biophys. Acta 1058, 312–328 [Google Scholar]
- 46.Zhang L., Yu L., Yu C. A. (1998) J. Biol. Chem. 273, 33972–33976 [DOI] [PubMed] [Google Scholar]
- 47.Muller F., Crofts A. R., Kramer D. M. (2002) Biochemistry 41, 7866–7874 [DOI] [PubMed] [Google Scholar]
- 48.Muller F. L., Roberts A. G., Bowman M. K., Kramer D. M. (2003) Biochemistry 42, 6493–6499 [DOI] [PubMed] [Google Scholar]
- 49.Springer B. A., Egeberg K. D., Sligar S. G., Rohlfs R. J., Mathews A. J., Olson J. S. (1989) J. Biol. Chem. 264, 3057–3060 [PubMed] [Google Scholar]
- 50.Wilson E. K., Bellelli A., Liberti S., Arese M., Grasso S., Cutruzzolà F., Brunori M., Brzezinski P. (1999) Biochemistry. 38, 7556–7564 [DOI] [PubMed] [Google Scholar]
- 51.Perutz M. F., Wilkinson A. J., Paoli M., Dodson G. G. (1998) Annu. Rev. Biophys. Biomol. Struct. 27, 1–34 [DOI] [PubMed] [Google Scholar]
- 52.Adachi S., Nagano S., Ishimori K., Watanabe Y., Morishima I., Egawa T., Kitagawa T., Makino R. (1993) Biochemistry 32, 241–252 [DOI] [PubMed] [Google Scholar]
- 53.Das T. K., Wilson E. K., Cutruzzolà F., Brunori M., Rousseau D. L. (2001) Biochemistry 40, 10774–10781 [DOI] [PubMed] [Google Scholar]
- 54.Allen J. F. (2004) Trends Plant Sci. 9, 130–137 [DOI] [PubMed] [Google Scholar]