

Abstract
Altered corticostriatal information processing associated with early dopamine systems dysfunction may contribute to attention deficit/hyperactivity disorder (ADHD). Mice with neonatal dopamine-depleting lesions exhibit hyperactivity that wanes after puberty and is reduced by psychostimulants, reminiscent of some aspects of ADHD. To assess whether the maturation of corticostriatal functional connectivity is altered by early dopamine depletion, we examined preadolescent and postadolescent urethane-anesthetized mice with or without dopamine-depleting lesions. Specifically, we assessed (1) synchronization between striatal neuron discharges and oscillations in frontal cortex field potentials and (2) striatal neuron responses to frontal cortex stimulation. In adult control mice striatal neurons were less spontaneously active, less responsive to cortical stimulation, and more temporally tuned to cortical rhythms than in infants. Striatal neurons from hyperlocomotor mice required more current to respond to cortical input and were less phase locked to ongoing oscillations, resulting in fewer neurons responding to refined cortical commands. By adulthood some electrophysiological deficits waned together with hyperlocomotion, but striatal spontaneous activity remained substantially elevated. Moreover, dopamine-depleted animals showing normal locomotor scores exhibited normal corticostriatal synchronization, suggesting that the lesion allows, but is not sufficient, for the emergence of corticostriatal changes and hyperactivity. Although amphetamine normalized corticostriatal tuning in hyperlocomotor mice, it reduced horizontal activity in dopamine-depleted animals regardless of their locomotor phenotype, suggesting that amphetamine modified locomotion through a parallel mechanism, rather than that modified by dopamine depletion. In summary, functional maturation of striatal activity continues after infancy, and early dopamine depletion delays the maturation of core functional capacities of the corticostriatal system.
Keywords: postnatal brain development, striatum, frontal cortex, dopamine, attention deficit/hyperactivity disorder, medium spiny neurons
Introduction
Higher brain functions require coordinated activity of neurons in the frontal cortex and striatum. The firing activity of the main striatal neuron class, the “medium spiny projection neurons,” is highly regulated in adults. Despite exhibiting rich near-threshold activity (“UP states”) driven by excitatory cortical inputs, these neurons are usually silent. It seems likely that medium spiny neurons filter out uncorrelated afferent activity and fire only in response to precisely synchronized inputs (Wilson, 1993; O'Donnell and Grace, 1995; Charpier et al., 1999; Kasanetz et al., 2006). Forebrain dopamine appears to increase the spatial and temporal selectivity of striatal neurons to cortical inputs (Rolls et al., 1984; DeFrance et al., 1985). Thus, abnormal dopamine function could result in uncoordinated corticostriatal activity and contribute to the clinical manifestations of Parkinson's disease, attention deficit/hyperactivity disorder (ADHD), and other neuropsychiatric conditions (Sagvolden et al., 2005; Hammond et al., 2007).
Rats undergo marked forebrain maturation during infancy. Corticostriatal synapses (Hattori and McGeer, 1973; Sharpe and Tepper, 1998), striatal UP states (Tepper et al., 1998), and dopamine receptors and nerve terminals (Stamford, 1989; Teicher et al., 1995) all become adult-like before puberty. In humans too, corticostriatal circuits and dopamine receptors experience marked postnatal adaptations (Seeman et al., 1987; Montague et al., 1999; Castellanos et al., 2002). Consistent with the view that cortical control of striatal activity and its regulation by dopamine are shaped during infancy, neonatal dopamine depleting lesions cause marked molecular adaptations in the rat forebrain (Bruno et al., 1998; Masuo et al., 2004; Brown and Gerfen, 2006). Moreover, during human infancy and adolescence, the volume of the caudate nucleus and prefrontal cortex is regulated by the dopamine transporter and dopamine D4 receptor genes (Durston et al., 2005). Factors modifying dopamine transmission during this period could therefore produce enduring alterations in corticostriatal functional connectivity. Although this hypothesis has little experimental support, current theories suggest that a state of dopamine deficiency contributes to ADHD [for review, see Biederman and Faraone (2005), Sagvolden et al. (2005), and Swanson et al. (2007)]. To determine whether corticostriatal functional connectivity relies on the postnatal availability of forebrain dopamine, we used infant [postnatal day 28 (PD28)–PD32] and adult (12–16 weeks old) anesthetized mice which had or had not undergone a neonatal dopamine-depleting brain lesion and evaluated the responsiveness of striatal neurons to cortical stimulation and the degree of phase locking between striatal firing and ongoing oscillations in the frontal cortex local field potential (LFP). Importantly, mice with such lesions exhibit hyperactivity that wanes after puberty and is reduced by psychostimulants and have been proposed as a model of ADHD (Avale et al., 2004b). Urethane anesthesia provided a good framework for studying corticostriatal functional connectivity, as the different frequency oscillations of the frontal cortex LFP have been related to the natural patterns of brain activity that drive UP states in striatal neurons (Kasanetz et al., 2006; Mahon et al., 2006).
Materials and Methods
Lesions and behavior.
CF-1 mice were maintained under a 12 h light:12 h dark cycle with ad libitum access to food and water and cared for in accordance with institutional (CICUAL, RS2079/2007, University of Buenos Aires) and government regulations (SENASA, RS617/2002, Argentina). Bilateral dopamine neuron depletion was induced as described by Avale et al. (2004b). Briefly, PD2 pups received injections of 6-hydroxydopamine (12.5 μg/1.5 μl; MP Biomedicals) or vehicle (0.1% ascorbic acid) in each lateral ventricle (1.1 mm below the skin, 0.6 mm from midline, and 1.5 mm anterior to the lambda), after desipramine pretreatment (20 mg/kg, s.c.), under hypothermal anesthesia. Injections were performed at a constant rate of 1.25 μl/min with a 30 gauge needle coupled to a 25 μl Hamilton syringe driven by a microinfusion pump (Bee syringe pump and controller, Bioanalytical Systems). In each litter, half of the pups received the toxin and the other half vehicle. After surgery, they were warmed up and returned to their home cages in groups of up to eight pups per breast-feeding mother until weaning (PD24). Thereafter, control and lesioned mice were housed together in the same cage in groups of six to eight until the electrophysiological experiment.
Behavioral, body growth, and endocrine studies suggests that, in rodents, adolescence could be considered to span from PD28 to PD42 (reviewed by Spear, 2000). All the mice that received neonatal injections underwent horizontal locomotor activity recordings between PD21 and PD25 (Fig. 1). A first set of 76 mice, of which 26 were selected for glass microelectrode recordings (see below), were examined for 30 min in a 30 × 30 cm white walls open field with a force plate actometer (Fowler et al., 2001). Twelve were used for electrophysiology at PD28–PD32 (for the sake of simplicity, “infant”); the remaining 14 were retested in the open field and used for electrophysiology during postnatal weeks 12–16 (“adults”). The force plate was coupled to four supporting force transducers, positioned at the corners, which sensed animal movement on the plate. Briefly, the forces were digitized and used to compute position on the plate, as described by Fowler et al. (2001), with a spatial resolution of <1 mm and a time resolution of 1 ms. A second set of PD21–PD25 mice (n = 35) was recorded with a video camera in an open-field arena (40 cm × 40 cm) with black walls for 30 min, then injected with amphetamine (4 mg/kg, i.p.) and recorded for further 30 min. At this dose amphetamine induces mild hyperactivity in controls but decreases hyperactivity in dopamine neuron-depleted mice (Avale et al., 2004b). Twelve of these mice were used for multisite electrophysiological recordings with silicon probes at PD28-PD32 (see below). After electrophysiological recordings, all animals were subjected to postmortem quantitative examination of tyrosine hydroxylase (TH) immunoreactivity in the mesencephalon and striatum.
Figure 1.

Mice depleted of mesencephalic dopamine neurons as neonates showed hyperlocomotion during infancy. A, Horizontal locomotor activity in response to a novel environment (square open field) was assessed during 30 min in PD21–PD25 control and dopamine neuron-depleted mice. One set was studied in 2006 (circles), the other in 2007 (diamonds). The distribution of locomotor scores of control mice, but not those of dopamine-depleted mice, could be fitted to a Gaussian function (left). We considered as hyperlocomotor those dopamine-depleted mice scoring above 3 SD of the average locomotor score of controls. Filled markers correspond to the animals used for electrophysiology. B, Twelve of 38 lesioned mice in set 1 were hyperlocomotor according to this criterion and were selected for electrophysiology. On average, the hyperlocomotor mice covered a 70% longer distance than controls (nSHAM = 14, n6-OHDA = 12; t test, #t(24) = 7.211; p < 0.001). Six hyperlocomotor and six control mice were used for electrophysiological recordings 1 week later (PD28–PD30). The remaining six hyperlocomotor and eight controls were retested in the open field several weeks later (when they were 11–15 weeks old) and used for electrophysiology 1 week after behavioral testing. The differences in locomotor activity were no longer visible in adulthood. Data are mean ± SEM.
Electrophysiology.
All recordings were performed under urethane anesthesia (1.2–1.5 g/kg i.p.). Local anesthetic (bupivacaine hydrochlorate solution, 5% v/v, Durocaine, AstraZeneca) was applied subcutaneously on the scalp (0.1 ml) and the mice were affixed by means of a custom-made mouse adapter to a stereotaxic frame (Stoelting). During the experiment, additional urethane (customarily 0.3 g/kg s.c. every 2–3 h) was given to maintain a constant level of anesthesia, which was assessed by online visual examination of the frontal cortex LFP (see text below) and by regularly testing the nociceptive hind limb withdrawal reflex. Body temperature was maintained at 36−37°C with a servo-controlled heating pad (Fine Science Tools). One concentric bipolar electrode (SNE-100, Better Hospital Equipment; outer contact diameter 0.25 mm, central contact diameter 0.1 mm, contact separation 0.75 mm, contact exposure 0.25 mm) placed in the lateral motor cortex was used to differentially record the LFP (0.1–300 Hz; Lab1 amplifier, Akonic) and two other electrodes placed in the prefrontal and medial motor cortices were used to deliver electrical stimulation pulses (Fig. 2A).
Figure 2.
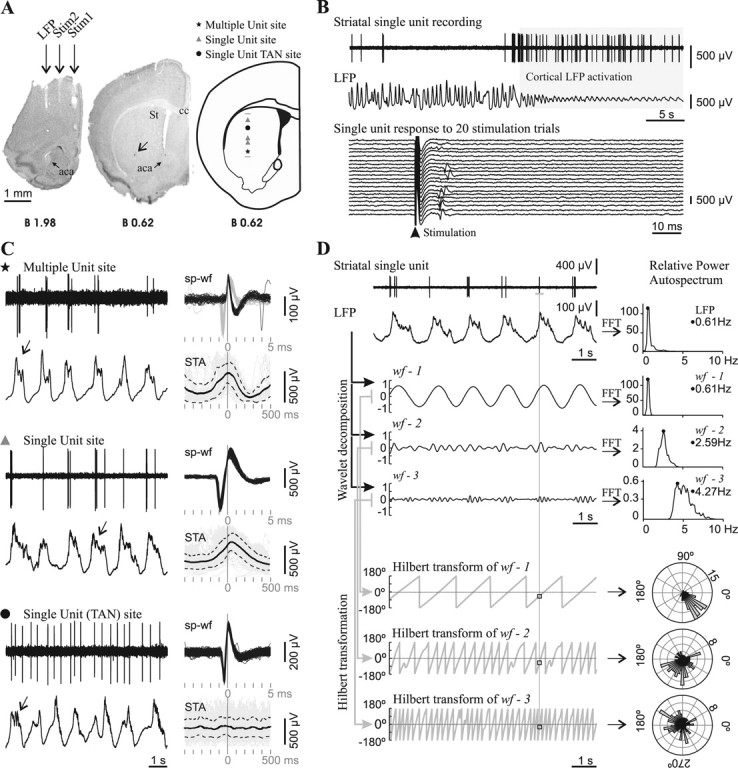
Spontaneous striatal spikes were phase locked to cortical field potential rhythms in adult control mice. A, Striatal neuronal discharge activity was recorded simultaneously with the motor cortex LFP in urethane-anesthetized mice. Photographs of coronal brain sections illustrate the position of electrodes in a representative experiment conducted in an adult mouse. The LFP electrode was positioned in the motor cortex area 1 [2 mm anterior to bregma, 1.5 mm lateral to midline, and 1.3 mm below cortical surface (Franklin and Paxinos, 2001)]. Two other concentric bipolar electrodes allowed electrical stimulation of the prefrontal cortex (Stim1: 2 mm anterior to bregma, 0.4 mm lateral, and 2.1 mm below cortical surface) and motor cortex area 2 (Stim2: 2 mm anterior to bregma, 1 mm lateral, and 1.3 mm below cortical surface). The arrow points to a Chicago Sky Blue deposit indicating the end of a glass microelectrode track in the striatum (St). Right, Schematic diagram of a coronal brain section (Franklin and Paxinos, 2001) illustrating the localization of all spontaneously active “single-unit” and “multiple-unit” type recording sites along a glass microelectrode track. aca, Anterior commissure, anterior; St, striatum; cc, corpus callosum. Scale bar, 1 mm. B, Mice striatal single units were modulated by the frontal cortex LFP and showed short latency excitatory responses to frontal cortex electrical stimulation. Top, The spontaneous activity of most striatal single units was modulated by slow-wave activity in adult mice. This striatal neuron fired exclusively during the positive part of the slow waves and showed increased spontaneous discharge during LFP activation. Bottom, The same unit fired a single spike with a short latency (10–20 ms) in response to electrical stimuli delivered to the motor cortex, in 35% of the trials at a stimulation current of 300 μA. C, Representative glass microelectrode recordings (left) taken from three sites along the track depicted in A, together with all spike waveforms (sp-wf) collected during a 3-min-long recording and the spike-triggered average (STA: solid black trace; SD: dotted traces) of LFP (right). Note that, in the two upper recordings, spikes occurred during the positive part of the slow wave, while in the recording site depicted at the bottom, spikes occurred throughout the slow wave. The presence of delta waves and spindles (arrows) on the top of the wave is an index of neuronal activity and indicates which is the “active part” of the slow wave (Kasanetz et al., 2006). The symbols indicate the position of the recording site along the track in A. TAN, Tonically active neuron. D, To assess phase locking of single-unit discharges to LFP oscillations (raw signals at upper left), the LFP was decomposed in three band-passed waveforms (wf-1, wf-2, wf-3) retaining different LFP frequencies, which were characterized by performing fast Fourier transforms (FFT) to compute relative power auto-spectra with 0.11 Hz resolution (right). wf-1 retained most of the power of the dominant slow-wave oscillation (range 0.4–1.5), wf-2 mainly reflected delta waves (1.5–4 Hz), and wf-3 retained frequency components related to spindles (4–8 Hz). The whole frequency range studied (0.4–10 Hz) encompassed >90% of the relative LFP spectral power when slow-wave activity was dominant. A Hilbert transformation (2 ms resolution) was performed to obtain the phase angle (gray sawtooth traces at bottom left) at every point of wf-1, wf-2, and wf-3, and to establish the instantaneous phase at the time of occurrence of every spike along the spike train (squares). Next, the number of spikes (radial axis) occurring at different phase angles (circular axis) of wf-1, wf-2, and wf-3 was depicted in circular plots (right; bin size: 7.2°). Phase locking of spike discharges is indicated by assessing deviation from uniformity and circular dispersion (see Materials and Methods). The illustrated neuron showed significant phase locking to slow-wave, delta, and spindle-related LFP activity.
In the first set of mice we performed extracellular spike recordings with glass electrodes filled with NaCl (2 m) and 1% Chicago Sky Blue (5–12 MΩ, in vitro at 1 kHz), and the recordings were amplified and bandpass filtered (0.3–3 kHz; Lab1, Akonic) and digitized at 10 kHz simultaneously with the LFP. Dopamine-depleted mice with the highest locomotor scores and randomly selected sham controls from the same litter were used. Spike recordings were taken along 1–2 mm vertical tracks (up to two per experiment, 500 μm apart) (Fig. 2A). The electrode was moved forward in steps of 10–20 μm per min until reaching a spontaneously active site (biphasic or triphasic waveforms, signal-to-noise ratio 2:1 or higher) (Fig. 2B,C). Every neuron was recorded for at least 3 min during ongoing slow-wave activity. Then, sets of 20–30 constant current pulses (0.3 ms duration at 0.5 Hz, 100–500 μA; Iso-Flex and Master 8, AMPI) were successively delivered to the prefrontal and medial motor cortices (Fig. 2B). Striatal units were considered “responsive” to cortical stimulation if they discharged in >20% of stimulation trials at the maximal current tested (500 μA) (Fig. 2B). For many units we could test several intensities and determine the current evoking discharges in ∼50% stimulation trials (I50%). Spontaneously active sites were classified as “single-unit” (a sole, constant shape and amplitude waveform having >5 ms refractory period) or “multiple-unit” type (Fig. 2C). Single units were separated from noise by amplitude discrimination.
In the second set of mice, 12 of 16 channels of a silicon probe electrode (200 μm vertical site spacing and 500 μm horizontal shank spacing; NeuroNexus Technologies) were used for simultaneous recordings of multiunit striatal ensembles (300–3000 Hz, digitized at 20 kHz). As the behavioral scores of the first set of mice revealed that >50% of the dopamine-depleted animals cannot be considered hyperlocomotor, these experiments were done in three animal groups: (1) randomly chosen sham operated mice; (2) dopamine-depleted mice showing the highest levels of locomotor activity; and (3) dopamine-depleted mice with locomotor scores that matched those in the sham-operated control group. The multielectrode array was positioned within the striatum with an angle of 20° in the coronal plane (0.8–1.0 mm anterior to bregma, 2.5–3.0 mm lateral to the midline at the skull level, and 3.0 mm below cortical surface). After repeatedly recording spontaneous activity during cortical LFP slow-wave activity (three blocks 3 min each) the mice were injected with amphetamine (same dose used for the behavioral testing) and further blocks of spontaneous activity were recorded during the following 30 min. Multielectrode recordings and spike sorting with Wave_Clus (Quiroga et al., 2004) were performed as described by Zold et al. (2007).
To study phase locking (Fig. 2D), the LFP was wavelet transformed as described by Kasanetz et al. (2006) to obtain three bandpass-filtered waveforms (wf) having maximal spectral powers at ∼0.8 (wf-1), ∼2.2 (wf-2), and ∼4.4 (wf-3) Hz. We then obtained the phase of wf-1, wf-2, and wf-3 (by performing a Hilbert transform) at the time of every action potential during a spike train (resolution: 2 ms). The resulting collections of wf phases were depicted in circular frequency plots, where the circular axis was wf phase angle. If spiking occurred randomly, wf phases should distribute uniformly. We used the Rayleigh probability and circular dispersion to estimate deviation from circular uniform distribution and concentration of spike discharges at certain phase angles (Fisher, 1993), and both as indexes of phase-locked activity (Siapas et al., 2005).
Histology.
At the end of each experiment mice received a lethal dose of urethane and were transcardially perfused with 10 ml of cold saline solution followed by 20 ml of paraformaldehyde (4% w/v) in 0.1 m PBS. Brains were removed, immersed for 30–45 min in the same fixative at room temperature, and stored in 0.1 m PBS containing 15% sucrose at 4°C for 24–72 h. Coronal brain sections (40 μm thick) were Nissl stained to verify the good placement of recording and stimulation sites, and underwent TH immunohistochemistry to assess dopamine neuron depletion (Avale et al., 2004b). In the second set of mice we examined compensatory hyperinnervation by serotoninergic fibers (Avale et al., 2004a) in sections adjacent to those used for TH immunohistochemistry, with a rabbit polyclonal antibody against the serotonin transporter (SERT) (PC177L, Calbiochem). The final position of the glass electrode for each track was marked by iontophoretic application of Chicago Sky Blue and confirmed in the histological sections. Location of the multielectrode array was also determined postmortem by visualization of fluorescent material left along the electrode track (DiI, Molecular Probes).
For the second set of mice, we did a quantitative assessment of the lesions. The sections were visualized with a Nikon Eclipse 50i microscope equipped with a high-quality video camera (Optronics MicroFire mod. S99808). First, we counted the number of TH-immunoreactive (TH+) neurons in the substantia nigra and ventral tegmental area in three sections per mouse (rostral −3.00, central −3.28, and caudal −3.52 mm from bregma). All TH+ cell bodies showing a nucleus and at least one process were counted with a semiautomatic system that allows tagging the position of the counted cells within the outlined structure (Mercator Pro, Explora Nova). As dopamine-depleted animals also exhibit TH+ neurons in the striatum (Huot and Parent, 2007), we counted the number of TH+ cells within 0.16 mm2 squares in the dorsolateral and ventromedial striatum. Second, we measured the optical density (OD) in the dorsal striatum, nucleus accumbens, and different areas of the frontal cortex, in both TH- and SERT-stained sections. Structures' outlines were adopted from the atlas of Franklin and Paxinos (2001). We did measures in two striatal sections for TH and two adjacent sections for SERT immunohistochemistry, and averaged the OD measures taken from both sides of the brain. Optical density was transformed in gray levels using a 256-grade scale (NIH ImageJ software). Background staining was measured in the corpus callosum and subtracted from regions of interest. In addition, we measured the immunoreactive area occupied by TH+ and SERT+ structures in different areas of the frontal cortex. However, the fact that three concentric electrodes were placed in rostral areas of the frontal cortex precluded examining more comprehensively TH and SERT cortical immunoreactivity.
Results
Neonatal dopamine depletion caused hyperlocomotion in some but not all the lesioned animals
In rodents, neonatal depletion of mesencephalic dopamine neurons produces increased locomotor activity that wanes after adolescence (Shaywitz et al., 1976a; Miller et al., 1981; Avale et al., 2004b). Dopamine depletion must be incomplete (80–90%) to induce the hyperlocomotor phenotype, because more extensive damage induces akinesia (Miller et al., 1981; Avale et al., 2004b). Figure 1A shows the horizontal activity recorded at PD21–PD25 in the two sets of animals used for the present study. The horizontal distances traveled by control mice could be fitted with a Gaussian function. In contrast, the scores of dopamine-depleted mice showed a bimodal or strongly skewed distribution (Kolmogorov–Smirnov normality test, p < 0.01), with 31% to 43% (sets 1 and 2, respectively) of the mice exhibiting “hyperlocomotor” scores (>3 SD from the control group mean) (Fig. 1A). Furthermore, in line with previous findings (Shaywitz et al., 1976a; Miller et al., 1981; Avale et al., 2004b), the hyperlocomotor mice showed scores similar to those of controls when retested as adults (Fig. 1B). Another well known effect of neonatal dopamine depletion, reduced body weight (Shaywitz et al., 1976a; Miller et al., 1981; Avale et al., 2004b), was independent of the hyperlocomotor phenotype (sham: 20.4 ± 0.9 g; nonhyperlocomotor 15.4 ± 0.8 g; hyperlocomotor 14.6 ± 1.2 g; Tukey post hoc test p < 0.001 between sham and any lesioned group after significant one-way ANOVA). Thus, the data are in general agreement with published findings (van der Kooij and Glennon, 2007).
Synchronization of striatal firing to cortical rhythmic activity in mice
The relationship between cortical and striatal activity has not been thoroughly investigated in mice. In urethane-anesthetized adult mice, the cortical LFP displayed prevalent slow-wave activity (∼0.8 Hz) associated with robust delta and spindle activity and sporadic episodes of “activation” (Fig. 2B). A majority of the striatal neurons showed a very low spontaneous firing rate (typically <1 spike per second) and fired exclusively during the active part of slow waves (Fig. 2C,D). This is in good agreement with reports on the activity of striatal medium spiny projection neurons in rats (Wilson and Groves, 1981; Charpier et al., 1999; Mahon et al., 2001; Kasanetz et al., 2006; Mahon et al., 2006). A few mouse striatal neurons (4 of 187 single units recorded through glass microelectrodes for the present study) displayed sustained firing throughout the slow wave (Fig. 2C), reminiscent of the “tonically active interneurons” previously described in other species. Thus, mouse corticostriatal physiology closely resembles that of rats.
Spontaneous striatal activity decreased after infancy more steeply in control than in dopamine neuron-depleted mice
Striatal activity increases steeply during the first three postnatal weeks in rats (Napier et al., 1985; Tepper et al., 1998). It is likely that it decreases thereafter, because striatal medium spiny neurons show very low firing rates in adults. However, to our knowledge, there are no reports comparing striatal activity between infant and adult rodents, or studying the influence of the dopamine pathways on the development of corticostriatal functional connectivity.
In a first experiment, we simultaneously recorded the frontal cortex LFP and striatal neuronal activity in infant (PD28–PD32) and adult (12–16 week-old) control and dopamine-depleted mice that exhibited hyperlocomotor activity when tested at PD21–PD25 but not when they were adults (Fig. 1B). Because the activity of striatal neurons is distinctively modulated by global brain state (Fig. 2B), special care was taken to perform all recordings under comparable slow-wave activity. Interestingly, the average discharge rate of striatal neurons did not differ between treatments (see also Luthman et al., 1993) or across stages of development (Fig. 3A). Because of the very low discharge rate and bursty activity of striatal neurons, the density of active sites could be a better index of spontaneous discharge across the striatum than the firing rate of individual neurons. Control infant mice showed high levels of spontaneous activity (Fig. 3B) (16.5 ± 1.9 active sites per mm, mean ± SEM), considerably greater than in adult controls (3.45 ± 0.5; p = 0.0002, Tukey post hoc within controls after significant age per treatment interaction). Moreover, the proportion of active sites with multiple units was also higher in infants than in adult controls (29/150 vs 31/49, χ2 = 33.85, p < 0.00001), indicating that striatal spontaneous activity declined with increasing age.
Figure 3.

The normal ontogenetic decrease of spontaneous striatal activity occurring after infancy is regulated by dopamine pathways. Recordings of striatal neuron discharges and the frontal cortex LFP were performed in dopamine-depleted and control mice, during infancy (PD28–PD32) or adulthood (12–16 weeks after birth), with glass microelectrodes under urethane anesthesia. The locomotor activity of these animals is shown in Figure 1B. A, The average discharge rate of striatal neurons was assessed from 3-min-long signal segments, under slow-wave cortical activity. Data are the median (thick line), 25–75% (box), 10–90% (whiskers), and 5–95% (circles) of the distribution. The number of neurons in each group is given between parentheses. Discharge rates did not differ across treatments or along development (Kruskal–Wallis ANOVA). B, The density of spontaneously active striatal sites per mm of recording track decreased with age regardless of treatment (data are mean ± SEM; two-way ANOVA main effect of age, F(1,27) = 68.1, p < 7.3 × 10−9), but it decreased more steeply in controls than in lesioned mice (interaction between treatment and age, F(1,27) = 17.9, p = 0.0002; #Tukey post hoc test within adulthood, p < 0.0005). C, Further proof that striatal activity decreased with age was that the proportion of single-unit type sites (dark gray bars) increased between infancy and adulthood, in both controls (χ2 = 33.85, p < 0.00001) and dopamine-depleted mice (χ2 = 15.68, p < 0.0001). However, the proportion of single-unit sites did not differ between groups at either age (χ2 = 3.13, p = 0.07 within infancy; χ2 = 0.62, p = 0.4 within adulthood). D, Schematic drawings of coronal brain sections showing the localization of recording tracks and sites, in the four experimental groups [distance to bregma in millimeters, estimated according to the atlas of Franklin and Paxinos (2001)].
The density of spontaneously active sites in infant hyperlocomotor mice was similar to that of age-matched controls (14.5 ± 0.9 sites per mm; Tukey post hoc within infant mice p = 0.62) and higher than in dopamine neuron-depleted adults (10.3 ± 0.9 sites/mm; Tukey post hoc within lesion p = 0.048). Moreover, within lesioned mice, the proportion of “single-unit” sites was higher in adulthood (45/80 and 35/123, respectively, χ2 = 15.68, p < 0.0001). Thus, striatal discharge activity decreased after infancy despite neonatal depletion of dopamine neurons. However, the density of spontaneously active sites was higher in adult dopamine neuron-depleted mice than in adult controls despite similar locomotor scores at this age (p < 0.0005, Tukey post hoc within adulthood).
Striatal responsiveness to cortical stimulation decreased after infancy and was impaired in infant hyperlocomotor mice
As a first attempt to functionally characterize the maturation of corticostriatal connectivity, we studied striatal single units' responsiveness to electrical stimulation of the motor and prefrontal cortices in the same set of mice (Fig. 4A). Striatal neurons were more responsive in infant than adult control mice. This was supported by a lower I50% (Fig. 4B) (239 ± 23 vs 330 ± 34 μA in adults, mean ± SEM, n = 29 and 13 single units, respectively; p < 0.03, Tukey post hoc within controls after significant interaction) and a smaller proportion of “nonresponsive” units (12/35 vs 8/16, respectively; χ2 = 6.91, p < 0.04) in infant mice. In hyperlocomotor mice, instead of increasing with age, I50% was similarly high in infants and adults (Fig. 4B) (429 ± 27 μA, n = 21 vs 389 ± 24 μA, n = 25 single units, respectively; Tukey post hoc p > 0.7), and higher than in infant controls (p < 0.0003, Tukey post hoc within infants. Also, the proportion of “nonresponsive” neurons was similarly high at both ages in hyperlocomotor mice (23/41 for infant, 20/34 for adults) and higher than in infant controls (χ2 = 16.3, p < 0.0003).
Figure 4.
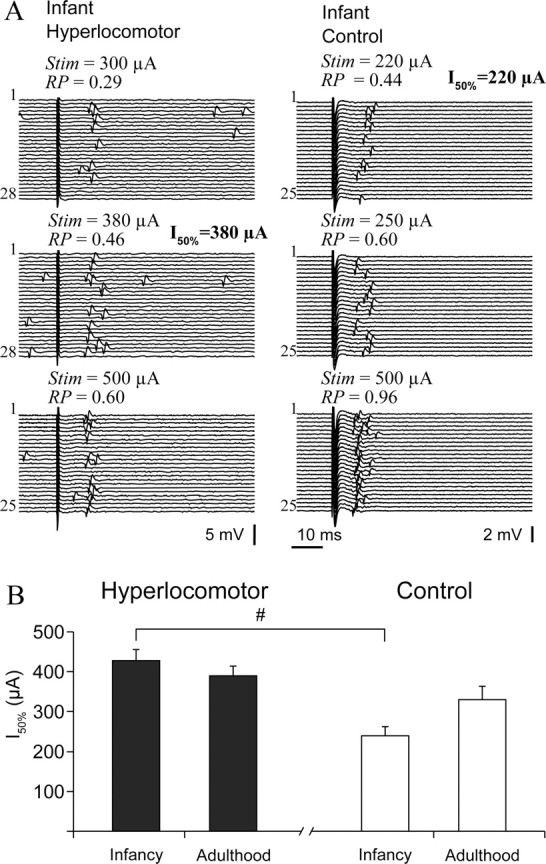
Impaired striatal responsiveness to cortical electrical stimulation in infant hyperlocomotor mice. A, Typical responses of striatal single units to cortical stimulation in infant hyperlocomotor (left) and control (right) mice, at increasing stimulation currents (Stim). Response probability (RP) at different stimulation currents was estimated on the basis of 20–30 stimulation trials, as illustrated. B, The current needed to drive spike discharges in ∼50% of the trials (I50%) was different between infant hyperlocomotor and control mice (significant interaction in a two-way ANOVA, F(1,84) = 5.71, p = 0.019; #Tukey post hoc test p < 0.0003), but not in adulthood. Note that, because the relationship between stimulation current and probability of driving spikes did not seem linear, and only a limited number of current intensities could be tested for each neuron, the current we call I50% is in fact the tested current that drove spikes in approximately 50% of the trials. The average response probability corresponding to the intensities computed for the I50% parameter was as follows (mean ± SEM): 6-OHDA lesioned infant hyperlocomotor: 0.51 ± 0.04, infant controls: 0.55 ± 0.04, 6-OHDA lesioned adults: 0.56 ± 0.04, adult controls: 0.58 ± 0.06. Response probability at I50% was not different between groups (two-way ANOVA). For neurons that were responsive to both cortical areas, only the minor I50% was used to study striatal sensitivity to cortical stimulation. These data correspond to mice of set 1.
Spatial segregation of cortical input across striatal space is a core feature of corticostriatal connectivity in adult animals (Alexander et al., 1986). Convergent responses were frequent in infant controls, with 60% striatal neurons responding to both motor and prelimbic cortex stimulation (21/35 neurons). This proportion was 25% in adult controls (4/16 neurons, χ2 = 6.91, p < 0.04). Conversely, in lesioned mice convergent responses were as low in infants as in adults (17%, 7/41 and 6/34, respectively), and lower in hyperlocomotor than in infant controls (χ2 = 16.3, p < 0.0003).
In summary, striatal responsiveness to cortical stimulation decreased and functional segregation of cortical inputs increased after infancy, paralleling an ontogenetic decrease in spontaneous discharge activity. Moreover, striatal neurons were less responsive and more spatially selective in hyperlocomotor mice.
Phase locking of striatal spikes to cortical rhythms increased after infancy and was less stringent in infant hyperlocomotor mice
Cortical slow waves induced by anesthesia provide a more patterned input to the striatum than cortical stimulation and closely resemble natural slow-wave sleep (Steriade, 2000). Indeed, slow waves drive transitions to the UP state in striatal neurons (Kasanetz et al., 2006; Mahon et al., 2006), which is the physiological condition during which medium spiny neurons fire action potentials. Moreover, it is believed that spontaneous cortical rhythms sculpt the wiring of neural circuits during postnatal development (Penn and Shatz, 1999). Therefore, it is important to assess whether cortical LFP rhythms entrain striatal discharges with similar strength in control and hyperlocomotor mice. To separately assess phase locking of striatal spikes to different cortical oscillations, we generated three nonoverlapping band-passed waveforms, wf-1, wf-2, and wf-3, retaining different main frequencies of the LFP corresponding to the slow oscillation, delta waves, and spindles, respectively (Fig. 2D). Then, we computed the phase of each waveform at the time of occurrence of each spike within a spike train (Fig. 2D). If spikes are not related to the LFP, they will occur with equal probability at every phase angle of cortical oscillations, providing uniform circular frequency plots. Conversely, condensation of spikes around a given phase angle will result in nonuniform distributions with little circular dispersion (Fig. 5A). The collection of p values indexing deviation from uniformity across the whole sample of recorded spike trains and the circular dispersion of these distributions contain information about the influence of age and neonatal dopamine depletion on the cortical control of striatal activity.
Figure 5.
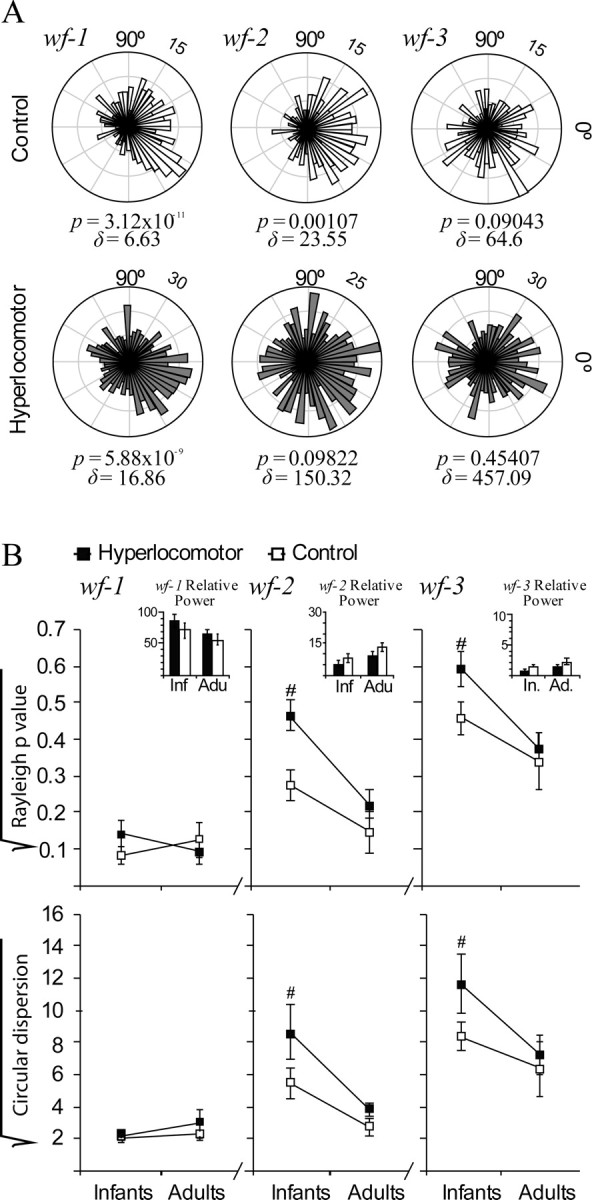
Striatal neuron spiking was less phase locked to cortical local field potential in infant hyperlocomotor mice. To assess phase locking of striatal discharges to cortical rhythms, we used wavelet decomposition to generate three nonoverlapping band-passed LFP waveforms, wf-1, wf-2, and wf-3, retaining different main frequency components of the LFP corresponding to the slow oscillation, delta waves, and spindles, respectively (see Fig. 2D). Phase locking was defined as a significant deviation from uniformity (Rayleigh p < 0.05) in circular plots depicting the number of spike occurrences (radial axis) at every waveform angle (circular axis; bin size: 7.2 degrees). Circular dispersion was taken as an additional index of locking strength. A, Circular plots corresponding to representative control (above) and hyperlocomotor (below) infant mice striatal neurons. The control mouse neuron was significantly locked to wf-1, wf-2, and wf-3, whereas the neuron from a hyperlocomotor mouse was locked to wf-1 only. Rayleigh p values (p) and circular dispersion (δ) are provided below each plot. B, Average Rayleigh p value and circular dispersion for control and dopamine neuron-depleted mice, in infancy and adulthood, depicted separately for wf-1, wf-2, and wf-3 (mean ± SEM; values were square root transformed for normalization). Statistical comparisons were made with three-way ANOVAs, separately for Rayleigh p values and circular dispersion, with two between-group factors (treatment and age) and one within-group factor (band). There were significant main effects of age (p = 0.001) with significant band-per-age interactions (p < 0.001). Therefore, we computed the simple effects of age, regardless of treatment, which indicated that circular distributions within wf-2 and wf-3 were more skewed in adult than in infant mice (Tukey post hoc test, p < 0.01 for either wf-2 or wf-3). Also, there were significant main effects of lesion or lesion-per-band interaction, because of lesion effects within wf-2 and wf-3 at infancy (p < 0.05 in Tukey post hoc comparisons within the different bands and ages). Insets depict the power (mean ± SEM) of the corresponding LFP band for each group and age, showing that, for the segments of signal studied, there were no differences in LFP power. Data are from set 1. Number of neurons per group: infant controls, 53, infant hyperlocomotor, 49, adult controls, 20, lesioned adults 37. There were no between group differences in the number of spikes per neuron used for the computations (mean ± SEM for the whole population of neurons, 194.7 ± 23.1).
Significant phase locking to slow-wave activity (p < 0.05 in the Rayleigh test) occurred in >90% striatal spike trains, regardless of age and lesion (Fig. 5B; see also Fig. 2C). This could be expected because medium spiny neurons cannot fire spikes unless they are in the UP state (Wilson and Groves, 1981), which takes place concomitantly with the slow wave (Kasanetz et al., 2006), and these cells account for 90% of all striatal neurons (Wilson, 1993). Phase locking to higher frequency LFP components retained in wf-2 and wf-3 could be indicative of finer cortical control of striatal firing during UP states. Both in control and dopamine neuron-depleted mice, phase locking to wf-2 and wf-3 was higher in adulthood (i.e., circular distributions were more skewed), indicating a more stringent cortical control of striatal firing as age increases (Fig. 5B) (simple effects of age on Rayleigh scores and circular dispersion, p < 0.001 for wf-2 and wf-3, after significant effect of age × band interaction in the main ANOVA). Moreover, hyperlocomotor mice showed less locking of spikes to wf-2 and wf-3. Post hoc comparisons showed that the lesion only affected phase locking to wf-2 and wf-3 when animals were young (p < 0.05, Tukey comparisons). Importantly, the effects of age or lesion cannot be explained by differences in the pattern of resting cortical activity (Fig. 5B, inset).
Amphetamine enhanced phase locking of striatal neuron firing to LFP rhythms in hyperlocomotor mice
As only a fraction of the neonatally lesioned animals exhibited hyperlocomotion, the remaining question was whether the above electrophysiological findings were related to dopamine depletion in general or more specifically to dopamine depleting lesions inducing hyperactivity. Determining this would require assessing corticostriatal connectivity in “nonhyperlocomotor” dopamine-depleted mice. Additionally, amphetamine induces a reduction of activity in rodents rendered hyperlocomotor by neonatal dopamine depletion, instead of the increase commonly observed in controls (Shaywitz et al., 1976b; Avale et al., 2004b). Importantly, the most common treatment for ADHD is psychostimulants, which supposedly act by improving corticostriatal circuit regulation of behavior and attention (Kuczenski and Segal, 2005; Volkow et al., 2005; Arnsten, 2006). Thus, demonstrating that certain electrophysiological changes occur specifically in hyperlocomotor mice and are reduced by amphetamine would strengthen the notion that these changes are related to the behavioral phenotype. To answer these questions, we did additional experiments in three groups of mice (selected among those of set number two in Fig. 1A): sham controls (n = 4), hyperlocomotor dopamine-depleted mice (n = 4), and dopamine-depleted mice with locomotor scores matching those of the control group (n = 4). First, the mice were challenged with amphetamine (4 mg/kg, i.p.) in the open field between PD21 and PD25 (Fig. 6A). Consistent with previous reports (Avale et al., 2004b), amphetamine produced a mild increase in horizontal activity in controls (35%; p < 0.03 vs baseline) but reduced activity by 50% and 75% in nonhyperlocomotor and hyperlocomotor mice, respectively (p < 0.007 and p < 0.001; Tukey test after significant drug × lesion interaction in the main ANOVA). Thus, rather than a specific effect in hyperactive mice, amphetamine reduced horizontal activity in all dopamine-depleted animals.
Figure 6.

Amphetamine effects on locomotor activity and corticostriatal physiology. A, Effect of amphetamine (4 mg/kg, i.p.) on horizontal locomotor activity in PD21–PD25 mice of set 2. Amphetamine increased activity in control mice (*p = 0.03) but reduced traveled distance in both hyperlocomotor (*p < 0.001) and nonhyperlocomotor (*p = 0.007) mice (Tukey post hoc after p < 0.001 animal group × drug effect interaction in a two-way ANOVA for repeated measures within independent groups; n = 4 animals per group). B, The mice were then used for electrophysiological recordings of striatal activity with multielectrode arrays between PD28 and PD32. The multielectrode array was positioned in the striatum as seen in the schematic drawings of coronal sections and photographs (DiI fluorescence over a transmitted light image) taken from a representative experiment. Bottom, Representative segment of signal obtained through the multielectrode array and recorded simultaneously with slow waves in the frontal cortex LFP under baseline (left) and amphetamine (right). C, Effect of amphetamine on the timing of striatal spikes relative to cortical rhythms, as assessed by means of the Rayleigh test and circular dispersion, in representative recordings taken before (baseline) and 15 min after amphetamine administration in a hyperlocomotor mouse. Bottom, Circular plots, Rayleigh p values, and circular dispersions corresponding to 3-min-long recordings including the signal segments illustrated above. Spike discharges were always highly correlated with the slow oscillation (wf-1). Phase locking to wf-2 and wf-3 was increased by the drug. D, Baseline recordings of spontaneous activity confirmed the main finding in mice of set 1, that is, phase locking of striatal spikes to wf-2 and wf-3 is higher in control (empty squares) than hyperlocomotor (filled squares) mice (#p < 0.05, Tukey test). Nonhyperlocomotor dopamine-depleted animals (triangles) scored in between control and hyperlocomotor mice, differing from the latter group only at the highest frequencies and only in one of the measures (*p < 0.05, Tukey test for circular dispersion). Amphetamine (Amph) strengthened the association between striatal discharges and cortical rhythms represented in wf-2 and wf-3 in hyperlocomotor mice (p < 0.05 vs baseline, Tukey test) without showing effects in control and nonhyperlocomotor mice. Statistical analysis was performed with separate two-way ANOVAs for each frequency band, with animal group and drug effect as between and within factors, where significant group × drug effect interactions (p < 0.05) allowed performing post hoc comparisons. All data are mean ± SEM.
Then, the animals were used for multielectrode striatal recordings at PD28–PD32 (Fig. 6B). These recordings cannot be readily compared with those made with glass electrodes because the multielectrode array was positioned so as to maximize the number of spontaneously active channels with a good signal-to-noise ratio and spike sorting is biased against the selection of units with very low firing rates. Having said that, the average firing rate and proportion of channels showing spontaneous activity (Table 1) were similar in the three animal groups and in general agreement with findings in the first set of mice. Moreover, confirming findings in the first set, phase locking of striatal spikes to cortical rhythms was more stringent in control than hyperlocomotor mice, across a range of frequencies extending from 2 to 10 Hz (Fig. 6C) (p < 0.05, Tukey post hoc test). Importantly, the nonhyperlocomotor dopamine-depleted animals resembled the control mice, differing from the hyperlocomotor group only at the highest frequencies (4–10 Hz; p < 0.05). Thus, the data show that mice rendered hyperlocomotor by neonatal dopamine depletion exhibit a singular pattern of corticostriatal connectivity involving a less stringent locking of striatal activity to cortical rhythms.
Table 1.
Spontaneous striatal activity in control and dopamine-depleted mice as recorded with multielectrode arrays
| Average firing rate(spikes/s) | Spontaneously active/totalnumber of channels | Units identified through sorting | |
|---|---|---|---|
| Sham controls | 1.08 ± 0.07 | 0.76 ± 0.08 | 51 |
| Hyperlocomotor dopamine-depleted | 0.92 ± 0.05 | 0.76 ± 0.07 | 59 |
| Nonhyperlocomotor dopamine-depleted | 0.96 ± 0.06 | 0.69 ± 0.07 | 54 |
Data are mean ± SEM.
As reported by others (Rebec, 2006), amphetamine induced changes in resting cortical activity and striatal firing rates (data not shown). Because our interest was to study functional coupling between striatal discharges and cortical rhythms, we selected recordings with comparable preamphetamine and postamphetamine slow-wave activity and estimated phase locking of spike discharges to different frequency bands in the cortical LFP. No effect on coupling to the lowest LFP frequencies driving UP states was observed (Fig. 6C) although amphetamine did markedly tighten phase locking to rhythms of higher frequencies in the hyperlocomotor mice (p < 0.01 vs baseline, Tukey test). Consequently, under the effect of amphetamine, entrainment of striatal firing by cortical rhythms was similar in the three animal groups.
The relative abundance of serotoninergic hyperinnervation could account for the different behavioral and electrophysiological outcomes
As dopamine-depleted and control mice belonged to the same litters and grew up together in the same cage, different degrees or patterns of dopamine depletion could result in the different behavioral and electrophysiological outcomes. To examine this possibility, instead of measuring the degree of striatal dopamine depletion (Shaywitz et al., 1976a; Miller et al., 1981; Avale et al., 2004b; Huot and Parent, 2007), we assessed the extent of the lesion in both hyperlocomotor and nonhyperlocomotor mice (Fig. 7). The neonatal lesion induced an extensive loss of TH+ neurons in the substantia nigra and a less severe but marked decrease in the ventral tegmental area. This was accompanied by severe depletion of TH immunoreactivity in the dorsal striatum and less severe reduction in the ventral striatum. Quantitative assessments of TH immunostained sections failed to reveal any differences between hyperlocomotor and nonhyperlocomotor dopamine-depleted mice, except for a slightly more marked loss of TH immunoreactivity in the medial striatum in the nonhyperlocomotor mice (Fig. 7A–C). Concerning the frontal cortex, no differences in TH or SERT immunoreactivity were found (Fig. 7D). However, as the number of sections available for immunohistochemistry was small because of the positioning of electrodes in the frontal cortex, we cannot completely rule out the occurrence of changes in rostral cortical areas.
Figure 7.
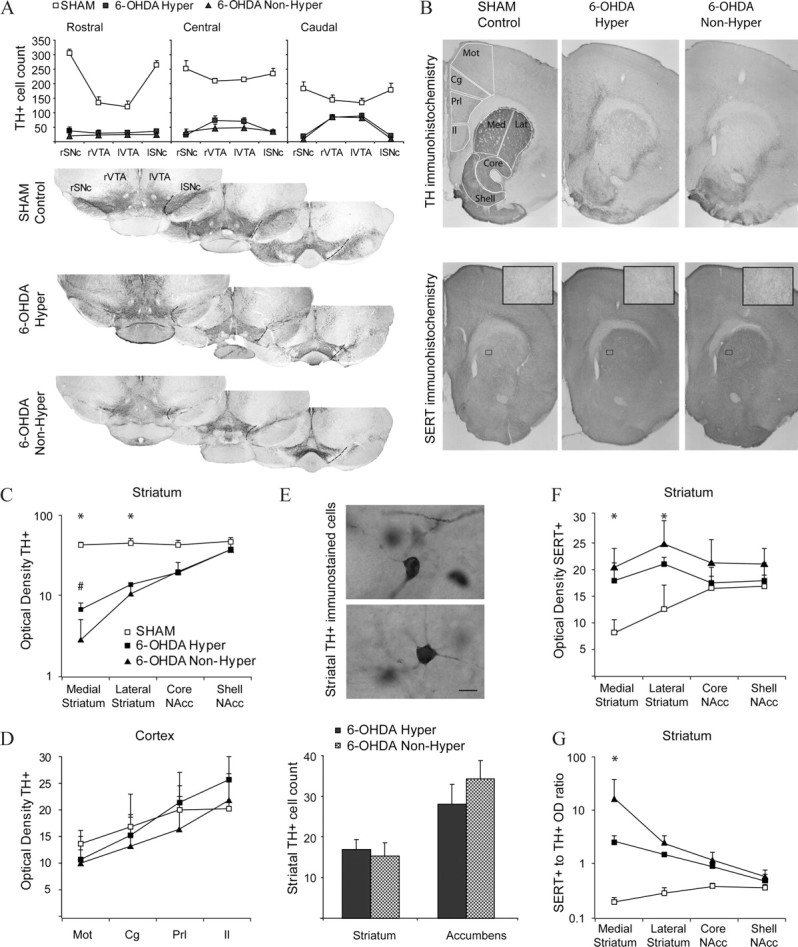
Lesion extent and compensations in hyperlocomotor and nonhyperlocomotor mice. A, TH-immunoreactive neurons remaining in the right (r) and left (l) substantia nigra (SNc) and ventral tegmental area (VTA) in sham (empty squares), hyperlocomotor (filled squares), and nonhyperlocomotor (triangles) mice. Cell counts were performed in three coronal sections per animal, as illustrated for representative animals of each group (rostral: bregma −3.00; central: bregma −3.28; caudal: bregma −3.52 mm). B, Photographs spanning the striatum and medial prefrontal cortex in a representative sham (left), hyperlocomotor (middle), and nonhyperlocomotor (right) mice, immunostained for TH (upper line) or SERT (bottom line; insets: high-magnification detail of SERT+ fibers in the striatum). Outlines illustrate the areas used for optical density measurements (Mot, motor cortex; Cg, cingulate cortex; Prl, prelimbic cortex; Il, infralimbic cortex; Med, medial striatum; Lat, lateral striatum; Core, nucleus accumbens core; Shell, nucleus accumbens shell) and cell counts (the frontal cortex was also sampled at a more rostral level, and the striatum at more caudal level; not shown for simplicity). C, TH immunohistochemistry: relative optical densities in the medial and lateral dorsal striatum and in the core and shell of nucleus accumbens, in the three animal groups. *p < 0.05 for sham versus lesioned, #p < 0.05 for nonhyperlocomotor versus any other group, Tukey post hoc within striatal region after p < 0.001 interaction in a two-way ANOVA. D, TH immunohistochemistry: relative optical densities in the motor, cingulate, prelimbic, and infralimbic frontal cortex, in the three animal groups. Measures of the area occupied by immunoreactive structures provided similar results (data not shown). E, Counts of TH+ cells in the striatum. The high intensity of TH staining in the control striatum precluded individualization of cell bodies. Scale bar, 10 μm. F, SERT immunohistochemistry: relative optical densities in the medial and lateral dorsal striatum and in the core and shell of nucleus accumbens, in the three animal groups. *p < 0.05 Tukey post hoc for sham versus any other group within the dorsal striatum, after p < 0.001 interaction in a two-way ANOVA. G, Ratio of SERT optical density to TH optical density. *p < 0.05 Tukey post hoc for any comparison within the medial striatum, after p < 0.001 interaction in a two-way ANOVA. Data are mean ± SD.
Moreover, we examined two compensatory mechanisms that follow dopamine depleting brain lesions, the appearance of TH+ cells in the striatum (Huot and Parent, 2007), and the sprouting of serotonergic fibers into the striatum (Avale et al., 2004a; Brown and Gerfen, 2006). Hyperlocomotor and nonhyperlocomotor mice exhibited similar numbers of TH+ neurons in the striatum (Fig. 7E), and a marked hyperinnervation by SERT+ fibers of the dorsal striatum (Fig. 7F) (p < 0.01, Tukey post hoc test for sham vs either group of dopamine-depleted mice, after significant region per lesion interaction in a two-way ANOVA). Finally, the dopamine-depleted nonhyperlocomotor mice showed a severalfold higher ratio of SERT-OD to TH-OD than the hyperlocomotor animals (Fig. 7G). Thus, the data suggest that subtle differences in the extent of striatal dopaminergic denervation and the resulting serotoninergic hyperinnervation could have induced the different behavioral and electrophysiological phenotypes. However, as a complete screening of all the possible compensatory mechanisms is beyond the scope of this study, it remains possible that other mechanisms could have contributed to the phenotypic differences.
Discussion
Corticostriatal functional maturation continues after infancy
In adults, medium spiny neurons filter out uncorrelated inputs and only fire in response to precisely synchronized incoming activity occurring during UP states (Wilson, 1993; O'Donnell and Grace, 1995; Charpier et al., 1999; Tseng et al., 2001; Kasanetz et al., 2006). Moreover, the topographical organization of corticostriatal projections allows information processing to occur in partially segregated channels (Alexander et al., 1986; Voorn et al., 2004; Kasanetz et al., 2008). Thus, striatal neurons encode information through spatial selectivity and spike timing relative to afferent oscillatory activity (Alexander and DeLong, 1985; West et al., 1990; Berke et al., 2004; Pennartz et al., 2004; Fujii and Graybiel, 2005). There is evidence that this capacity is shaped after birth. Morphological studies indicate that medium spiny neurons and corticostriatal synapses do not attain adult properties until adolescence (Hattori and McGeer, 1973; Adinolfi, 1977; Difiglia et al., 1980; Sharpe and Tepper, 1998; Uryu et al., 1999). Concomitantly, resting striatal activity increases until PD28 (Napier et al., 1985), which is approximately when medium spiny neurons first exhibit mature UP states (Tepper et al., 1998). We extend these observations by demonstrating that marked changes in striatal activity occur after infancy: (1) neurons showing spontaneous discharge and convergent responses to cortical inputs decrease in number; (2) the timing of spikes relative to ongoing cortical oscillations becomes more stringent. Thus, striatal neurons may encode cortical input within wider spatial and temporal frames in infants. The finding that functional corticostriatal maturation continues after infancy is in line with studies showing that cortical activity undergoes substantial changes during adolescence (Casey et al., 1997; O'Donnell et al., 2002; Tseng and O'Donnell, 2005, 2007).
Some of our findings (reduced responsiveness and convergence) could be accounted for by a contraction of striatal projection fields of cortical neurons with increasing age. After completion of coarse morphological development during infancy, further functional tuning could stem from selective pruning of synapses or changes in synaptic efficacy. Indeed, rearing conditions influence spine density in striatal neurons (Comery et al., 1995) and corticostriatal synapses undergo developmental changes in synaptic efficacy (Tang et al., 2001). However, as little attention has been paid to late corticostriatal maturation, the ultimate mechanisms underlying the protracted changes reported here remain speculative. Interestingly, the human caudate nucleus (primate homolog of rodent striatum) experiences progressive volume reduction during adolescence (Castellanos et al., 2002; Valera et al., 2007), which has been speculatively attributed to synaptic pruning (Toga et al., 2006; Swanson et al., 2007).
Corticostriatal dysfunction in hyperlocomotor mice
We hypothesized that dopamine shapes the functional maturation of corticostriatal connectivity. Similar to rats (Shaywitz et al., 1976a; Miller et al., 1981), mice with neonatal dopamine depleting lesions often display hyperlocomotor behavior that wanes after puberty and is reduced by psychostimulants (Avale et al., 2004b), and thus they have been proposed as a model of ADHD (van der Kooij and Glennon, 2007). Although dopamine neurons do not degenerate in ADHD, it is postulated that a state of functional dopamine deficiency could play a role in the disease (Biederman and Faraone, 2005; Nigg and Casey, 2005; Sagvolden et al., 2005; Swanson et al., 2007). Additionally, changes in norepinephrine in the prefrontal cortex could contribute to or even be the main dysfunction in ADHD (Nigg and Casey, 2005; Sagvolden et al., 2005; Arnsten, 2006). Although ADHD could also involve changes in several other transmitter systems, simplified models can provide insight into how single transmitter dysfunctions could contribute to this condition.
Striatal neurons from infant hyperlocomotor mice were less responsive to cortical command as revealed by the amount of current needed to elicit spike responses and the reduced tuning of striatal discharges to cortical rhythms. The fact that striatal activity was robustly modulated by slow waves despite dopamine depletion indicates that, as in normal mice, discharges were confined to UP states (Tepper et al., 1998; Kasanetz et al., 2006). Thus, striatal neurons are driven by the frontal cortex after dopamine depletion, but become less sensitive to spatially and temporally refined cortical inputs. One possibility is that corticostriatal connections have narrower projection fields in dopamine-depleted mice, explaining both why more current is needed to drive striatal responses and the reduced convergence and temporal tunelessness of spontaneous activity (as phase synchronization could depend on spatial correspondence between recording sites). Similarly, reduced connectivity within the frontal cortex could limit the spread of activity locally and contribute to the observed outcome. Thus, changes at multiple levels could have given rise to the functional changes reported here.
Importantly, siblings with very similar lesions and raised in the same cages with hyperlocomotor mice can show normal horizontal activity and corticostriatal functioning. Published studies do not provide detailed information about those animals that do not become hyperactive, which are assumed to have lesions which are either incomplete or too large. In our study, the nonhyperlocomotor phenotype was associated with a slightly stronger depletion of TH+ fibers and a relatively higher abundance of SERT over TH immunoreactivity in the dorsal striatum. However, we cannot exclude the possibility that changes in the monoaminergic innervation of frontal cortical areas, which could not be studied in enough detail in the present study, also contribute to the differences between hyperlocomotor and nonhyperlocomotor mice. Thus, our data show marked alterations in the core functional capacities of the corticostriatal system in association with a singular profile of monoaminergic innervation of the dorsal striatum in hyperlocomotor mice.
Amphetamine reduces locomotion in dopamine-depleted mice
Psychostimulants reduce hyperactivity in rodents with neonatal dopamine depletion (Shaywitz et al., 1976b; Avale et al., 2004b). However, whether this reflects selective “anti-hyperlocomotor” actions remains unclear, partly because psychostimulant effects in nonhyperlocomotor dopamine-depleted animals are seldom reported. In our experiments, amphetamine reduced horizontal activity in dopamine-depleted animals regardless of baseline locomotor scores. Together with findings showing psychostimulant calming effects in dopamine transporter knock-out mice (Gainetdinov et al., 1999), this suggests that reduced availability of presynaptic dopamine transporters unmasks amphetamine-induced hypolocomotor actions mediated by other monoamine transporters. Consistent with this, selective norepinephrine and serotonin transporter inhibitors reduce hyperactivity in neonatally dopamine-depleted rats (Davids et al., 2002).
In parallel, amphetamine increased corticostriatal tuning in hyperlocomotor mice. Although we cannot rule out amphetamine effects on spared dopamine terminals, the number of TH+ fibers remaining in the striatal region from which recordings were taken was scarce. Thus, the acute effects of amphetamine could involve other monoamine terminals. Along these lines, recent studies show that norepinephrine transmission strengthens functional connectivity within the frontal cortex (Wang et al., 2007). We favor the view that, rather than selectively reversing the effects of chronic dopamine depletion, acute amphetamine acts via parallel mechanisms located downstream of the dopamine neurons.
Corticostriatal activity shows incomplete and delayed maturation in dopamine deficiency
Hyperactivity wanes after puberty in mice with neonatal lesions of the dopamine system. In parallel, corticostriatal synchronization shows a developmental trajectory that resembles that seen in controls, changing even more markedly in lesioned mice, so that by adulthood its control levels are reached. In contrast, the density of spontaneously active sites decreases through maturation despite dopamine depletion, but remains significantly elevated compared with control animals. Other measures of corticostriatal function (responsiveness to focal inputs and convergence) evolve only in controls, but in a direction (diminution) that reduces the gap with the lesioned mice. Thus, although control and dopamine-depleted mice look more alike in adulthood, some parameters of corticostriatal function remain static or show little development after infancy in dopamine-depleted animals. Importantly, some behavioral alterations that outlast the waning of hyperactivity, like the lack of preference for the protected arms of a plus maze (Archer et al., 1988; Avale et al., 2004b), could be related to the altered maturation of corticostriatal function. Thus, compensatory capacity through serotonin or other mechanisms seems to be incomplete, allowing the persistence of functional alterations in adulthood.
Implications for ADHD
Our findings in mice with neonatal dopamine-depleting lesions parallel remarkably well some observations in ADHD, such as the delayed maturation of caudate volume (Castellanos et al., 2002), the striatal hypoactivity revealed in brain imaging studies (Rubia et al., 1999; Teicher et al., 2000; Durston, 2003), and the reduced span of corticocortical connectivity as seen in EEG studies (Murias et al., 2006). However, in addition to the etiologic validity of the mouse model, several issues remain to be addressed before our findings can be linked to ADHD. Namely, what are the relative contributions of frontal cortex and striatum, of medial and lateral corticostriatal circuits, and of other transmitter systems to ADHD-like behavior. Perhaps, hyperactivity is only one of the possible outcomes of neonatal dopamine depletion rather than a marker of a “successful” lesion. More exhaustive analysis of hyperactive and nonhyperactive animals, indexing separately impulsivity, attention, and perseverance at different ages, could reveal a spectrum of parallel electrophysiological and behavioral outcomes that evolve as the animal develops. In summary, this first study of corticostriatal functional connectivity in a putative animal model of ADHD reveals that early dopamine depletion produces dramatic long-term effects in the responsiveness of striatal neurons to stereotyped, yet physiologically meaningful, cortical commands.
Footnotes
This work was supported by National Institutes of Health Research Grant R03 TW6282 (Patricio O'Donnell, University of Maryland School of Medicine) funded by the Fogarty International Center and the National Institute on Mental Health, and the following Argentine institutions: Secretaría de Ciencia, Tecnología e Innovación Productiva, Fondo para la Investigación Científica y Tecnológica (PICT 2002 05-11012 and PICT 2007 05-1000), Universidad de Buenos Aires (UBACYT M050), and Consejo Nacional de Investigaciones Científicas y Técnicas (PIP 5890). M.G.M. was also supported by the J. S. Guggenheim Foundation. We thank Dr. Kuei Tseng (Rosalind Franklin University, Chicago Medical School) and the anonymous reviewers for helpful comments on an early version of this manuscript, Elena Avale and Marcelo Rubinstein (Instituto de Investigaciones en Ingeniería Genética y Biología Molecular–Consejo Nacional de Investigaciones Científicas y Técnicas) for help with the implementation of lesion procedures in our lab and helpful discussions, Luis Riquelme (Universidad de Buenos Aires) for advice about signal analysis, Carola Rodriguez (Universidad de Buenos Aires) for help with animal care, and James McCutcheon for checking the use of language.
References
- Adinolfi AM. The postnatal development of the caudate nucleus: a Golgi and electron microscopic study of kittens. Brain Res. 1977;133:251–266. doi: 10.1016/0006-8993(77)90762-4. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR. Microstimulation of the primate neostriatum. II. Somatotopic organization of striatal microexcitable zones and their relation to neuronal response properties. J Neurophysiol. 1985;53:1417–1430. doi: 10.1152/jn.1985.53.6.1417. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Archer T, Danysz W, Fredriksson A, Jonsson G, Luthman J, Sundström E, Teiling A. Neonatal 6-hydroxydopamine-induced dopamine depletions: motor activity and performance in maze learning. Pharmacol Biochem Behav. 1988;31:357–364. doi: 10.1016/0091-3057(88)90358-9. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Stimulants: therapeutic actions in ADHD. Neuropsychopharmacology. 2006;31:2376–2383. doi: 10.1038/sj.npp.1301164. [DOI] [PubMed] [Google Scholar]
- Avale ME, Nemirovsky SI, Raisman-Vozari R, Rubinstein M. Elevated serotonin is involved in hyperactivity but not in the paradoxical effect of amphetamine in mice neonatally lesioned with 6-hydroxydopamine. J Neurosci Res. 2004a;78:289–296. doi: 10.1002/jnr.20245. [DOI] [PubMed] [Google Scholar]
- Avale ME, Falzone TL, Gelman DM, Low MJ, Grandy DK, Rubinstein M. The dopamine D4 receptor is essential for hyperactivity and impaired behavioral inhibition in a mouse model of attention deficit/hyperactivity disorder. Mol Psychiatry. 2004b;9:718–726. doi: 10.1038/sj.mp.4001474. [DOI] [PubMed] [Google Scholar]
- Berke JD, Okatan M, Skurski J, Eichenbaum HB. Oscillatory entrainment of striatal neurons in freely moving rats. Neuron. 2004;43:883–896. doi: 10.1016/j.neuron.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366:237–248. doi: 10.1016/S0140-6736(05)66915-2. [DOI] [PubMed] [Google Scholar]
- Brown P, Gerfen CR. Plasticity within striatal direct pathway neurons after neonatal dopamine depletion is mediated through a novel functional coupling of serotonin 5-HT2 receptors to the ERK 1/2 map kinase pathway. J Comp Neurol. 2006;498:415–430. doi: 10.1002/cne.21034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno JP, Sandstrom MI, Arnold HM, Nelson CL. Age-dependent neurobehavioral plasticity following forebrain dopamine depletions. Dev Neurosci. 1998;20:164–179. doi: 10.1159/000017311. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Trainor RJ, Orendi JL, Schubert AB, Nystrom LE, Giedd JN, Castellanos FX, Haxby JV, Noll DC, Cohen JD, Forman SD, Dahl RE, Rapoport JL. A developmental functional MRI study of prefrontal activation during performance of a Go-No-Go task. J Cogn Neurosci. 1997;9:835–847. doi: 10.1162/jocn.1997.9.6.835. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Lee PP, Sharp W, Jeffries NO, Greenstein DK, Clasen LS, Blumenthal JD, James RS, Ebens CL, Walter JM, Zijdenbos A, Evans AC, Giedd JN, Rapoport JL. Developmental trajectories of brain volume abnormalities in children and adolescents with attention-deficit/hyperactivity disorder. JAMA. 2002;288:1740–1748. doi: 10.1001/jama.288.14.1740. [DOI] [PubMed] [Google Scholar]
- Charpier S, Mahon S, Deniau JM. In vivo induction of striatal long-term potentiation by low-frequency stimulation of the cerebral cortex. Neuroscience. 1999;91:1209–1222. doi: 10.1016/s0306-4522(98)00719-2. [DOI] [PubMed] [Google Scholar]
- Comery TA, Shah R, Greenough WT. Differential rearing alters spine density on medium-sized spiny neurons in the rat corpus striatum: evidence for association of morphological plasticity with early response gene expression. Neurobiol Learn Mem. 1995;63:217–219. doi: 10.1006/nlme.1995.1025. [DOI] [PubMed] [Google Scholar]
- Davids E, Zhang K, Kula NS, Tarazi FI, Baldessarini RJ. Effects of norepinephrine and serotonin transporter inhibitors on hyperactivity induced by neonatal 6-hydroxydopamine lesioning in rats. J Pharmacol Exp Ther. 2002;301:1097–1102. doi: 10.1124/jpet.301.3.1097. [DOI] [PubMed] [Google Scholar]
- DeFrance JF, Sikes RW, Chronister RB. Dopamine action in the nucleus accumbens. J Neurophysiol. 1985;54:1568–1577. doi: 10.1152/jn.1985.54.6.1568. [DOI] [PubMed] [Google Scholar]
- Difiglia M, Pasik P, Pasik T. Early postnatal development of the monkey neostriatum: a Golgi and ultrastructural study. J Comp Neurol. 1980;190:303–331. doi: 10.1002/cne.901900207. [DOI] [PubMed] [Google Scholar]
- Durston S. A review of the biological bases of ADHD: what have we learned from imaging studies? Ment Retard Dev Disabil Res Rev. 2003;9:184–195. doi: 10.1002/mrdd.10079. [DOI] [PubMed] [Google Scholar]
- Durston S, Fossella JA, Casey BJ, Hulshoff Pol HE, Galvan A, Schnack HG, Steenhuis MP, Minderaa RB, Buitelaar JK, Kahn RS, van Engeland H. Differential effects of DRD4 and DAT1 genotype on fronto-striatal gray matter volumes in a sample of subjects with attention deficit hyperactivity disorder, their unaffected siblings, and controls. Mol Psychiatry. 2005;10:678–685. doi: 10.1038/sj.mp.4001649. [DOI] [PubMed] [Google Scholar]
- Fisher NI. Statistical analysis of circular data. New York: Cambridge UP; 1993. [Google Scholar]
- Fowler SC, Birkestrand BR, Chen R, Moss SJ, Vorontsova E, Wang G, Zarcone TJ. A force-plate actometer for quantitating rodent behaviors: illustrative data on locomotion, rotation, spatial patterning, stereotypies, and tremor. J Neurosci Methods. 2001;107:107–124. doi: 10.1016/s0165-0270(01)00359-4. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Fujii N, Graybiel AM. Time-varying covariance of neural activities recorded in striatum and frontal cortex as monkeys perform sequential-saccade tasks. Proc Natl Acad Sci U S A. 2005;102:9032–9037. doi: 10.1073/pnas.0503541102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 1999;283:397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- Hammond C, Bergman H, Brown P. Pathological synchronization in Parkinson's disease: networks, models and treatments. Trends Neurosci. 2007;30:357–364. doi: 10.1016/j.tins.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Hattori T, McGeer PL. Synaptogenesis in the corpus striatum of infant rat. Exp Neurol. 1973;38:70–79. doi: 10.1016/0014-4886(73)90008-3. [DOI] [PubMed] [Google Scholar]
- Huot P, Parent A. Dopaminergic neurons intrinsic to the striatum. J Neurochem. 2007;101:1441–1447. doi: 10.1111/j.1471-4159.2006.04430.x. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Riquelme LA, O'Donnell P, Murer MG. Turning off cortical ensembles stops striatal Up states and elicits phase perturbations in cortical and striatal slow oscillations in rat in vivo. J Physiol. 2006;577:97–113. doi: 10.1113/jphysiol.2006.113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasanetz F, Riquelme LA, Della-Maggiore V, O'Donnell P, Murer MG. Functional integration across a gradient of corticostriatal channels controls UP state transitions in the dorsal striatum. Proc Natl Acad Sci U S A. 2008;105:8124–8129. doi: 10.1073/pnas.0711113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Stimulant actions in rodents: implications for attention-deficit/hyperactivity disorder treatment and potential substance abuse. Biol Psychiatry. 2005;57:1391–1396. doi: 10.1016/j.biopsych.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Luthman J, Friedemann M, Bickford P, Olson L, Hoffer BJ, Gerhardt GA. In vivo electrochemical measurements and electrophysiological studies of rat striatum following neonatal 6-hydroxydopamine treatment. Neuroscience. 1993;52:677–687. doi: 10.1016/0306-4522(93)90416-d. [DOI] [PubMed] [Google Scholar]
- Mahon S, Deniau JM, Charpier S. Relationship between EEG potentials and intracellular activity of striatal and cortico-striatal neurons: an in vivo study under different anesthetics. Cereb Cortex. 2001;11:360–373. doi: 10.1093/cercor/11.4.360. [DOI] [PubMed] [Google Scholar]
- Mahon S, Vautrelle N, Pezard L, Slaght SJ, Deniau JM, Chouvet G, Charpier S. Distinct patterns of striatal medium spiny neuron activity during the natural sleep–wake cycle. J Neurosci. 2006;26:12587–12595. doi: 10.1523/JNEUROSCI.3987-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuo Y, Ishido M, Morita M, Oka S, Niki E. Motor activity and gene expression in rats with neonatal 6-hydroxydopamine lesions. J Neurochem. 2004;91:9–19. doi: 10.1111/j.1471-4159.2004.02615.x. [DOI] [PubMed] [Google Scholar]
- Miller FE, Heffner TG, Kotake C, Seiden LS. Magnitude and duration of hyperactivity following neonatal 6-hydroxydopamine is related to the extent of brain dopamine depletion. Brain Res. 1981;229:123–132. doi: 10.1016/0006-8993(81)90750-2. [DOI] [PubMed] [Google Scholar]
- Montague DM, Lawler CP, Mailman RB, Gilmore JH. Developmental regulation of the dopamine D1 receptor in human caudate and putamen. Neuropsychopharmacology. 1999;21:641–649. doi: 10.1016/S0893-133X(99)00062-7. [DOI] [PubMed] [Google Scholar]
- Murias M, Swanson JM, Srinivasan R. Functional connectivity of frontal cortex in healthy and ADHD children reflected in EEG coherence. Cereb Cortex. 2007;17:1788–1799. doi: 10.1093/cercor/bhl089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier TC, Coyle S, Breese GR. Ontogeny of striatal unit activity and effects of single or repeated haloperidol administration in rats. Brain Res. 1985;333:35–44. doi: 10.1016/0006-8993(85)90121-0. [DOI] [PubMed] [Google Scholar]
- Nigg JT, Casey BJ. An integrative theory of attention-deficit/hyperactivity disorder based on the cognitive and affective neurosciences. Dev Psychopathol. 2005;17:785–806. doi: 10.1017/S0954579405050376. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell P, Lewis BL, Weinberger DR, Lipska BK. Neonatal hippocampal damage alters electrophysiological properties of prefrontal cortical neurons in adult rats. Cereb Cortex. 2002;12:975–982. doi: 10.1093/cercor/12.9.975. [DOI] [PubMed] [Google Scholar]
- Penn AA, Shatz CJ. Brain waves and brain wiring: the role of endogenous and sensory-driven neural activity in development. Pediatr Res. 1999;45:447–458. doi: 10.1203/00006450-199904010-00001. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Lee E, Verheul J, Lipa P, Barnes CA, McNaughton BL. The ventral striatum in off-line processing: ensemble reactivation during sleep and modulation by hippocampal ripples. J Neurosci. 2004;24:6446–6456. doi: 10.1523/JNEUROSCI.0575-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga RQ, Nadasdy Z, Ben-Shaul Y. Unsupervised spike detection and sorting with wavelets and superparamagnetic clustering. Neural Comput. 2004;16:1661–1687. doi: 10.1162/089976604774201631. [DOI] [PubMed] [Google Scholar]
- Rebec GV. Behavioral electrophysiology of psychostimulants. Neuropsychopharmacology. 2006;31:2341–2348. doi: 10.1038/sj.npp.1301160. [DOI] [PubMed] [Google Scholar]
- Rolls ET, Thorpe SJ, Boytim M, Szabo I, Perrett DI. Responses of striatal neurons in the behaving monkey. 3. Effects of iontophoretically applied dopamine on normal responsiveness. Neuroscience. 1984;12:1201–1212. doi: 10.1016/0306-4522(84)90014-9. [DOI] [PubMed] [Google Scholar]
- Rubia K, Overmeyer S, Taylor E, Brammer M, Williams SC, Simmons A, Bullmore ET. Hypofrontality in attention deficit hyperactivity disorder during higher-order motor control: a study with functional MRI. Am J Psychiatry. 1999;156:891–896. doi: 10.1176/ajp.156.6.891. [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Johansen EB, Aase H, Russell VA. A dynamic developmental theory of attention-deficit/hyperactivity disorder (ADHD) predominantly hyperactive/impulsive and combined subtypes. Behav Brain Sci. 2005;28:397–419. doi: 10.1017/S0140525X05000075. discussion 419–468. [DOI] [PubMed] [Google Scholar]
- Seeman P, Bzowej NH, Guan HC, Bergeron C, Becker LE, Reynolds GP, Bird ED, Riederer P, Jellinger K, Watanabe S, Tourtellotte WW. Human brain dopamine receptors in children and aging adults. Synapse. 1987;1:399–404. doi: 10.1002/syn.890010503. [DOI] [PubMed] [Google Scholar]
- Sharpe NA, Tepper JM. Postnatal development of excitatory synaptic input to the rat neostriatum: an electron microscopic study. Neuroscience. 1998;84:1163–1175. doi: 10.1016/s0306-4522(97)00583-6. [DOI] [PubMed] [Google Scholar]
- Shaywitz BA, Yager RD, Klopper JH. Selective brain dopamine depletion in developing rats: an experimental model of minimal brain dysfunction. Science. 1976a;191:305–308. doi: 10.1126/science.942800. [DOI] [PubMed] [Google Scholar]
- Shaywitz BA, Klopper JH, Yager RD, Gordon JW. Paradoxical response to amphetamine in developing rats treated with 6-hydroxydopamine. Nature. 1976b;261:153–155. doi: 10.1038/261153a0. [DOI] [PubMed] [Google Scholar]
- Siapas AG, Lubenov EV, Wilson MA. Prefrontal phase locking to hippocampal theta oscillations. Neuron. 2005;46:141–151. doi: 10.1016/j.neuron.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Stamford JA. Development and ageing of the rat nigrostriatal dopamine system studied with fast cyclic voltammetry. J Neurochem. 1989;52:1582–1589. doi: 10.1111/j.1471-4159.1989.tb09212.x. [DOI] [PubMed] [Google Scholar]
- Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101:243–276. doi: 10.1016/s0306-4522(00)00353-5. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Kinsbourne M, Nigg J, Lanphear B, Stefanatos GA, Volkow N, Taylor E, Casey BJ, Castellanos FX, Wadhwa PD. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol Rev. 2007;17:39–59. doi: 10.1007/s11065-007-9019-9. [DOI] [PubMed] [Google Scholar]
- Tang K, Low MJ, Grandy DK, Lovinger DM. Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc Natl Acad Sci U S A. 2001;98:1255–1260. doi: 10.1073/pnas.031374698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher MH, Andersen SL, Hostetter JC., Jr Evidence for dopamine receptor pruning between adolescence and adulthood in striatum but not nucleus accumbens. Brain Res Dev Brain Res. 1995;89:167–172. doi: 10.1016/0165-3806(95)00109-q. [DOI] [PubMed] [Google Scholar]
- Teicher MH, Anderson CM, Polcari A, Glod CA, Maas LC, Renshaw PF. Functional deficits in basal ganglia of children with attention-deficit/hyperactivity disorder shown with functional magnetic resonance imaging relaxometry. Nat Med. 2000;6:470–473. doi: 10.1038/74737. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Sharpe NA, Koós TZ, Trent F. Postnatal development of the rat neostriatum: electrophysiological, light- and electron-microscopic studies. Dev Neurosci. 1998;20:125–145. doi: 10.1159/000017308. [DOI] [PubMed] [Google Scholar]
- Toga AW, Thompson PM, Sowell ER. Mapping brain maturation. Trends Neurosci. 2006;29:148–159. doi: 10.1016/j.tins.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Post-pubertal emergence of prefrontal cortical up states induced by D1-NMDA co-activation. Cereb Cortex. 2005;15:49–57. doi: 10.1093/cercor/bhh107. [DOI] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex. 2007;17:1235–1240. doi: 10.1093/cercor/bhl034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Kasanetz F, Kargieman L, Riquelme LA, Murer MG. Cortical slow oscillatory activity is reflected in the membrane potential and spike trains of striatal neurons in rats with chronic nigrostriatal lesions. J Neurosci. 2001;21:6430–6439. doi: 10.1523/JNEUROSCI.21-16-06430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Butler AK, Chesselet MF. Synaptogenesis and ultrastructural localization of the polysialylated neural cell adhesion molecule in the developing striatum. J Comp Neurol. 1999;405:216–232. doi: 10.1002/(sici)1096-9861(19990308)405:2<216::aid-cne6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Valera EM, Faraone SV, Murray KE, Seidman LJ. Meta-analysis of structural imaging findings in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2007;61:1361–1369. doi: 10.1016/j.biopsych.2006.06.011. [DOI] [PubMed] [Google Scholar]
- van der Kooij MA, Glennon JC. Animal models concerning the role of dopamine in attention-deficit hyperactivity disorder. Neurosci Biobehav Rev. 2007;31:597–618. doi: 10.1016/j.neubiorev.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Ding YS. Imaging the effects of methylphenidate on brain dopamine: new model on its therapeutic actions for attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1410–1415. doi: 10.1016/j.biopsych.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM. Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci. 2004;27:468–474. doi: 10.1016/j.tins.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- West MO, Carelli RM, Pomerantz M, Cohen SM, Gardner JP, Chapin JK, Woodward DJ. A region in the dorsolateral striatum of the rat exhibiting single-unit correlations with specific locomotor limb movements. J Neurophysiol. 1990;64:1233–1246. doi: 10.1152/jn.1990.64.4.1233. [DOI] [PubMed] [Google Scholar]
- Wilson CJ. The generation of natural firing patterns in neostriatal neurons. Prog Brain Res. 1993;99:277–297. doi: 10.1016/s0079-6123(08)61352-7. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Groves PM. Spontaneous firing patterns of identified spiny neurons in the rat neostriatum. Brain Res. 1981;220:67–80. doi: 10.1016/0006-8993(81)90211-0. [DOI] [PubMed] [Google Scholar]
- Zold CL, Ballion B, Riquelme LA, Gonon F, Murer MG. Nigrostriatal lesion induces D2-modulated phase-locked activity in the basal ganglia of rats. Eur J Neurosci. 2007;25:2131–2144. doi: 10.1111/j.1460-9568.2007.05475.x. [DOI] [PubMed] [Google Scholar]