

Abstract
Background:
Abnormal neuronal inclusions composed of the TAR DNA binding protein 43 (TDP-43) are the characteristic neuropathological lesions in sporadic and familial forms of amyotrophic lateral sclerosis (ALS). This makes TARDBP, the gene encoding for TDP-43, an interesting candidate gene for genetic screening in ALS.
Objective:
To investigate the presence and frequency of TARDBP mutations in ALS.
Design:
Genetic analysis
Participants:
One hundred thirty-four patiens with sporadic ALS, 31 patients with familial non-SOD1-ALS, and 400 healthy control subjects.
Results:
We identified two missense mutations in TARDBP (G348C and the novel N352S) in two small kindreds with a hereditary form of ALS with early spinal onset resulting in fatal respiratory insufficiency without clinical relevant bulbar symptoms or signs of cognitive impairment. The mutations located in the C-terminus of TDP-43 were absent in 400 Caucasian control individuals. The novel identified N352S mutation is predicted to increase TDP-43 phosphorylation while the G348C mutation might interfere with normal TDP-43 function by forming intermolecular disulfide bridges.
Conclusion:
Mutations in TARDBP are a rare cause of familial non-SOD1-ALS. The identification of TARDBP mutations provides strong evidence for a direct link between TDP-43 dysfunction and neurodegeneration in ALS.
Keywords: TDP-43, ALS, TARDBP
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder leading to degeneration of upper and lower motor neurons in the brain and spinal cord. Clinical hallmarks are spasticity, brisk tendon reflexes, pyramidal signs and progressive atrophy and weakness of the skeletal musculature. The patients die within a few years after onset, usually from respiratory failure 1. Most cases are sporadic (sALS), but about 10% are familial (fALS). About 15-20% of the autosomal dominant fALS patients show mutations in the superoxide dismutase gene (SOD1) 2 while mutations in other genes including sentaxin 3, dynactin 1 4, and vesicle-associated membrane protein B 5 are described as rare causes of fALS.
Recently, TAR-DNA binding protein 43 (TDP-43) was identified as major disease protein in the abnormal inclusions in neurons and glial cells in sALS and fALS, with the exception of fALS forms due to SOD1 mutations6,7. TDP-43 is a highly conserved 414-amino acid nuclear protein first cloned as a protein capable of binding to the transactive response DNA element of human immunodeficiency virus type 1 and later identified as part of a complex involved in splicing of the cystic fibrosis transmembrane conductance regulator gene 8,9. TDP-43 contains two RNA recognition motifs and a glycine-rich C-terminal region. Described functions include involvement in transcription regulation, exon skipping and a role as scaffold for nuclear bodies through an interaction with survival motor neuron protein10. These findings make TARDBP, the gene on chromosome 1p36.22 encoding TDP-43, an auspicious candidate gene for fALS. Indeed very recently, while this study was in progress, 13 different mutations in TARDBP have been reported in fALS (G290A, G298S, A315T, M337V, A382T) and sALS cases (D169G, G287S, G294A, Q331K, G348C, R361S, N390D, N390S)11,12,13,14. In this study we present the genetic analysis data on TARDBP in a German cohort of 134 sALS cases and 31 index patients with non-SOD1-fALS in order to further define the spectrum and frequency of TARDBP mutations. While no mutations were found in sALS, two heterozygous missense mutations, G348C and the novel N352S mutation were identified in fALS. The occurrence of TARDBP mutations in 6.4 % of our cohort of non-SOD1-fALS does not only underline the direct role of TDP-43 dysfunction and pathogenesis in ALS, but implicates that screening for TARDBP mutations should be considered in all non-SOD1-fALS cases.
Material and Methods
Subjects
DNA from 134 patients with sALS (mean age at onset 57.7±11.9 years) and 31 index patients with SOD1-negative fALS (mean age at onset 46.5±13.2 years) were included in the study. All patients were neurologically examined at the Department of Neurology, University of Ulm and diagnosed as probable or definite ALS according to El Escorial criteria15. Additional family members of the two families with identified TARDBP mutations were tested. DNA sampling and genetic analysis was approved by the local Ethics committee. Written informed consent for genetic analysis was obtained from each individual. In all fALS cases, mutations in SOD1, DCNT1 and VAPB were excluded prior to their inclusion in the present study.
Control samples were obtained from the following sources: 276 controls from the Coriell Institute (Neurologically Normal Caucasian control panels, Camden, NJ, mean age 70), 63 clinical controls from the Alzheimer Disease Center at UPenn (n=47, mean age 76) or from University of Ulm (n=16, mean age 49) and 61 brain autopsy samples without evidence of neurodegenerative diseases from UPenn (n=41, mean age 69) or from ZNP (n=20, mean age 71).
Genetic analysis
Genomic DNA was extracted from blood or frozen brain using standard procedures. The coding region of TARDBP, Exons 2-5 and the first 528 nucleotides of exon 6 were amplified by PCR using primers from adjacent intronic/noncoding regions. PCR products were sequenced using the BigDye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) and were run on an ABI3130 capillary sequencer (Applied Biosystems, Foster City, CA).
SNP genotyping of TARDBP variants
A total of 400 control samples were analyzed for TARDBP variants NM_007375: c.1176G>T (p.G348C) and c.1189A>G (p.N352S) by a TaqMan chemistry-based allelic discrimination assay with “Assay by Design” (Applied Biosystems, Foster City, CA) probes on an Applied Biosystems 7900 followed by analysis with Sequence Detection System 2.2.1 software (Applied Biosystems) or by sequencing.
Results
The genetic analysis of 31 index patients from families with ALS led to the identification of two heterozygous missense mutations in exon 6 of TARDBP (G348C, N352S) in two small German kindreds. Clinical information on family members of both kindreds is summarized in Table 1.
Table 1.
Clinical features of TARDBP mutation families
| Age at onset (y) |
Disease duration (y) |
Site of onset |
Disease course | Bulbar involvement |
Cognitive impairment |
Electrophysiology (age performed) |
|
|---|---|---|---|---|---|---|---|
| Family A (G348C) |
|||||||
| II/1 / F | 31 | 13 | Right distal upper extremity |
Progressive, asymmetric, flaccid tetraparesis, slow progression, arms before legs; gait disturbance, wheelchair-dependent after 6 years, death from respiratory failure |
No | No | NA |
| III/1 / F | 55 | 3 | Right distal upper extremity |
Progressive, asymmetric, flaccid tetraparesis, arms before legs; wheelchair- dependent, death from respiratory failure |
No | No |
NCV: Normal SNAP amplitudes, normal sensory and motor conduction times (56) EMG: PSW/fibrillations in arms and legs; elongated, large motor unit potentials (56) MEP: normal to M. tibialis ant. and M. abductor digiti quinti (56) |
|
Family B (N352S) |
|||||||
| II/1 / F | 68 | 4 | Right distal upper extremity |
Progressive, asymmetric, flaccid tetraparesis; arms before legs, gait disturbance, but preserved ability to walk, death from respiratory failure |
No | No | NA |
| II/3 / F | 50 | 4 | Distal upper extremity |
Progressive, asymmetric, flaccid tetraparesis; arms before legs, wheelchair- dependent, death from respiratory failure |
No | No | NA |
| III/1 / F | 40 | 7* | Right distal upper extremity |
Progressive, asymmetric, flaccid tetraparesis; arms before legs, wheelchair- dependent after 4 years, respiratory insufficiency, clinically stable since initiation of NIV |
No | No |
NCV: Normal SNAP amplitudes, normal sensory and motor conduction times (40) EMG: PSW/fibrillations in arms and legs; elongated, large motor unit potentials (40) MEP: normal to M. tibialis ant and M. abductor digiti quinti (46) |
Abbreviations: F, female; y, years; NIV, NIV, non-invasive ventilation, SNAP, sensory nerve action potential; PSW, positive sharp waves; NCV, nerve conductance velocity; EMG, electromyography; MEP, motor evoked potentials
patient alive
The mutations were absent in 400 control samples. Except for a synonymous mutation at amino acid 66 (A66A) in one sALS case, no variants were detected in TARDBP in the other 133 sALS cases or controls (n=36) were all exons were sequenced.
Family A
The G348C mutation was found in the index patient (III/1) of family A (Fig. 1A and B). She presented with pareses of her right hand at the age of 55 years which spread to proximal muscles and to the opposite arm and both lower limbs leaving her wheelchair-dependent after two and a half years. Electromyography showed acute and chronic changes in distal and proximal muscles of upper and lower limbs. CSF and routine blood parameters as well as brain and spine MRI were normal. She died from respiratory insufficiency three years after disease onset. The daughter (IV/1) of the index patient is currently 38 years old and healthy. The proband's mother (II/1) presented with a progressive lower motor neuron disease (MND) at the age of 31 years. The disease was classified as multiple sclerosis, though she had only slowly progressive motor symptoms. The site of onset was the right hand. Over the course of her disease she progressed to have a gait disturbance. After 5 years she suffered from an asymmetric tetraparesis and one year later she was wheelchair-dependent. She died after a disease course of 13 years from respiratory insufficiency. Only limited information is available about the proband's grandfather (I/1). He was wheelchair-dependend in the last few years of his life and died at the age of 54 years. His wife (I/2) died early at the age of 45 years without clinical signs of MND or dementia. The proband has two siblings currently 67 (III/2) and 64 (III/3) years old. DNA was available from the non-affected III/3 family member, who did not show the G348C mutation. DNA from other affected or unaffected older family members was not available. No autopsy was performed in the deceased family members.
Figure 1.
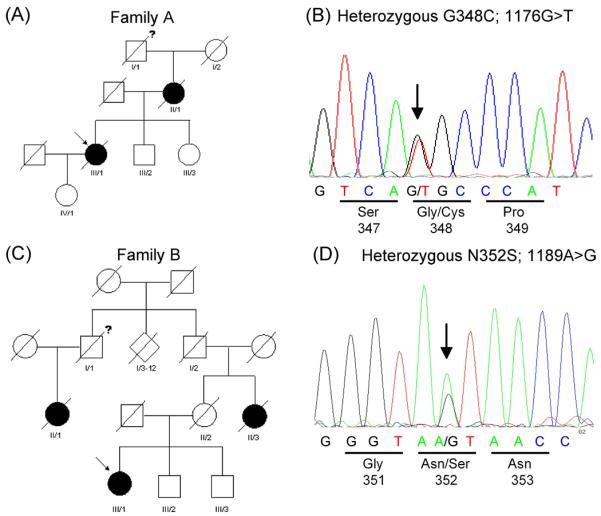
(A) Pedigree diagram of family A with the G348C mutation in TARDBP. (B) Chromatograms of part of exon 6 of the TARDBP gene showing G348C mutation in the index patient.
(C) Pedigree diagram of family B with the N352S mutation in TARDBP. (D) Chromatograms of part of exon 6 of the TARDBP gene showing N352S mutation in the index patient.
Filled symbols indicate affected individuals, “?” indicates possibly affected subjects. Arrow indicates index patients.
Family B
The N352S mutation was found in the index patient of family B (Fig. 1 C and D). This subject (III/1) showed first clinical signs at the age of 40 years with impairment of the fine motor skills of the right hand. Motor and sensory nerve conduction was normal, but electromyography showed acute and chronic changes in distal and proximal muscles of upper and lower limbs. CSF, routine blood parameters as well as brain and cervical MRI were normal. Non-invasive ventilation was necessary four years after disease onset. After 5 years she developed severe tetraparesis and became wheelchair-dependent. No bulbar impairment, autonomic dysfunctions or incontinence of bladder or bowel are present at the time of this analysis, 7 years after onset. There are no subjective complaints or evident cognitive deficits recognized during interviews, however no formal neuropsychologic testing was performed. The index patient's mother (II/2) showed no clinical signs of MND or dementia until she died at the age of 72 years from stroke. However, the index patient's aunt (II/3) had MND with onset in the distal upper limbs at an age of about 50 years. Over the course of 3-4 years, severe tetraparesis developed and she died from respiratory insufficiency. The proband's grandfather (I/2) died at the age of 80 years due to stroke without clinical signs of MND. The grandmother died at the age of 65 years due to heart failure. I/2 had 11 siblings born between 1890 and 1910. Many of these siblings died early of unknown reasons. MND was reported for one woman (II/1) whose father (I/1) was a brother of I/2.
She died at the age of 72 years of respiratory insufficiency after a 3-4 years course of progressive MND with spinal onset. Her father, I/1, died early at the age of 30 years without clinical signs of MND. III/2 and III/3 are brothers of the index patient, currently 56 and 52 years old and not reporting signs of MND. DNA was only available from III/3. He carries the N352S mutation, however, due to the current age of this subject in the range of disease-onset in this family this data is uninformative. No autopsy was performed in deceased individuals.
Predicted effects of G348C and N352S mutations
Both amino acid exchanges affect highly conserved residues of TDP-43 (Fig. 2A). Glycine at codon 348 is fully conserved in TDP-43 across the phylogenic spectrum from Homo sapiens to Danio rerio, whereas asparagine at codon 352 is conserved across all examined mammals and Gallus gallus.
Figure 2.

(A) Sequence alignment of amino acids 340-360 of TDP-43 from diverse vertebrate species. Mutation sites are bold.
(B) Effects of TARDBP mutation G348C and N352S on protein structure and function predicted using PolyPhen software program and on phosphorylation site prediction using NetPhos 2.0 program. *The lower the score, the more benign the substitution. ** The higher the score, the higher the probability for phosphorylation. Phopshorylation sites for predicted scores are bold.
Using the PolyPhen software program (http://coot.embl.de/PolPhen/) to predict effects on protein structure or function, the G348C mutation was predicted to have a probably damaging function whereas no deleterious effect was predicted for the N352S mutation (Fig. 2B). NetPhos2.0 was utilized to predict putative effects on phosphorylation sites (http://www.cbs.dtu.dk/services/NetPhos). The N352S mutations does not only introduce a new serine residue at position 352 but also leads to an increased phosphorylation prediction score for serine residues at position 347 and 350 of TDP-43 (Fig. 2B).
Discussion
We report two kindreds with a familial form of ALS with autosomal dominant inheritance due to the G348C and the novel N352S missense mutations in the TARDBP gene. These mutations were not found in 400 control samples and not reported in >1000 Caucasian controls sequenced in other reports 12,14. Since no DNA was available from other affected family members of either kindred, we cannot definitively prove that the mutations co-segregate with the disease in the families. However, as discussed below, their critical locations and predicted functional changes, together with the family history and their absence in numerous control samples strongly support the idea that both mutations are pathogenic.
The clinical phenotype in both families with spinal onset and predominance of lower motor neuron signs with absence of bulbar signs or evidence for cognitive impairment is in accordance with previous findings reported in TARDBP mutation cases. So far, spinal onset is described in 77% of TARDBP mutation cases, in 39% LMN signs were predominant and absence of cognitive impairment is a consistent finding11,12,13,14. However, this clinical phenotype does not allow separating TARDBP mutation cases from other forms of ALS, with similar features being reported in sALS and SOD-1 fALS 16,17.
Disease onset in our two kindreds is within the range of described disease onset of 30-83 years among other reported TARDBP mutation cases 11,12,13,14. However, it is notably that the G348C mutation, which has also been described very recently in a single sALS case 14, leads to the earliest disease onset among all TARDBP mutation cases with onset at 31 years (this study) and 30 years 14, suggesting that this amino acid exchange evokes severe functional changes of TDP-43.
Interestingly, with the exception of the D169G mutation, all other TARDBP gene mutations, including the G348C and the novel N352S mutation reported in this study, are located in exon 6 encoding for the C-terminus of TDP-43. The importance of the glycine-rich C-terminal domain of TDP-43 in mediating its exon skipping and splicing inhibitory ability has been demonstrated and has been observed to correlate with its ability to interact with other members of the hnRNP A/B protein families with well known splicing inhibitory properties 10.
The introduction of a cysteine residue due to the G348C mutation in the C-terminal region of TDP-43 is predicted to affect protein function (Fig. 2B and 14) possibly by the formation of intermolecular disulfide bridges which might interfere with protein-protein interaction or increase the aggregation tendency of TDP-43. The most likely effect of the N352S mutation is increased TDP-43 phosphorylation. This might lead to impaired nuclear-cytoplasmic transport and/or protein-protein interaction thereby leading to TDP-43 accumulation. Abnormal phosphorylation of TDP-43, either by introducing new threonine or serine residues or increasing the probability of phosphorylation of adjacent serine sites, is discussed as putative effect for several other TARDBP mutations 12,13,14.
So far, the functional analysis of TARDBP mutations are limited and need to be investigated in detail in future studies including the generation of transgenic animal models. However, preliminary functional data on the M337V and Q331K mutation suggest, that the mutated TDP-43 might fragment more readily and leads to increased apoptotic cell death in chick embryos 12 or as reported for the G348C, R361S and N390D mutation might lead to increased aggregation properties of TDP-43 14.
In summary, the identification of two kindreds with fALS due to TARDBP mutations, including the novel N352S mutation, extends the spectrum of TARDBP mutations. Moreover, the occurrence of TARDBP mutations in 6.4% (2/31) of our non-SOD1-fALS cohort, similar to described frequencies of 5.1% in another study 13 does not only underline the direct role of TDP-43 dysfunction and neurodegeneration in ALS but also implicates that screening for TARDBP mutations should be considered in all non-SOD1-fALS cases.
Acknowledgement
This work was funded by the Friedrich-Baur Stiftung (grant 0017/2007) and the National Institute of Health (AG 17586, AG 10124).
References
- 1.Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet. 2007;369(9578):2031–2041. doi: 10.1016/S0140-6736(07)60944-1. [DOI] [PubMed] [Google Scholar]
- 2.Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology. 2008;70(2):144–152. doi: 10.1212/01.wnl.0000296811.19811.db. [DOI] [PubMed] [Google Scholar]
- 3.Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74(6):1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 5.Nishimura AL, Mitne-Neto M, Silva HC, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75(5):822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 7.Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61(5):427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 8.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20(7):1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008:13867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- 11.Gitcho MA, Baloh RH, Chakraverty S, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008 Mar 30; doi: 10.1002/ana.21344. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008 Apr 4; doi: 10.1016/S1474-4422(08)70071-1. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008 Mar 30; doi: 10.1038/ng.132. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 15.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 16.Ravits J, Paul P, Jorg C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology. 2007;68(19):1571–1575. doi: 10.1212/01.wnl.0000260965.20021.47. [DOI] [PubMed] [Google Scholar]
- 17.Battistini S, Giannini F, Greco G, et al. SOD1 mutations in amyotrophic lateral sclerosis. Results from a multicenter Italian study. J Neurol. 2005;252(7):782–788. doi: 10.1007/s00415-005-0742-y. [DOI] [PubMed] [Google Scholar]