

Abstract
A novel approach to integrating biochip and microfluidic devices is reported in which microcontact printing is a key fabrication technique. The process is performed using an automated microcontact printer that has been developed as an application-specific tool. As proof-of-concept the instrument is used to consecutively and selectively graft patterns of antibodies at the bottom of a glass channel for use in microfluidic immunoassays. Importantly, feature collapse due to over compression of the PDMS stamp is avoided by fine control of the stamp’s compression during contact. The precise alignment of biomolecules at the intersection of microfluidic channel and integrated optical waveguides has been achieved, with antigen detection performed via fluorescence excitation. Thus, it has been demonstrated that this technology permits sequential microcontact printing of isolated features consisting of functional biomolecules at any position along a microfluidic channel and also that it is possible to precisely align these features with existing components.
Keywords: Microcontact printing, Biochip, Microfluidics, Optical Waveguide, Antibody
1. Introduction
Point-of-care diagnostics is an emerging application area for microarray-based immunoassays. To realise such devices it will be necessary to integrate biochip with lab-on-a-chip (LOC) technologies. The former provides functions associated with biomolecular recognition, e.g. specificity, sensitivity and multi-parametric analysis [1, 2], whilst the latter allows miniaturisation and integration of different components necessary to manipulate biological fluids: filters, channels, pumps and mixers [3]. Combined, it will be possible to automatically perform the various complex and time consuming processing steps, e.g. filtering, labelling, and PCR detection, while at the same time, optimise the use of high value analytes, reduce reagent consumption and improve the overall device reliability [4]. The need to elaborate a fabrication platform that is capable of merging both technologies is therefore evident.
Biochips can be manufactured by grafting biomolecular probes onto well demarcated areas on glass, silicon, or polymer substrates by means of photochemistry, spotting [5], projection without contact [6, 7], microfluidics [8], dip-pen lithography [9] or microcontact printing (μCP) [10-15]. Oligonucleotide or oligopeptide probes can also be synthesized in situ on the substrate via photochemistry [16], spotting [17], projection without contact [6, 18], microfluidics [19] or μCP [20].
Among these techniques, μCP [21] attracts strong interest as a promising, low cost technique, capable of patterning biomolecular features over a range of areas from several mm down to 50 nm [22]. Conversely, the resolution of spotting, projection and microfluidic methods are limited to approximately 200 μm, 30 μm and 10 μm features respectively. Dip-pen lithography and μCP are the highest resolution patterning methods but unlike dip-pen lithography, μCP is carried out in a collective manner. In comparison with photochemistry, μCP is more flexible as it does not require specific photoactive components: μCP is compatible with all kinds of biomolecules, e.g. DNA, antibodies, proteins and enzymes [11-13, 15, 23] and also chemical products used for in situ synthesis [20]. Therefore, for manufacturing biochips, it is anticipated that μCP will be of very high interest.
However, to date the widespread implementation and application-oriented development of μCP have been impeded by the lack of a fully dedicated, low cost and versatile instrument. We have recently reported on the development of an automated microcontact printer [24] and as the first example of application with this instrument, the fabrication of microfluidic structures on glass substrates has been described [25]. The objective of this present work is to show the potential of this machine in the field of nanobiotechnology and more precisely, to demonstrate its ability to print biomolecules inside previously etched microfluidic channel. In contrast to a recently described method [26], which focused on the same objective but deposited biomolecules in a fairly non-specific manner (i.e. not only in the recessed areas but also on the mesa regions of the device), our goal is to graft individual spots of antibodies with biomolecular recognition capability at very precise locations within the channel. Moreover, in order to use a stamp inked with only one kind of biomolecule, our intention is to perform the grafting sequentially, that is to say, in a consecutive manner, one spot after the other with the resulting advantage that contamination of the stamp with different biomolecules is impossible.
In this article, two proof-of-concept experiments are reported. In the first, isolated microcontact features of antibodies were sequentially grafted by μCP at the bottom of a microfluidic glass channel. Here, feature collapse due to excess compression of the PDMS stamp was avoided by fine control of the load on the stamp during contact. A fluorescent antigen was delivered to the resultant biomolecular spots by pressure driven flow or electroosmotic flow, so as to detect the biomolecular interaction. In the second experiment, in addition to the printing of antibodies at the bottom of the microfluidic channel, spatial localisation of the printed features is demonstrated to an accuracy that in combination with integrated optical components permits a LOC’s surface to be interrogated at numerous, strictly predefined locations. This makes the implementation of biorecognition on LOCs feasible.
2. Microcontact printer overview
The microcontact printer is an automated, compact and low cost machine that has been especially designed to perform complex surface chemistry operations. Since a detailed explanation of the machine has already been published [24], only those features pertinent to the current work will be described here.
Figure 1 provides an overview of the microcontact printer, which is composed of the following components:
-
-
Computer controlled pneumatic actuator to push or pull the stamp holder.
-
-
Mobile head to carry the stamp holder, which has a 48 × 23 mm working stamp area.
-
-
Substrate holder with a substrate in place (we shall report here only on the use of microscope slides).
-
-
CCD camera.
-
-
PC running LabView and Vision software packages from National Instruments.
Figure 1.

View of the microcontact printer. The LOC substrate (microscope slide) is placed in the substrate holder, which has three degrees of freedom (two translations along the Y and Z axis and one rotation θX around the X axis) for alignment. The PDMS stamp is positioned in the stamp holder, which has two degrees of freedom (two rotations θY and θZ, respectively around the Y and Z axis) to make the stamp parallel to the substrate. The stamp is guided towards the substrate by a linear slide, actuated with a pneumatic jack. The stamp is released from the substrate by reversing the direction of air flow in the piston.
A key feature of the microcontact printer is that the stamp’s movement is actuated by a computer controlled pneumatic actuator until it makes contact with the substrate. Since the actuator’s piston is not sufficiently stable to achieve reproducible movement in the micrometric range, the stamp holder is mounted on a mobile head that is guided by a high accuracy, miniature linear slide. A flexible coupler is placed between this mobile head and the piston to transmit the movement and to compensate for any misalignment between the piston and the axis of the slide. Such automation of the stamping procedure greatly simplifies the process and improves reproducibility, allowing for the microprinting of adjacent or superimposable patterns in a reproducible manner. This is the key for successful grafting of different biomolecular patterns in a consecutive manner along a microfluidic channel, as shown later in the text.
The substrate holder of the microcontact printer is designed in such a way that two sides of any given microscope slide can be aligned with respect to a notch in the machine, thus constituting the common reference for all substrates. Hence, subsequent slides can be loaded and repeatedly stamped without changing the machine parameters. In this manner, the exchange of substrates is a quick and simple procedure, and a series of microscope slides can be microprinted in only a few minutes.
To achieve alignment between the substrate and stamp, the substrate holder has three degrees of freedom, with two manual translation stages in the directions of the Y and Z-axes, which possess a 2 μm resolution, and a manual rotation stage θX around the X-axis, with 0.1 mrad resolution. Alignment is performed with a CCD camera placed behind the microscope slide substrate that allows the operator to observe the contact between the stamp and substrate, and also aids quality control during serial μCP of substrates since all contacts can be recorded and analyzed by image processing.
The stamp holder has also been designed with a guiding fixture to facilitate rapid and reproducible installation into the microcontact printer, thanks to a precisely machined cavity into which the stamp holder locates itself. Hence, an exchange of stamps is also a quick and simple procedure. This makes microprinting with different stamps (and/or products, if the same stamp is successively inked with different products with intermediate washes) possible.
An important concern with the μCP technology is that the top of stamp can be unparalleled to the substrate for several reasons (e.g. variation of stamp thickness and orientation) and that it is necessary to flatten the whole working area of the stamp against the substrate. Also, the mechanical link between the stamp and the machine may have some inherent flexibility that is difficult to control during contact. Therefore, to solve all these problems, a head holding the stamp has been implemented in the machine that can position the stamp in parallel to the substrate with 0.003° of resolution by means of micrometric screws. In addition, the head has an internal mobile part, a semifree connection with three degrees of freedom, which allows for compensation of the remaining difference in parallelism between stamp and substrate (if necessary up to 14° yaw and pitch) [24].
3. μCP at the bottom of a microfluidic channel
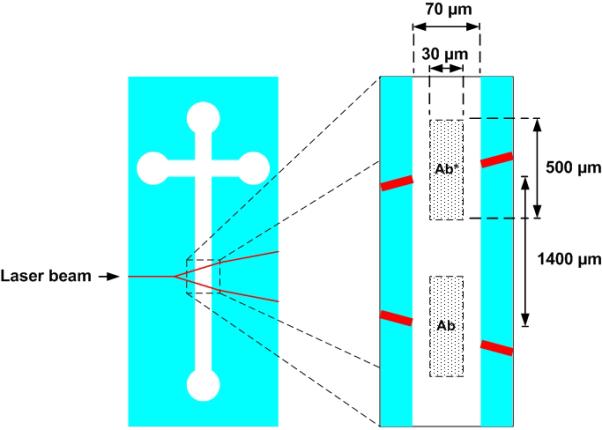
In this work, the PDMS stamp has a special design in order to print at the bottom of a microfluidic channel. The stamp geometry is defined in Fig. 2a, along with those parameters important to the process, e.g. the initial stamp thickness L and the feature’s height h. The stamp’s dimensions are 48 × 23 × 1 mm with a parallelepiped feature located centrally on one face, possessing dimensions of 30 × 500 μm and a height of 25 μm, i.e. an isolated feature. The whole stamp is inked with a specific antibody as explained below but only the parallelepiped feature is put into contact with the bottom of the channel, as shown in Fig. 2b. To do so, it is necessary to fine-tune the load exerted on the stamp. Indeed, this is a general concern for μCP in order to avoid collapse of the stamp relief during contact [27, 28]. The microcontact printer addresses this point by utilising the two adjustable micrometric screws shown in Fig. 2b that are set manually by the operator to a common length D (with reference to the substrate holder), thereby, limiting the stamp’s compression during contact. The load applied by the piston is distributed between the two screws and the stamp. In turn, the effective load on the stamp is fixed by the value of D and is therefore, independent from the pressure in the piston or any fluctuation of the applied load. The principle of operation is as follows. The stamp is pushed by the piston until the displacement of the stamp holder is stopped by the two screws. If L is the initial stamp thickness and e is the substrate thickness, then D must be adjusted so that e ≤ D ≤ e + L; if L’ is the thickness of the compressed stamp during contact then we have L’= D - e. From an experimental point-of-view, stamp compression D is adjusted by the operator when putting the stamp into contact with the substrate and checking the contact area with the camera of Fig. 1. The length D is adjusted so that the compression L-L’ is sufficient to provide contact between the parallelepiped’s top and channel bottom, but low enough to avoid any contact between the stamp base and the substrate’s mesa (in practice, this is performed with an an uninked stamp as explained in section 5).
Figure 2.
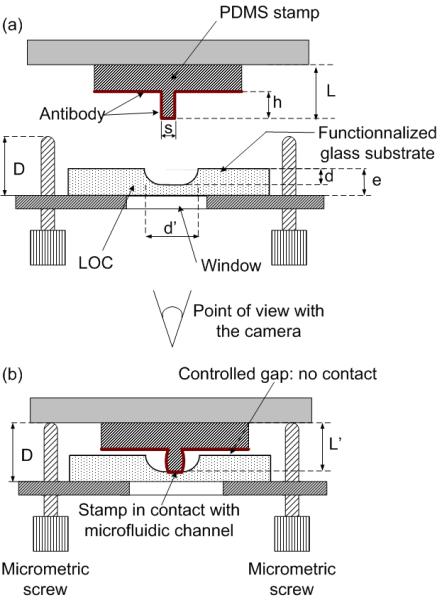
Principle of printing at the bottom of a microfluidic channel with a parallelepiped PMDS feature raised on the stamp. (a) Inking of the stamp with one kind of antibody. (b) Transfer of the antibody onto the bottom side of the functionalised microfluidic channel. The geometrical parameters are as follows: h = 25 μm; s = 30 μm; L = 1 mm; e = 1 mm; d = 20 μm; d’ = 70 μm. The length D of the screws out of the stamp holder is adjusted so that L - L’ ∼ 3 of +/- 1 μm.
In this work, the process of μCP is carried out on a glass substrate with microfluidic channels 70 μm wide and 20 μm deep Considering compression of the parallelepiped feature with a height of 25 μm, then the machine is set up so that L - L’ ∼ 3 ± 1 μm and thus, the transfer of antibodies is realised solely inside the microfluidic channel corresponding to the region in contact with the parallelepiped’s outside surface, i.e. only one spot of antibodies is grafted inside the microfluidic channel and nothing is transferred to the mesa region thanks to the remaining gap (Fig. 2.b).
4. Experimental
A flow chart of the experimental procedure is presented in Figure 4. Steps “a” and “b” illustrate fabrication of a microfluidic system with integrated optical waveguides. Separately, the stamp used for μCP was prepared (steps “c” and “d”). Next, biomolecules were grafted in the microchannel by μCP (step “e”), and the device sealed with a PDMS cover (step “f”). The device was now ready for characterisation (step “g”). Below, each of these steps is described in greater detail.
Figure 4.
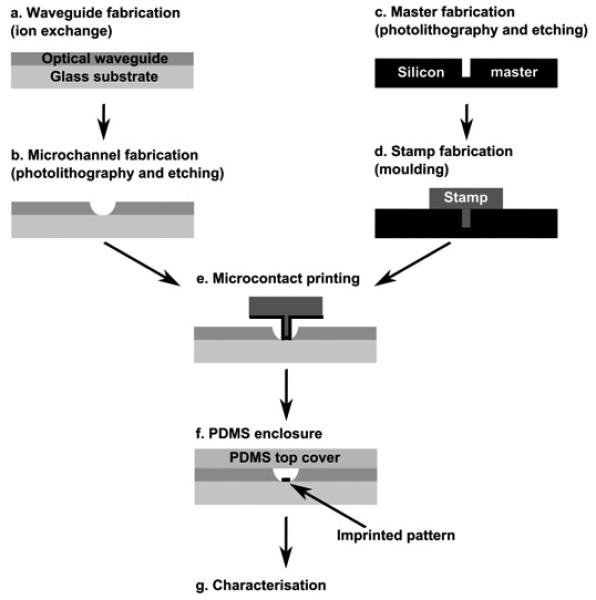
Flow chart of the fabrication technology.
4.1. LOC fabrication
Chip fabrication starts with the creation of optical waveguides in the soda lime substrate [29] (Corning #2947) (Fig. 4a). Briefly, a localised change in the refractive index of glass is realised by the Na+/K+ ion-exchange method and hence, the waveguiding effect is achieved. The process is carried out at a temperature of 380° C, in an environment of molten KNO3 salt and its duration depends upon the required size of the resultant waveguide. For this work, waveguides with a width and depth of approximately 40 μm and 30 μm respectively, were used. In those experiments that did not require integrated waveguiding optics, step “a” was omitted.
The microfluidic system was fabricated utilising photolithography and wet chemical etching methods [29]. In brief, a chromium/photoresist layer was used as a masking film and a mixture of buffered oxide etchant (BOE), hydrochloric acid (37%) and water in the proportion of (1:2:2) was used as an etching agent. The resultant microfluidic channels were 70 μm wide and 20 μm deep (Fig. 3b), and formed a cross with a 60 mm long sample separation channel, intersected with a 10 mm long sample injection channel. All channels terminated with a cylindrical reservoir of diameter 250 μm. Prior to μCP, the microfluidic glass substrate was cleaned in a Piranha solution (H2O2:H2SO4 1:3) for 10 minutes and then dried with nitrogen. The substrate was functionalised by immersing it into a mixture containing 95% acetone, 3% H2O and 2% aminopropyltriethoxysilane (APTES). The substrate was incubated in this solution overnight at room temperature, then cleaned with acetone and dried under nitrogen.
Figure 3.
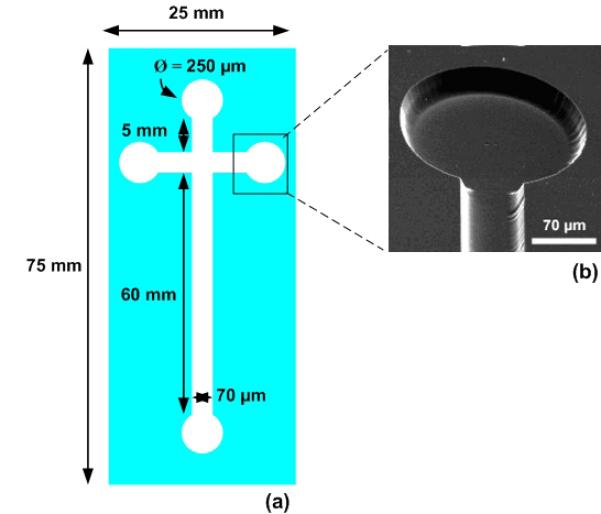
View of the LOC etched in a microscope slide. (a) Typical dimensions of the microfluidic channels. (b) SEM image of a microfluidic channel and reservoir. The channel is 70 μm wide and 20 μm deep. The reservoir diameter is 250 μm.
4.2. Master fabrication and stamp moulding
A 4-inch <100> silicon wafer (Siltronix) with a 2 μm thermal SiO2 layer was cleaned in a Piranha solution for 2 minutes, rinsed with deionised water for 10 minutes, dried with nitrogen gas and baked at 100° C. A photoresist layer (AZ5214E) was deposited by spin coating at 5,000 rpm for 30 seconds and then baked at 120° C for 2 minutes 30 seconds. The resin was exposed to UV and the wafer then dipped into developer AZ726 MIF for 50 seconds, dried under nitrogen and baked at 120° C for 2 minutes 30 seconds. The SiO2 layer was etched with BOE 7:1. The silicon was etched in RIE plasma (Alcatel Nextral 110 system, Cl2 20 sccm, 250 W, 100 mTorr), with the etch rate of 20 nm/s.
Prior to stamp moulding, the master was immersed in 10-3 M octadecyltrichlorosilane for 10 minutes to facilitate demoulding. The PDMS stamp was then prepared from Rhodia, RTV 3255 kit by mixing 5 g of PDMS prepolymer with 0.5 g of its curing agent and pouring the degassed mixture onto the master. The stamp was baked for 36 hours at 40° C, rinsed with ethanol and dried under a stream of nitrogen.
4.3. Preparation of antibodies and antigen
Performance of the LOC device was evaluated with an application concerning the detection of cystein proteinases from Porphyromonas gingivalis. The Gram-negative anaerobic bacterium, P. gingivalis, plays an important role in the development and progression of periodontal disease [30, 31]. For example, it is an important etiologic agent for adult periodontitis, a common inflammatory disease of tissues supporting the teeth that ultimately leads to tooth loss [32, 33]. A monoclonal antibody that recognises specifically one of the gingipains, RgpB, has been previously produced and employed to detect infections with P. gingivalis [31]. For proof-of-concept, two patterns of this monoclonal antibody were printed inside the LOC channel: one with fluorescent Cy3-labelled monoclonal antibody (Ab*) and the second with unlabelled monoclonal antibody (Ab).
Prior to μCP, Ab*, Ab and the Cy3-labelled antigen (Ag*), must first be activated. This was achieved using 42.5 μL of the selected protein with 5 μL of N-Hydroxysuccinimide (NHS) at 10 mM, and 2.5 μL of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDAC) at 20 mM. The solutions were left to rest for one hour at room temperature. The LOC was printed with Ab and Ab* according to the protocol discussed in section 5. The substrate was then washed with Phosphate Buffered Saline (PBS) (diluted ten times) for 5 minutes and rinsed with deionised water to remove any unfixed protein. Next, the substrate was dipped in a blocking solution of bovine serum albumin (BSA) to saturate the NH2 sites on the functionalised glass. Without this step, non-specific binding of the fluorescent antigen to the substrate’s surface would occur resulting in the entire channel fluorescing. The substrate was then washed to remove surplus BSA. To confirm that the antibodies had been successfully grafted at the desired locations, the substrate was dried and a fluorescence image was acquired (Genepix Personal 4100A Axon Scanner) with an excitation at a wavelength of 532 nm.
4.4. LOC enclosure
Following substrate functionalisation and deposition of the biomolecules via μCP, the LOCs channels were enclosed with a thin PDMS sheet [34] (Fig. 4f). Sample reservoirs were made over each reservoir with a hole-punch of approximately 2 mm outer-diameter. The device was now complete and ready for use.
4.5. Immunoassay protocol
To perform the proof-of-concept immunoassays, the LOC’s channels were filled with the fluorescent antigen: either by pressure driven flow with a syringe used to pump the antigen from one reservoir and through the channel to its opposite reservoir; or via electrokinetic flow by introducing a drop of antigen into a reservoir and displacing it along the length of the channel using an electric field of 350 V/cm. Following an incubation time of 30 minutes, the substrate was washed with PBS for 10 minutes, then with a PBS-Tween (0.1 %) solution for 10 minutes. The LOC device was then opened by simply pealing off the PDMS cover, the channels washed and dried. For LOC devices without integrated waveguides, a second fluorescence image was acquired using the fluorescent scanner at a wavelength of 532 nm. For LOC devices with integrated optical waveguides, the protocol for printing antibodies Ab* and Ab into the microfluidic channel was followed as described above, however the method of fluorescence excitation and detection was different. Here, the LOC was placed in a fluorescent BXFM Olympus microscope equipped with a 100 mW doubled YAG laser excitation source (wavelength 532 nm) and the resultant fluorescence image collected with a digital camera. The laser’s excitation beam was coupled with a monomode optical fibre (core diameter of 3.5 μm) and then, with the use of the x-y-z positioner, aligned and injected into the waveguide of the chip. The beam was divided at a Y-junction and guided to the microfluidic channel, where the fluorescent light was collected at right angles with respect to the excitation beam.
5. Results
In this work, the patterning of Ab and Ab* was performed sequentially, that is to say, in two printing operations: the first print with Ab* and the second print with Ab. The same stamp was used for the two prints with an intermediate stamp washing. To separate the patterns the substrate was translated along the microfluidic channel between the two printing operations by means of the Y-Z translation stages of the substrate holder (travel range of 5 mm), and its rotation θx around the X axis [24]. Importantly, before each printing, it was necessary to laterally align the stamp’s feature with the microfluidic channel of the substrate and to adjust the stamp compression during contact, thereby ensuring that the stamp feature was only in contact with the bottom of the channel. This was performed via observation of the contact aided by the CCD camera, during several back and forth stamping cycles [24]. To do so, the machine was set up with an uninked dry stamp to avoid contamination of the substrate according to the following steps (Fig. 1):
Alignment in Y, Z and θx of the substrate’s features with the stamp.
Coarse adjustment in θx and θY of the stamp-substrate parallelism.
Adjustment of the contact load on the stamp by means of the screws depicted in Fig. 2 (with the pressure on the piston set at 2 bars in this work).
Next, the stamp was inked with a droplet of Ab* whilst still positioned in the microcontact printer, dried with nitrogen, and automatically pushed with the actuator against the substrate, so as to transfer the first pattern of biomolecules. The contact time was determined by the operator (30 s). By reversing the air flow in the piston, the stamp was released from the substrate and sent back to the starting position. The stamp was then removed from the microcontact printer and cleaned. The machine was set up for the second time with the same uninked stamp, but the next contact point was translated along the microfluidic channel by means of the substrate holder’s displacement stages. The stamp was inked with a droplet of Ab and dried with nitrogen. Printing of the second pattern of biomolecules was performed as described previously.
5.1. μCP of two antibodies inside a microfluidic channel
In this experiment, a plain substrate (i.e. no optical waveguide) was used. Figure 5 depicts the nominal position of the printed features relatively to one another and to the microfluidic channel. The distance between the two imprinted areas was 250 μm. As expected, prior to execution of the immunoassay, only fluorescence from Ab* was detected and the presence of Ab was not revealed (Fig. 6a). This latter biomolecule was only detected after introduction of the fluorescent antigen, Ag*, due to the subsequent generation of Ab/Ag* and Ab*/Ag* complexes (Fig. 6b). Significantly, the absence of fluorescence in the non-printed area of the channel was indicative that the washing protocol was efficient enough to remove any non-specific absorbed Ag* and also that the antibodies were sufficiently well grafted to the substrate to withstand such rinsing. Hence, it has been shown that the microcontact printer allows the sequential transfer of individual stamp features within microfluidic channels, without collapse of the stamp.
Figure 5.
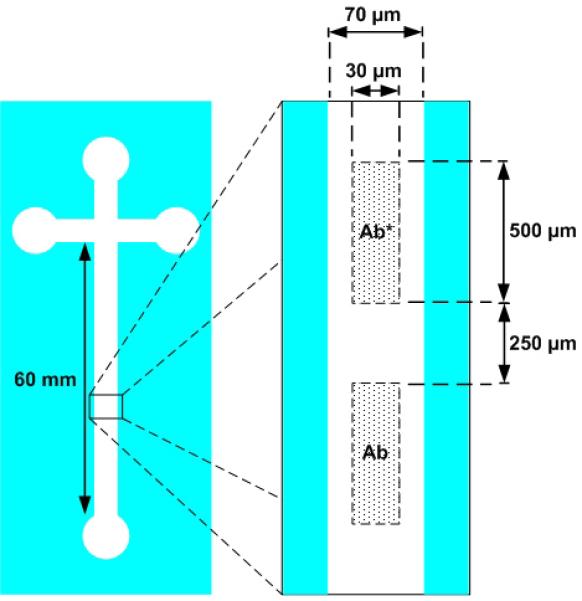
Nominal position of antibodies Ab and Ab* relatively to one another and to the microfluidic channel.
Figure 6.
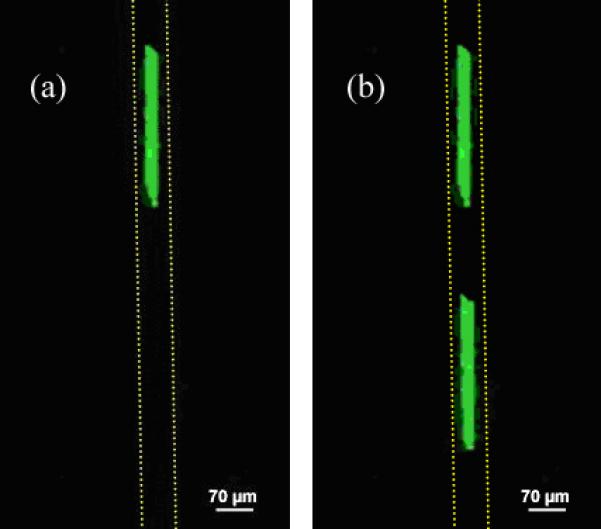
(a) Fluorescence image of the microfluidic channel containing Ab* and Ab sites grafted inside the channel by μCP, following rinsing but The grafted Ab* stamped control pattern is visible. Channel walls are indicated as a dotted line. (b) Fluorescence image of the same microfluidic channel following introduction of the fluorescent antigen and rinsing. The Ab/Ag* complex is now visible (Genepix Personal 4100A Axon Scanner).
5.2. μCP of two antibodies inside a LOC channel at the intersection with two optical waveguides
In the previous experiment, spots of antibodies were printed at arbitrary locations along the microfluidic channel’s axis. Without using the microcontact printer, the “traditional” approach would have been to print the biomolecules on a flat substrate (e.g. microscope slide) by manual μCP and to assemble a microfluidic chamber made of a PDMS cover with microfluidic channel on top of the device. Here, our objective was to print one feature of Ab* and one feature of Ab within the existing microchannel, precisely in front of two optical waveguide intersections that originate from a Y-junction. This is an example of a device which cannot be easily obtained by a combination of printing and assembling since the optical waveguides are integrated in the glass substrate and intersect the microfluidic channels. Figure 7 shows a plan view of a LOC’s microfluidic channel and waveguide intersection area. The distance between waveguide intersections along the channel was approximately 1400 μm. Figure 8 depicts the nominal position of the printed features relatively to one another, the microfluidic channel and the Y-junction waveguide intersections.
Figure 7.

Plan view of two optical waveguides originating from a Y-junction and intersecting the microfluidic channel (optical microscope).
Figure 8.
Nominal position of antibodies Ab and Ab* relatively to one another, the microfluidic channel and the optical waveguides. A 100 mW doubled YAG laser laser is injected at the input of the Y junction.
As shown in Fig. 9a, only one fluorescent spot corresponding to Ab* was observed immediately following μCP. Following the application of Ag* via electrokinetic flow and the subsequent washing steps, two fluorescent spots were visible, the second spot corresponding to the Ab/Ag* complex (Fig. 9b).
Figure 9.

(a) Fluorescence image of the microfluidic channel with intersecting optical waveguides and containing Ab* and Ab sites grafted inside the channel by μCP, following rinsing but prior to the introduction of the fluorescent antigen (Ab invisible). Channel walls are indicated as a dotted line. The grafted Ab* control pattern is visible in front of the Y-junction’s upper waveguide. (b) Fluorescence image of the same microfluidic channel following introduction of the fluorescent antigen and rinsing. The Ab/Ag* complex positioned in fromt of the Y-junction’s lower waveguide is now visible (fluorescent BXFM Olympus microscope with a 100 mW doubled YAG laser excitation source).
This result confirms the successful printing of two discrete patterns of antibodies inside the same microfluidic channel and in precisely defined locations, so that an antibody-antigen immunoreaction can be initiated on-chip and detected by fluorescence excitation. Moreover, the excitation beam originates from a single light source that has been redistributed across the LOC via integrated optical waveguides. Interestingly, fluorescence from biomolecules deposited at the bottom of the microfluidic channel is easily excited by light coming from the lateral intersecting waveguides, without the need for sophisticated optical coupling between the waveguide and microfluidic channel.
6. Concluding remarks
Among the several strategies that are possible to print different biomolecules on the same substrate [24], the one consisting in sequential μCP was demonstrated in this work. The main advantage of this solution is that it makes possible printing with the same stamp without cross contamination between the different biomolecules at the surface of the stamp. To do so, the machine’s set up is performed with an uninked stamp. The stamp is then inked with only one kind of biomolecule and the printing operation performed. An identical process is repeated for each ink, with the setup of the machine being easily and quickly made. This is feasible because the movement of the microcontact printer’s stamp is higly reproducible (1 μm STD as reported previously [24], including the degrees of freedom provided by the mobile head holding the stamp). Importantly, feature collapse due to overload of the PDMS stamp is avoided via fine control of the stamp’s compression during contact. Moreover, thanks to the stamp’s particular geometry and its regulated compression, a gap between the stamp and substrate is generated in regions where no printing is required. This is achieved with the microcontact printer by merit of the screws’ 2 μm resolution of adjustment in length [24], which simultaneously controls the load, compression of the stamp and the gap distance.
Proof-of-concept experiments have demonstrated grafting of adjacent patterns of biomolecules into a microfluidic channel as well as printing at the intersections of this channel with integrated optical waveguides. Antibody-antigen recognition was also reported with the developed device (quantitative data on the microfabricated devices will be provided in a forthcoming paper). This is an encouraging result for the integration of biochips, microfluidics and integrated optics. In contrast to a recently described method [35] that allows the detection of a single type of a biomolecule, the μCP approach described herein will permit the detection of multiple types of biomolecules by grafting different probes inside a channel.
Optical integration is a general strategy intended to simplify instrumentation requirements to operate LOCs [29, 36] and redistribution of a single excitation beam to multiple locations on the LOC, via a network of integrated waveguides, satisfies this criterion. This approach is complicated by the necessity of depositing specific biomolecules in front of pre-existing components. Therefore, we predict that the solution presented here will greatly facilitate future development of such devices. In addition, it is anticipated that this approach to μCP will also be relevant for patterning biomolecules in microsystems with other pre-exisiting elements than waveguides such as electrodes, microcantilevers and microlenses.
Acknowledgments
This work was supported in part by the National Institute of Health through grant DE 09761. Jan Potempa is a recipient of the award SUBSYDIUM PROFESORSKIE from the Foundation for Polish Science (FNP, Warszawa). Colin Mansfield acknowledges the support of the European Community under a Marie Curie Intra-European Fellowship.
Biographies
Elie Bou Chakra received an engineering degree from the University of Beirut (Lebanon). He received his Ph.D. in micro and nanotechnology in 2007 from the Institut des Nanotechnologies de Lyon. He is currently a postdoctoral research fellow at the Laboratoire de Tribologie et Dynamique des Systèmes (Ecully, France). His research interests are in instrumentation, nanomechanics and nanotribology.
Benjamin Hannes received his M.Sc. in Microelectronics and Microsystems from the Institut National des Sciences Appliquées of Lyon (INSA) in 2004. He is currently a Ph.D. student at the Institut des Nanotechnologies de Lyon (Ecully, France). His main interests are in new fabrication technologies for microsystems, in particular, the development of microsystems for biomedical applications.
Julien Vieillard received his M.Sc. in pharmacology from the University of Nice and received his Ph.D. degree from the Institut des Nanotechnologies de Lyon in 2006. He is currently a postdoctoral research fellow at the Laboratoire d’Analyse des Systèmes Organiques Complexes (Evreux, France) at the University of Rouen. His research interests involve biosensors, separation systems and micro analytical systems.
Colin Mansfield received his Ph.D. from the University of Southampton in the field of biomedical physics in 2000. He subsequently joined the National Research Council Canada’s Institute for Biodiagnostics as a research associate for 5-years, his research interests during this time including the integration of microfluidics with infrared spectroscopy of biofluids. He is currently engaged with furthering his experience in nanobiotechnologies, by merit of a Marie Curie Intra-European Fellowship, at the Institut des Nanotechnologies de Lyon, France.
Radoslaw Mazurczyk received his Ph.D. in electrical engineering in 2002 from the Technical University of Lodz in Poland. Between 2003 and 2005 he was a postdoctoral fellow at Laboratoire d’Electronique, Optoélectronique et Microsystèmes (Ecole Centrale de Lyon, France). Currently, he is a research engineer at the Institut des Nanotechnologies de Lyon. His domains of research comprise semiconductor and microsystems technology, surface and thin film science and technology.
Jan Potempa is Research Professor at the Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University in Kraków (Poland) and is Senior Research Scientist in the Department of Biochemistry and Molecular Biology at the University of Georgia in Athens (USA). He received his Ph.D. in 1982 from the Jagiellonian University. His current research interests are in host and bacterial proteolytic enzymes and their inhibitors in homeostasis and diseases and as targets for therapeutic intervention.
Stanislas Krawczyk received his Ph.D. in Physics and Technology of Electronic Devices and has over 20 years of research experience. Since 1995, he has been Research Director at the Centre National de la Recherche Scientifique and is currently at the Institut des Nanotechnologies de Lyon. He is recognised as specialist in the field of semiconductor materials, microsystems and sensors. He initiated a research program aimed at the development of lab-on-a-chip microsystems for medical diagnostics.
Michel Cabrera is a researcher at the CNRS since 1986 and is currently at the Institut des Nanotechnologies de Lyon. He received an engineering degree from the Ecole Supérieure d’Electricité (Orsay, France) and his Ph.D. degree in 1986 in Chemical Engineering from the Institut National Polytechnique de Lorraine. He is recognised as specialist in instrumentation and is co-founder of several start-up companies. His current interests are in the development of non-conventional rapid machining techniques for microfluidics and biosensor devices including microcontact printing and micro nano electrical machining.
References
- [1].Southern EM. DNA chips: analysing sequence by hybridization to oligonucleotides on a large scale. Trends in Genetics. 1996;12:110–115. doi: 10.1016/0168-9525(96)81422-3. [DOI] [PubMed] [Google Scholar]
- [2].Zhu H, Snyder M. Protein chip technology. Current Opinion in Chemical Biology. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- [3].Reyes DR, Iossifidis D, Auroux PA, Manz A. Micro total analysis systems. 1. Introduction, theory, and technology. Anal Chem. 2002;74:2623–36. doi: 10.1021/ac0202435. [DOI] [PubMed] [Google Scholar]
- [4].Liu RH, Yang J, Lenigk R, Bonanno J, Grodzinski P. Self-Contained, Fully Integrated Biochip for Sample Preparation, Polymerase Chain Reaction Amplification, and DNA Microarray Detection. Analytical Chemistry. 2004;76:1824–1831. doi: 10.1021/ac0353029. [DOI] [PubMed] [Google Scholar]
- [5].Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- [6].Cabrera M, Jaber M, Dugas V, Broutin J, Vnuk E, Cloarec JP, Souteyrand E, Martin JR. Implementation of DNA chips obtained by microprojection for diagnostic and personalized medicine. Cell Mol Biol (Noisy-le-grand) 2004;50:225–32. [PubMed] [Google Scholar]
- [7].Okamoto T, Suzuki T, Yamamoto N. Microarray fabrication with covalent attachment of DNA using bubble jet technology. Nat Biotechnol. 2000;18:438–41. doi: 10.1038/74507. [DOI] [PubMed] [Google Scholar]
- [8].Bernard A, Michel B, Delamarche E. Micromosaic Immunoassays. Analytical Chemistry. 2001;73:8–12. doi: 10.1021/ac0008845. [DOI] [PubMed] [Google Scholar]
- [9].Demers LM, Ginger DS, Park SJ, Li Z, Chung SW, Mirkin CA. Direct patterning of modified oligonucleotides on metals and insulators by dip-pen nanolithography. Science. 2002;296:1836–8. doi: 10.1126/science.1071480. [DOI] [PubMed] [Google Scholar]
- [10].Bernard A, Renault JP, Michel B, Bosshard HR, Delamarche E. Microcontact Printing of Proteins. Advanced Materials. 2000;12:1067–1070. [Google Scholar]
- [11].Delamarche E. Nanobiotechnology. Wiley Verlag GmbH; Weinheim: 2005. Microcontact Printing of Proteins; pp. 31–52. [Google Scholar]
- [12].Inerowicz HD, Howell S, Regnier FE, Reifenberger R. Multiprotein Immunoassay Arrays Fabricated by Microcontact Printing. Langmuir. 2002;18:5263–5268. [Google Scholar]
- [13].Lange SA, Benes V, Kern DP, Horber JK, Bernard A. Microcontact printing of DNA molecules. Anal Chem. 2004;76:1641–7. doi: 10.1021/ac035127w. [DOI] [PubMed] [Google Scholar]
- [14].Renault JP, Bernard A, Bietsch A, Michel B, Bosshard HR, Delamarche E, Kreiter M, Hecht B, Wild UP. Fabricating Arrays of Single Protein Molecules on Glass Using Microcontact Printing. J. Phys. Chem. B. 2003;107:703–711. [Google Scholar]
- [15].Thibault C, Le Berre V, Casimirius S, Trévisiol E, François J, Vieu C. Direct microcontact printing of oligonucleotides for biochip applications. Journal of Nanobiotechnology. 2005;3 doi: 10.1186/1477-3155-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D. Light-directed, spatially addressable parallel chemical synthesis. Science. 1991;251:767–73. doi: 10.1126/science.1990438. [DOI] [PubMed] [Google Scholar]
- [17].Frank R. The SPOT-synthesis technique Synthetic peptide arrays on membrane supports- principles and applications. Journal of Immunological Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- [18].Blanchard AP, Kaiser RJ, Hood LE. High-density oligonucleotide arrays. Biosensors and Bioelectronics. 1996;11:687. [Google Scholar]
- [19].Moorcroft MJ, Meuleman WR, Latham SG, Nicholls TJ, Egeland RD, Southern EM. In situ oligonucleotide synthesis on poly(dimethylsiloxane): a flexible substrate for microarray fabrication. Nucleic Acids Res. 2005;33:e75. doi: 10.1093/nar/gni075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xiao PF, He NY, Liu ZC, He QG, Sun X, Lu ZH. In situ synthesis of oligonucleotide arrays by using soft lithography. Nanotechnology. 2002:756–762. [Google Scholar]
- [21].Xia Y, Whitesides GM. Soft lithography. Angewandte Chemie International Edition. 1998;37:550–575. doi: 10.1002/(SICI)1521-3773(19980316)37:5<550::AID-ANIE550>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- [22].Li HW, Muir BVO, Fichet G, Huck WTS. Nanocontact Printing: A Route to Sub-50-nm-Scale Chemical and Biological Patterning. Langmuir. 2003;19:1963–1965. [Google Scholar]
- [23].Bernard A, Delamarche E, Schmid H, Michel B, Bosshard HR, Biebuyck H. Printing Patterns of Proteins. Langmuir. 1998;14:2225–2229. [Google Scholar]
- [24].Bou-Chakra E, Hannes B, Dilosquer G, Mansfield CD, Cabrera M. A new instrument for automated microcontact printing with stamp load adjustment. Review of Scientific Instruments. 2008;79:064102. doi: 10.1063/1.2936259. [DOI] [PubMed] [Google Scholar]
- [25].Hannes B, Vieillard J, Bou Chakra E, Mazurczyk R, Mansfield CD, Potempa J, Krawczyk S, Cabrera M. The etching of glass patterned by microcontact printing with application to microfluidics and electrophoresis. Sensors and Actuators B: Chemical. 2008;129:255–262. [Google Scholar]
- [26].Foley J, Schmid H, Stutz R, Delamarche E. Microcontact Printing of Proteins Inside Microstructures. Langmuir. 2005;21:11296–11303. doi: 10.1021/la0518142. [DOI] [PubMed] [Google Scholar]
- [27].Bietsch A, Michel B. Conformal contact and pattern stability of stamps used for soft lithography. Journal of Applied Physics. 2000;88:4310–4318. [Google Scholar]
- [28].Huang YY, Zhou W, Hsia KJ, Menard E, Park JU, Rogers JA, Alleyne AG. Stamp Collapse in Soft Lithography. Langmuir. 2005;21:8058–8068. doi: 10.1021/la0502185. [DOI] [PubMed] [Google Scholar]
- [29].Mazurczyk R, Vieillard J, Bouchard A, Hannes B, Krawczyk S. A novel concept of the integrated fluorescence detection system and its application in a lab-on-a-chip microdevice. Sensors and Actuators B. 2006;118:11–19. [Google Scholar]
- [30].O’Brien-Simpson NM, Veith PD, Dashper SG, Reynolds EC. Antigens of bacteria associated with periodontitis. Periodontoly 2000. 2004;35:101–34. doi: 10.1111/j.0906-6713.2004.003559.x. [DOI] [PubMed] [Google Scholar]
- [31].Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontoly 2000. 2000;24:153–92. doi: 10.1034/j.1600-0757.2000.2240108.x. [DOI] [PubMed] [Google Scholar]
- [32].Haffajee AD, Socransky SS. Microbiology of periodontal diseases: introduction. Periodontology 2000. 2005;38:9–12. doi: 10.1111/j.1600-0757.2005.00112.x. [DOI] [PubMed] [Google Scholar]
- [33].Imamura T, Travis J, Potempa J. The biphasic virulence activities of gingipains: activation and inactivation of host proteins. Curr Protein Pept Sci. 2003;4:443–50. doi: 10.2174/1389203033487027. [DOI] [PubMed] [Google Scholar]
- [34].Mansfield CD, Mazurczyk R, Vieillard J. A foolproof method for sealing glass-based LOC devices for electrophoresis. Chips & Tips (an online supplement to Lab on a Chip) 2008 January 16; www.rsc.org/Publishing/Journals/lc/Chips_and_Tips/PDMS_seal.asp.
- [35].Xiang Q, Hu G, Gao Y, Li D. Miniaturized immunoassay microfluidic system with electrokinetic control. Biosensors and Bioelectronics. 2006;21:2006–2009. doi: 10.1016/j.bios.2005.09.019. [DOI] [PubMed] [Google Scholar]
- [36].Hunt H, Wilkinson J. Optofluidic integration for microanalysis. Microfluidics and Nanofluidics. 2008;4:53–79. doi: 10.1007/s10404-007-0223-y. [DOI] [PMC free article] [PubMed] [Google Scholar]