

Abstract
An RNA editing reaction that is both essential and specific to the trypanosomatid parasites is an attractive target for new drug development. Although high-throughput screening of chemical libraries is a powerful strategy often used to identify new drugs, the available in vitro editing assays do not have the necessary sensitivity and format for this approach to be feasible. A ruthenium labeled reporter RNA is described here that overcomes these limitations as it can both detect edited product in the low femtomole range and is ideal for high-throughput format. The reporter RNA consists of an RNA editing substrate linked to a streptavidin-binding aptamer that is initially held within an inactive conformation. An in vitro selection strategy optimized the linkage so that the streptavidin-binding aptamer is only activated by an editing-induced conformational change. An electrochemiluminescent signal results from the ruthenium label when the reporter is bound to the bottom of a streptavidin-coated microtiter plate where it can be stimulated by a carbon electrode. Chemical probing, mutagenesis, and binding affinity measurements were used to characterize the reporter. The highly sensitive assay could be adapted to a broad range of RNA processing reactions.
Keywords: electrochemiluminescence, high-throughput, RNA editing, Leishmania, trypanosomatid
INTRODUCTION
Several members of the trypanosomatid family of protozoa are causative agents of human diseases including leishmaniasis and African sleeping sickness (for review, see Stuart et al. 2008). Most of the trypanosomatid mitochondrial mRNAs are edited through uridylate (U) insertions and deletions (Benne et al. 1986). These reactions are catalyzed by three distinct complexes that share a common subset of proteins (Panigrahi et al. 2006; Carnes et al. 2008). Guide RNAs that are complementary to correctly edited mRNA sequence direct the location of editing and the number of deletions and insertions (Blum et al. 1990; Kable et al. 1996; Seiwert et al. 1996). Since the reactions are both essential and unique to the parasites, the editing complexes are attractive targets for novel drug development (Schnaufer et al. 2001).
There are currently not any drugs available that specifically inhibit the trypanosomatid editing reactions. An in silico screening strategy was previously used to identify novel drug-like compounds that can be docked to the known crystal structure of KREL1, one of the RNA ligases of the editing complexes (Amaro et al. 2008). Although this approach is highly promising, it could be limited by differences in structure or drug accessibility that result from the study of individual proteins outside the context of the intact multi-protein complexes. The approach also does not fully exploit the large drug-binding landscape that would potentially be presented by the intact complexes. High-throughput screening of chemical libraries is an alternative strategy to identify novel drugs inhibiting the editing reaction, but it also has limitations. These are primarily related to the in vitro editing assays not having the sensitivity and format necessary for high-throughput screening to be practical and economically feasible (Byrne et al. 1996; Kable et al. 1996; Seiwert et al. 1996; Wang et al. 2002; Pai et al. 2003). A novel assay is described here that can detect edited product in the low femtomole range and is ideal for high-throughput format. The assay exploits a reaction that generates electrochemiluminescence (ECL) as a result of an editing-responsive conformational change within an RNA reporter. The conformational change induced by the editing is analogous to the conformational changes induced by small ligand binding to aptamers that have been exploited as in vitro sensors (for review, see Rajendran and Ellington 2002). The described assay could be adapted to a broad range of RNA processing reactions.
RESULTS AND DISCUSSION
Development of the assay
The editing assay outlined in Figure 1 was designed to maximize detection sensitivity and also to be compatible with high-throughput format. The assay is performed within streptavidin-coated microtiter plates that have carbon electrodes running through the bottom of the wells (Best et al. 2005). The reporter RNA for the editing reaction is labeled with a ruthenium complex that can generate ECL only when held in proximity to these electrodes. Editing causes a conformational change in the reporter that activates a streptavidin-binding aptamer. This results in the immobilization of the RNA at the bottom of the microtiter well and the generation of ECL after electrode stimulation. In the absence of editing, the reporter RNA is in a conformation that inhibits streptavidin binding, and as a result pre-edited RNA does not generate appreciable ECL.
FIGURE 1.

Development of a high-throughput assay for an editing reaction. In response to the insertion of three U's (red) by the in vitro editing reaction, the reporter RNA undergoes a conformational change that activates the streptavidin-binding aptamer (blue). The reporter RNA is labeled with a ruthenium complex. Upon immobilization of the RNA at the bottom of a streptavidin-coated microtiter plate and electrical stimulation, the ruthenium complex will generate an ECL signal. Nonimmobilized RNAs do not generate a signal.
The major challenge in developing the assay was to obtain a reporter RNA with the appropriate properties. For a significant fraction of the edited RNA to become bound to the streptavidin-coated plate, the edited RNA is required to have a relatively good affinity for streptavidin (Kd ≤ 1 × 10−7 M). At the same time, the RNA in its pre-edited state is required to have minimal affinity since any binding will lead to a high background signal and the deterioration of assay sensitivity. The reporter is further required to be an efficient substrate for the editing reaction. Satisfying all three criteria presents a potential complication because sequence that significantly influences editing efficiency can have conflicting constraints related to streptavidin binding and the switching mechanism.
An in vitro selection strategy was used to obtain a reporter RNA with the desired properties (Fig. 2; Table 1; Ellington and Szostak 1990; Tuerk and Gold 1990). The starting RNA contained elements of the previously described S1 streptavidin-binding aptamer (Srisawat and Engelke 2001), and this was linked to part of an aptamer that had been selected to be an efficient substrate for insertional editing (Pai et al. 2003). The editing aptamer was previously demonstrated to have many of the attributes associated with natural mRNA substrates; these include a dependence on guiding nucleotides, similar sequence and secondary structural constraints, and the copurification of the editing activities. Twenty-one positions within the starting RNA were randomized for the selection of the switching function (Fig. 2A). PCR mutagenesis also introduced variation so that additional sites could be optimized during the selection.
FIGURE 2.

In vitro selection of the reporter RNA. (A) The predicted secondary structure of the parental molecule used for the in vitro selection. Part of a streptavidin-binding aptamer (blue) is linked to part of an aptamer selected to be a good editing substrate (underlined). The 21 positions that were randomized for the selection are in lowercase, and the three U's that are guided into the editing site are indicated in red. (B) Progression of the in vitro selection. Radiolabeled pre-edited RNAs from the indicated cycles of selection were treated in a streptavidin-coated microtiter plate with mitochondrial extract in the presence or absence of the indicated nucleotide cofactors. After washing the plate, eluted RNA was analyzed on a denaturing gel. The size corresponding to the three U insertions expected from accurate editing is indicated. (C) The sequence of the RNAs obtained from the selection. The number of identical clones is indicated in parentheses. The maroon bases represent positions that differ from the starting parental sequence, and the nucleotides that had been deleted during the selection are indicated by a maroon dash. Other colors are as described above. Nucleotides are numbered relative to the pre-edited sequence of RNA A.
TABLE 1.
In vitro selection of the reporter

Several different types of selection pressure were applied to the starting pool to enrich for the reporter (Table 1). First, positive binding selections were performed on the RNA in its edited state for those that bind to streptavidin with high affinity. Second, negative binding selections were performed on the RNA in its pre-edited state to remove those RNAs that are capable of binding streptavidin in the absence of editing. Interconversion of the RNA population between the pre-edited and edited states was accomplished using appropriate primers for the RT-PCR amplification that overlapped the editing site. Third, selection pressure was applied to ensure that the RNA population was a good substrate for the editing reaction. The final cycles of selection also required that both in vitro editing and streptavidin binding occur together within streptavidin-coated microtiter wells. Selection pressure was also applied to ensure that the switching function would be compatible with the ECL detection requirements.
The in vitro selection
The RNA population was monitored throughout the in vitro selection for the properties of the reporter required in the proposed assay (Fig. 2B). Radiolabeled pre-edited RNAs from different cycles of selection were treated within streptavidin-coated microtiter wells with a fractionated mitochondrial extract either in the presence or absence of ATP and UTP, which are essential cofactors for the editing reaction. After treatment with the extract, the microtiter wells were washed, and the bound RNA was eluted and analyzed on a denaturing gel. Only a small background binding of pre-edited RNA was detected at the start of the selection, but intense higher mobility bands are clearly visible by the later cycles (Fig. 2B). The predominant edited band is consistent with three U insertions being guided by the three A's depicted in Figure 2A, and is dependent on the inclusion of the nucleotide cofactors that are essential for editing. The 3′ end of the RNA substrate had been blocked prior to the assay through ligation to the 5′ end by treatment with RNA ligase. This circular substrate RNA ensured that the higher-mobility product did not result from the addition of U's to the 3′ end by a terminal uridyyl transferase (TUTase) activity that is present within the editing extract (Brown et al. 1999; Pai et al. 2003).
A minor but significant fraction of the selected RNAs have an incorrect number of U insertions during the reaction (Fig. 2B). This is not an intrinsic deficiency of the editing extract as other RNAs are accurately edited by the same in vitro reaction (Pai et al. 2003). Rather, it is most likely a reflection of the sequence that was selected upstream of the editing site, which was previously shown to significantly influence editing fidelity (Igo et al. 2002; Pai et al. 2003). There was not an explicit requirement during the selection to have completely accurate editing provided that any U insertions still triggered a conformational change that activated streptavidin binding. The upstream sequence would have had significant additional pressure on it related to streptavidin binding and the switching function, and it is possible that these constraints may have necessitated some loss in fidelity. This, however, does not invalidate the selected RNAs as reporters for the editing reaction since the overall insertional reaction is still being detected.
After the last cycle of selection, the RNA pool was amplified by RT-PCR for cloning and sequence analysis. Thirty-seven clones were sequenced, and all are very closely related but contain several changes relative to the parental sequence (Fig. 2C). The RT-PCR primers overlapped nucleotides 1–17 and 72–90, and as a result, variation at these sites would not be evident from the sequence analysis, nor could it have been selected. The two most abundant selected sequences (RNAs A and B) only differ from each other by a single C48U change that is present within the large loop of the streptavidin-binding aptamer in RNA A. This change is also present in all but two of the other sequenced clones.
Assessment of the selected RNAs as reporters for the assay
Both the edited and pre-edited forms of RNAs A and B were labeled with a ruthenium complex so that their potential as reporters in the ECL-based assay could be evaluated (Fig. 3A). To label the RNAs, a short T7 RNA polymerase transcript with incorporated 5-(3-aminoallyl)-uridylates was ligated to the 3′ end of the selected RNAs (Moore and Sharp 1992). The ligated RNA with aliphatic amine groups was then reacted with an activated ester of the ruthenium complex. Coupling conditions were chosen so that approximately two ruthenium complexes were incorporated per RNA. Although not essential for the ECL-based assay, the ligation of the aminoallyl-derivatized RNA also has the effect of inhibiting TUTase activity at the 3′ end (data not shown).
FIGURE 3.

Characterization of the ECL assay. (A) Labeling the selected RNAs with the ruthenium complex. An RNA containing 5-(3-aminoallyl)-uridylates (NH2) was ligated to the 3′-end of the selected RNAs using DNA ligase and the appropriate DNA splint (Moore and Sharp 1992). The RNA was subsequently reacted with an N-hydroxysuccinimide ester of the ruthenium complex. (B) The indicated quantities of edited RNA A (●), edited RNA B (▲), pre-edited RNA A (○),pre-edited RNA B (Δ) and the starting randomer (×) were incubated with the streptavidin-coated microtiter plates, and after washing and scanning, the ECL was detected (n = 4). (C) The signal-to-background ratio of the complete ECL editing reaction was determined for reporters A (n = 6) and B (n = 8) by treating the pre-edited RNAs with editing extract both in the presence (gray) and absence (white) of the essential nucleotide cofactors. The indicated ECL units were detected after washing and scanning of the plate.
The indicated quantities of the ruthenium-labeled RNAs were incubated within a streptavidin-coated microtiter plate containing the carbon electrodes for generating ECL (Fig. 3B; Best et al. 2005). The plate binding and washing conditions had to be extensively optimized in order to minimize nonspecific RNA binding. As suggested by the ECL signal from the randomer used for the selection, nonspecific binding makes up a major component of the small ECL signals arising from the pre-edited RNAs. The scanner used to detect the ECL also generates a background of 20–40 ECL units in the absence of any RNA (data not shown). This can make up a significant fraction of the ECL signals arising from the pre-edited RNAs and randomer, and it accounts for much of the nonlinearity of these plots. However, the ECL signals arising from the corresponding edited RNAs can be up to 100-fold stronger, and as a result the nonspecific components, as assessed by the randomer binding, make only a minor contribution to the assay signal. The ECL signals from the edited and pre-edited forms of RNA A are higher than those from the corresponding forms of RNA B, and these ECL differences are consistent with the apparent Kd values for streptavidin binding (Table 2).
TABLE 2.
Characterization of the selected RNAs

The pre-edited forms of RNAs A and B were also tested in the complete ECL assay outlined in Figure 1. The signal-to-background ratio for the assay was determined by performing the editing reaction both in the presence and absence of the essential nucleotide cofactors (Fig. 3C). This ratio was determined to be 8 ± 2 (n = 6) for RNA A and 17 ± 2 (n = 8) for RNA B. The ECL from the minus ATP/UTP reactions (Fig. 3C) is higher than the ECL from the pre-edited RNAs without editing extract (Fig. 3B). This suggests that the extract increases background binding, which was confirmed using radiolabeled RNA (data not shown). The effect of the extract on the nonspecific binding is higher with RNA A (minus ATP/UTP in Fig. 3C), which also has the lower apparent Kd for steptavidin binding (Table 2). Although the signal-to-background ratios are strongly suggestive that either RNA could be used as a reporter in high-throughput screens, the result emphasizes the fine balance that had to be achieved during the selection between the optimization of binding affinity of the edited RNA and the minimization of the background.
A mechanism for the function of the aptamer switch
The edited and pre-edited forms of the selected RNAs are predicted to fold into different conformations (Jaeger et al. 1989; Zuker 2003), and this is illustrated for RNA B (Fig. 4). The conformation that is predicted to form with the edited RNA is similar to the parental sequence and is in overall agreement with the previously characterized streptavidin aptamer (cf. edited conformation in Fig. 2A and Fig. 4). In fact, several features of the streptavidin aptamer were reselected including parts of the key helices and even the loop nucleotides G-40, A-41, and A-42 that were within the fixed primer sequence of the initial streptavidin aptamer selection (Srisawat and Engelke 2001). In the absence of editing, the streptavidin-binding conformation would be destabilized by ∼3 kcal/mol resulting from the loss of the three internal A-U base pairs formed between the inserted U's and guiding A's (Fig. 4). A further 3.5-kcal/mol destabilization is predicted to result from the internal bulged loop arising from the unpaired guiding A's (Jaeger et al. 1989). As a result, alternative conformations would become more favorable in the absence of editing. One possible alternative conformation could simply result from one strand of the helix sliding so that U-30 becomes paired with A-80 in the pre-edited RNA rather than with A-76 when the RNA is edited (Fig. 4, cf. edited and major pre-edited conformations). This change is also predicted to disrupt the large loop of the streptavidin-binding aptamer.
FIGURE 4.

A mechanism for the aptamer switch. The pre-edited RNA is proposed to exist in equilibrium between a major conformation that is inactive for streptavidin binding and a minor conformation in which the streptavidin-binding aptamer is fully functional. The insertion of three U's by the editing reaction stabilizes the streptavidin-binding conformation. Base pairs within the edited and major pre-edited conformations that are supported by chemical probing and/or the selection phylogeny are indicated (●). The minor pre-edited conformation was inferred from the known structural requirements of the RNA editing reaction. Colors are as described for Figure 2. The three U's that are inserted by editing are not numbered.
For the RNA to be able to function as a substrate for the editing reaction, the major pre-edited conformation must be able to interconvert to a less stable minor conformation that places the guiding nucleotides, A-27 to A-29, opposite the editing site between A-76 and G-77 (Fig. 4). It is possible that the editing complex may directly facilitate the interconversion, or alternatively the editing complex may recognize and act on a small quantity of the minor conformation that is in pre-existing equilibrium with the major conformation.
Chemical probing of the switch
The outlined switching mechanism proposes that the major pre-edited and edited conformations have discrete differences in their secondary structures (Fig. 4). Both the pre-edited and edited forms of RNA B were probed with the nucleic-acid-modifying reagents dimethyl sulfate (DMS) and 1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide metho-p-toluene sulfonate (CMCT) to test whether these structural differences were present (Krol and Carbon 1989). DMS modifies the N1 position of A's and the N3 position of C's not involved in secondary or tertiary interactions, and CMCT modifies the unprotected N1 position of G's and the N3 position of U's. Sites of modifications were detected by reverse transcriptase primer extension, and a representative set of reactions is indicated for both the edited and pre-edited RNAs (Fig. 5). The intensity of the bands resulting from reverse transcriptase termination at chemically modified nucleotides was corrected for modification-independent termination, and the ratio of the corrected intensity obtained under native conditions to that obtained under denaturing conditions was determined for each position. Mean and standard deviations were calculated from three independent sets of reactions and are plotted in Figure 6. A difference in chemical reactivity between two corresponding nucleotides within the edited and pre-edited RNAs was defined as significant only if it was at least twice the standard deviation.
FIGURE 5.

A representative set of chemical probing reactions. (A,B) The edited and (C,D) pre-edited forms of RNA B were probed with DMS and CMCT under native and denaturing conditions. Sites of modification result in RT terminations. A RT primer complementary to the 3′ extension illustrated in Figure 3A was used to detect modifications of nucleotides #36–#81 and an internal primer used for nucleotides 3–36. Sites of modification migrate one nucleotide faster than the corresponding position within the RT dideoxy sequencing ladder. The 10 nucleotides that have significantly different chemical reactivity within the edited and pre-edited RNAs are indicated. The three U's that are inserted by editing are not numbered.
FIGURE 6.

Alterations of chemical reactivity in response to editing. The chemical reactivity values and standard deviations are plotted for each nucleotide within both the pre-edited (black) and edited (red) RNAs (n = 3). Those positions that have a chemical reactivity altered by at least two standard deviations in response to editing are indicated.
Editing of RNA B significantly altered the chemical reactivity of only 10 nucleotides (nt) (Figs. 5, 6), and these changes are highly consistent with the proposed switching mechanism (Fig. 4). G-22, U-23, U-24, A-25, and C-26 are predicted to be base paired in the edited RNA but are bulged in the major pre-edited conformation, and this is consistent with the observed increased reactivity at these sites within the pre-edited RNA. The reactivity of A-21 within the pre-edited RNA is also higher, suggesting that it is more accessible to modification within the predicted 6-nt loop as opposed to the single nucleotide loop of the edited RNA. A-27 is more reactive in the pre-edited RNA even though it could potentially pair with U-83, but fraying of this base pair at the end of the predicted helix could also account for the increased reactivity. The observed decrease in reactivity of A-32 in the pre-edited RNA is consistent with it being in the proposed base pair with U-78 and not paired within the edited structure. The different reactivity of U-52 is consistent with a structural change within the large loop of the streptavidin-binding aptamer in response to editing. Finally, the increased reactivity of A-76 in the pre-edited RNA is highly consistent with both the internal loop proposed for the pre-edited structure and the proposed base pair within the edited RNA.
The proposed switching mechanism predicted changes in DMS or CMCT reactivity at several sites that were not detected. These include nucleotides G-34, C-37, U-38, G-39, and G-70. The base modification reactions, however, can be inhibited by noncanonical base pairs and tertiary interactions that are not considered by the proposed secondary structures (Fig. 4). In addition, changes in chemical reactivity could not be evaluated at sites of intense modification-independent RT termination and at sites in close proximity to the 3′ primer used for the RT extensions. These include A-15, G-16, C-44, G-74, G-75, G-77, and nucleotides 82–90. Although additional changes are most likely occurring outside the sensitivity and resolution of the chemical probing, all of the changes that were detected are highly supportive of the proposed switching mechanism (Fig. 4).
Mutations testing the proposed mechanism
The binding affinity of edited RNA B for immobilized streptavidin approximates that of the parental streptavidin aptamer (Table 2). This suggests that the selected differences between these two RNAs resulted primarily from pressure on the switching function rather than the improvement of binding affinity. As a result, some of the selected sequence differences should provide insight into the switching mechanism. This is illustrated by the selected deletions centered near nucleotide 20 that are predicted to destabilize the helix proposed for the minor pre-edited and edited conformations, as compared to the corresponding parental sequence (Figs. 2A, 4, cf. ΔC and ΔA). In the absence of editing, the helix destabilization is predicted to favor the major pre-edited conformation and thereby minimize streptavidin binding. To test this possibility, the deleted nucleotides were added back to RNA B, and this resulted in the predicted effect of significantly increasing binding affinity of the pre-edited RNA (Table 2, mutations 18+C, 21+A). The same mutation did not have a significant effect on the binding affinity of the edited RNA, and this was expected since the U's added by editing provide additional helix stability. The selected U-31 and A-32 changes are also predicted to favor the major pre-edited conformation by forming base pairs with A-79 and U-78, respectively (Fig. 4). Restoring the parental sequence at these positions increased binding affinity of the pre-edited RNA, consistent with the major conformation being less stable with the parental sequence (Fig. 4; Table 2, mutations U31A, A32C).
Relative to the parental sequence, all but two of the selected RNAs have an extra nucleotide within the streptavidin-binding aptamer, G-39, as well as the U-38, C-68, and U-69 substitutions (Fig. 2C). The four changes were always selected together, suggesting that the nucleotides could covary and possibly interact. Changing U-38 and G-39 or C-68 and U-69 back to the parental sequence inhibited streptavidin binding with the edited RNA (Table 2, mutations U38A, ΔG39, mutations C68G, U69G). However, the simultaneous change of all four positions to the parental sequence restored binding affinity of the edited RNA, consistent with some type of interaction among these nucleotides (Table 2, mutations U38A, ΔG39, C68G, U69G). The four simultaneous changes also had the effect of increasing binding affinity of the pre-edited RNA. This is consistent with the proposed switching mechanism because the selected U-38 and G-39 nucleotides are predicted to stabilize the major pre-edited conformation through pairing with G-73 and C-72, respectively (Fig. 4). Likewise, the selected C-68 and U-69 nucleotides could destabilize the minor conformation by disrupting the base-pairing between the parental G-68 and G-69 nucleotides with U-52 and C-37.
The ability of the four simultaneous mutations to rescue binding is suggestive of complex interactions within the streptavidin-binding aptamer. It is also suggestive that some of the changes selected to optimize the switching mechanism may have necessitated additional changes to compensate for what could otherwise be detrimental alterations of the streptavidin-binding aptamer. Mutation of G-71 and the C-53–G-67 base pair within RNA B to the corresponding parental sequences significantly inhibited streptavidin binding (Table 2), and are possibly indicative of this effect.
Some of the selected sequences were also clearly under pressure to optimize in vitro editing. In addition to decreasing background binding, the deletions centered on nucleotide 20 also increased editing efficiency as mutations that restored these nucleotides inhibited editing (Table 2, mutations 18+C, 21+A). The disruption of the helix by the selected deletions possibly makes the editing site more accessible to the editing complex. The sequence and structure upstream of the editing site have previously been shown to influence editing efficiency (Igo et al. 2002; Pai et al. 2003), and as a result it is not surprising that the mutations of U-31 and A-32 also have major effects on editing. All of the other mutations within the streptavidin-binding aptamer had some type of minor effect on editing efficiency, suggesting that alterations in overall tertiary structure could influence the reaction (Table 2). This was previously observed for other in vitro editing substrates (Oppegard et al. 2003).
Conclusion
An assay exploiting a conformational change linked to ECL detection was described that is sensitive to low femtomole quantities of edited products. The assay has a good signal-to-background ratio that is considered to be well suited for high-throughput drug screens, and it can be performed in a 12-μL volume, within 384-well microtiter plates, which minimizes reagents and makes large-scale screens practical. Drugs identified from these screens not only will have potential therapeutic value, but will also be important biochemical probes for the characterization of the editing reaction. Similar assay strategies could be adapted to a wide range of other RNA processing reactions.
MATERIALS AND METHODS
Preparation of the editing extract and the in vitro editing reactions
A 25 L culture of the Leishmania tarentolae UC strain was grown to a density of ∼1.5 × 108 cells/mL, and the cells were lysed as previously described (Oppegard et al. 2000). A mitochondrial fraction was enriched through differential centrifugation, and taken up in 200 mL of resuspension buffer (0.5 mM dithiothreitol, 1 mM EDTA, 10% glycerol, 25 mM HEPES at pH 7.5, 10 μg/mL leupeptin, 10 mM MgCl2, and 1 mg/mL Pefabloc). The material that was soluble in 1.0% TX-100 was loaded onto a 15-mL SP-Sepharose column. After the column was washed with 10 volumes of resuspension buffer containing 100 mM KCl, the editing activity was eluted with 200 mM KCl in resuspension buffer. The fractions containing editing activity were pooled, and the KCl concentration was reduced to 150 mM by dilution with resuspension buffer. The extract was loaded onto a 2 mL Q-Sepharose column and after washing with 10 column volumes of resuspension buffer containing 150 mM KCl, the column was eluted with 225 mM KCl in resuspension buffer. Approximately 100,000 units of editing activity are obtained from the fractionation, where 1 unit of editing activity is defined as the quantity of extract resulting in 1 fmol of correctly edited product within 1 h under previously defined conditions (Pai et al. 2003). Although the editing activity would be enriched further by other purification strategies (Rusche et al. 1997; Madison-Antenucci et al. 1998; Panigrahi et al. 2001; Aphasizhev et al. 2003), this streamlined fractionation is relatively free of RNases and other complicating activities, results in relatively high yields, and can be readily scaled to produce sufficient material for high-throughput screens.
For a 50 μL in vitro editing reaction, 1 pmol of pre-edited RNA was denatured for 5 min at 65°C in 10 μL of denaturing buffer containing 0.2 mM EDTA and 25 mM Tris (pH 8.0, 27°C). The RNA was then mixed with 25 μL of a 2× editing buffer (2 mM ATP, 2 mM UTP, 2 mM dithiothreitol, 10 μg/mL leupeptin, 12 mM MgCl2, and 1 mg/mL Pefabloc SC) and incubated for 10 min at room temperature. The editing reaction was initiated by the addition of 700 units of editing activity in a 15 μL volume and incubated for 1 h at 27°C.
The in vitro selection
The starting 94-nt RNA used for the in vitro selection contained 21 randomized positions and key elements of the streptavidin-binding (Srisawat and Engelke 2001) and editing substrate (Pai et al. 2003) aptamers (Fig. 2A). The five positions immediately upstream of the editing site were kept fixed to facilitate the annealing of primers that overlapped the editing site. This permitted the RNA pool to be interconverted between the edited and pre-edited forms by using either an edited reverse primer (5′-AAAGCTTAAATTACAAATCCCG-3′) or a pre-edited reverse primer (5′-AAAGCTTAAATTACTCCCG-3′) during the RT-PCR. The same forward primer (5′-TAATACGACTCACTATAGGAAAGAATCTTCCAGA-3′) was used for both reactions. The initial cycle of selection contained 300 pmol of the randomized RNA pool that was synthesized by T7 RNA polymerase from the corresponding DNA template. Assuming no other biases, this would have resulted in a 99% probability of having representation of any combination of 21 nt (Ciesiolka et al. 1996). PCR mutagenesis was performed during the ninth cycle of selection using GenMorph II random mutagenesis (Stratagene).
For the positive binding selections (Table 1), the edited RNA pool was denatured for 5 min at 65°C in the denaturing buffer. The solution was then adjusted to a final concentration of 1 mM DTT, 0.2 mM EDTA, 50 mM KCl, 4 mM MgCl2, and 5 mM Tris (pH 8.0, 27°C). The RNA was allowed to refold for 10 min at room temperature and then incubated with a streptavidin matrix for 1 h at 27°C with mixing. The matrix was washed four times over a period of 1 h with 100 μL of binding buffer (4 mM MgCl2 and 5 mM Tris at pH 8.0) containing 50 mM KCl. The stringency of washing was increased for the last three cycles by raising the KCl concentration of the binding buffer to 150 mM. Bound RNAs were removed from the matrix by treatment with elution buffer (3 mM EDTA, 0.3 M sodium acetate at pH 5.2, and 7 M urea) for 30 min. The stringency of the positive binding selections was also progressively increased by decreasing the amount of RNA and streptavidin as follows: P-1 selections contained 300 pmol of RNA in a 100 μL volume that was mixed with 20 μL of settled streptavidin-agarose (∼35 pmol/μL immobilized streptavidin; Thermo Scientific). P-2 selections contained 2 pmol of RNA in a 50 μL volume that was incubated in 384-well streptavidin-coated plates (∼15 pmol of streptavidin/well; Thermo Scientific). The P-3 selection contained 1 pmol of RNA in a 50 μL volume that was incubated in streptavidin-coated 96-well ECL plates (∼100 fmol of streptavidin/well; Meso Scale Discovery). The P-4 selection contained 1 pmol of RNA in a 50 μL volume that was incubated in the streptavidin-coated high-capacity 384-well ECL plate (∼30 fmol of streptavidin/well; Meso Scale Discovery). The microtiter plates were pre-blocked with Super Block buffer (Thermo Scientific) to limit nonspecific binding and washed three times prior to use with 0.05% Tween 20 in 50 mM NaCl and 25 mM Tris (pH 8.0, 27°C).
For the negative binding selections, the RNA pool in the pre-edited form was denatured and allowed to refold as for the positive binding selections. N-1 selections (Table 1) contained 10–200 pmol of RNA in a 100 μL volume that was incubated with streptavidin-agarose as for the P-1 positive selections. The nonbound RNA was collected and ethanol-precipitated. N-2 negative selections involved treating the resuspended nonbound RNA a second time with fresh streptavidin-agarose.
The E-1 editing selections consisted of the treatment of 1 pmol of the pre-edited RNA pool in a 50 μL volume as described for the in vitro editing reactions. After the reaction, the edited RNA was resolved from the nonreacted RNA on a 6% polyacrylamide 8 M urea gel. Slower migrating RNA, consistent with 2–5 U insertions, was excised and eluted for subsequent enrichment. The pre-edited RNA used for the E-1 editing selections was circularized prior to the assay by treatment with RNA ligase (Brown et al. 1999). This functioned to block the 3′-end from being a substrate for a TUTase activity that is within the editing extract. Such activity would have resulted in a deterioration of the selection efficiency, as nonedited RNA with 3′ U additions would have comigrated with the edited product. For the E-2 selection, the editing and positive binding selection pressures were coupled by performing the editing reaction within streptavidin-coated 96-well ECL plates for 2 h at 27°C. The wells were washed four times over 1 h with 150 mM KCl in binding buffer and then eluted as for the positive binding selections. For the E-3 selection, the pre-edited RNA pool was labeled with the ruthenium complex to increase the likelihood that the selected RNA would be compatible with the ECL detection requirements. The reaction was the same as described for E-2 with the exception that one additional wash was done with Read Buffer T (Meso Scale Discovery), which is required for ECL generation. An editing-specific primer (5′-AAAGCTTAAATTACAAA-3′) was used for the RT-PCR of the RNAs enriched from the editing selections. This provided an additional means of limiting the amplification of contaminating nonedited RNAs.
The ECL assay of the editing reaction
The short aminoallyl derivatized RNA indicated in Figure 3A was transcribed by T7 RNA polymerase from the corresponding single-stranded oligodeoxynucleotide template as previously described (Milligan and Uhlenbeck 1989). The two 5′-most nucleotides of the template were modified with 2′-methoxyl groups to limit template-independent additions to the 3′ end (Kao et al. 1999). The transcription reactions contained 4 mM ATP, 4 mM CTP, 5 mM GMP, 1 mM GTP, 4 mM 5-(3-aminoallyl) UTP, 5 mM dithiothreitol, 22 mM MgCl2, 2 mM spermidine, 40 mM Tris (pH 8.2, 20°C), 0.01% Triton X-100, and 0.5 units/μL of T7 RNA polymerase (Invitrogen). The DNA templates for the in vitro transcription of the reporter RNAs were synthesized with the PCR primers already indicated for the in vitro selections with the exception that the reverse PCR primers also had the 2′-methoxyl modifications. The transcription of the reporter RNAs was done without modified nucleotides and GMP. After gel purification, the aminoallyl-derivitized RNA was ligated to the 3′ end of the reporter RNA using DNA ligase and the appropriate DNA splints (Moore and Sharp 1992). The ligated RNA was purified by gel electrophoresis, and 200 pmol were then reacted with 50 nmol of the N-hydroxysuccinimide ester of the ruthenium complex (Sulfo-Tag NHS-Ester; Meso Scale Discovery) in a 40 μL volume containing 45 mM NaHCO3 (pH 9.0). After incubating for 1 h at room temperature with mixing, the ruthenium-labeled RNA was purified from the nonreacted ruthenium by ethanol precipitation, followed by two successive washes in 75% ethanol. The RNA was resuspended in dH2O, and the quantity of incorporated ruthenium was estimated by measuring the absorbance at 455 nm (E455 = 15,400 M−1 cm−1).
The ECL assays were performed in streptavidin-coated, standard-capacity 384-well ECL plates (∼6 fmol of streptavidin/well; Meso Scale Discovery) that had been pre-blocked and washed as described for the positive selections. The minimal volume that could be used in these plates was determined to be 12 μL, and this was limited by the ability to efficiently mix different components rather than by the sensitivity of the assay. For the ECL assay of the editing reaction, 240 fmol of the ruthenium-labeled pre-edited RNA were treated with 160 units of the editing extract within a 12 μL volume under the conditions described for the editing reactions. After incubation for 2 h at 27°C with mixing, the microtiter plate was washed four times over a 1-h period with binding buffer containing 150 mM KCl. Read Buffer T was then added to the wells, and the ECL signals were recorded using a SECTOR Imager 6000 Reader (Meso Scale Discovery). The quantity of RNA in the assay can be reduced to 100 fmol and the amount of editing extract to 80 units with no significant deterioration in the assay signal (data not shown). Binding curves of the randomer and the pre-edited and edited RNAs were generated similarly but in the absence of editing extract.
Affinity determination
The apparent Kd values for the interaction of the RNAs with streptavidin were determined by performing 8–10 binding reactions with different concentrations of radiolabeled RNA (Fig. 7). Reactions were performed in a 50 μL volume within 384-well streptavidin coated plates (∼15 pmol of streptavidin/well). The plate blocking and binding reactions were as described for the positive binding selections except that the washing of the bound RNA involved only two very brief rinses with binding buffer containing 50 mM KCl. The bound RNA was removed from the wells with elution buffer and quantified by scintillation counting. Apparent Kd values were determined by plotting the bound RNA concentration (B) as a function of the total RNA concentration (R) and fitting to the equation (Lebruska and Maher 1999):
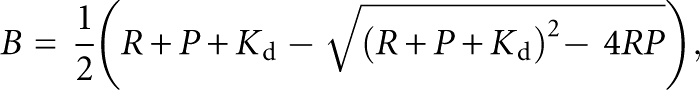 |
where P is the total effective streptavidin concentration on the plate, but it is not necessary to accurately know this value for the curve fitting, which was performed with Kaleidagraph software. Representative binding curves are indicated in Figure 7, and the standard deviations of the measured values from the fitted curves are indicated in Table 2. Because there are potential complications related to the immobilization of the streptavidin and the binding and washing conditions, the affinity measurements are referred to as apparent Kd values. The apparent Kd measured for the parental streptavidin aptamer is close to the previously published Kd value determined by a gel-shift assay (Table 2; Srisawat and Engelke 2001), which is suggestive that the potential complications are not having a major effect on the higher-affinity interactions. However, the lower-affinity interactions would be more vulnerable to small complicating effects.
FIGURE 7.

Representative binding curves used for the determination of apparent Kd values for the interaction of streptavidin with edited RNA B (▲), pre-edited RNA B (Δ), and the starting randomer (×). The RNA concentration points were all performed in duplicate, but because there is little variance, many of the duplicates are not resolved.
ACKNOWLEDGMENTS
We are grateful to Dr. Derek Hook of the University of Minnesota Institute for Therapeutics Discovery and Development for suggestions on the assay development. This work was supported by the U.S. Department of Defense, Grant W81XWH-06-1-0794, and by a grant from the Minnesota Medical Foundation.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1720209.
REFERENCES
- Amaro RE, Schnaufer A, Interthal H, Hol W, Stuart KD, McCammon JA. Discovery of drug-like inhibitors of an essential RNA-editing ligase in Trypanosoma brucei. Proc Natl Acad Sci. 2008;105:17278–17283. doi: 10.1073/pnas.0805820105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I, Nelson RE, Gao G, Simpson AM, Kang X, Falick AM, Sbicego S, Simpson L. Isolation of a U-insertion/deletion editing complex from Leishmania tarentolae mitochondria. EMBO J. 2003;22:913–924. doi: 10.1093/emboj/cdg083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benne R, Van den Burg J, Brakenhoff JP, Sloof P, Van Boom JH, Tromp MC. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell. 1986;46:819–826. doi: 10.1016/0092-8674(86)90063-2. [DOI] [PubMed] [Google Scholar]
- Best JD, Jay MT, Otu F, Ma J, Nadin A, Ellis S, Lewis HD, Pattison C, Reilly M, Harrison T, et al. Quantitative measurement of changes in Aβ40 in the rat brain and CSF following treatment with the γ-secretase inhibitor, N2-[(2S)-2-(3,5-Difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-dihydro-5Hdibenzo[b,d]azepin-7-yl]-L-alaninamide (LY-411575) J Pharmacol Exp Ther. 2005;313:902–908. doi: 10.1124/jpet.104.081174. [DOI] [PubMed] [Google Scholar]
- Blum B, Bakalara N, Simpson L. A model for RNA editing in kinetoplastid mitochondria: “Guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell. 1990;60:189–198. doi: 10.1016/0092-8674(90)90735-w. [DOI] [PubMed] [Google Scholar]
- Brown LM, Burbach BJ, McKenzie BA, Connell GJ. A cis-acting A-U sequence element induces kinetoplastid U-insertions. J Biol Chem. 1999;274:6295–6304. doi: 10.1074/jbc.274.10.6295. [DOI] [PubMed] [Google Scholar]
- Byrne EM, Connell GJ, Simpson L. Guide RNA-directed uridine insertion RNA editing in vitro. EMBO J. 1996;15:6758–6765. [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K. RNA editing in Trypanosoma brucei requires three different editosomes. Mol Cell Biol. 2008;28:122–130. doi: 10.1128/MCB.01374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesiolka J, Illangasekare M, Majerfeld I, Nickles T, Welch M, Yarus M, Zinnen S. Affinity selection-amplification from randomized ribooligonucleotide pools. Methods Enzymol. 1996;267:315–335. doi: 10.1016/s0076-6879(96)67021-9. [DOI] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Igo RP, Jr, Lawson SD, Stuart K. RNA sequence and base pairing effects on insertion editing in Trypanosoma brucei. Mol Cell Biol. 2002;22:1567–1576. doi: 10.1128/mcb.22.5.1567-1576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger JA, Turner DH, Zuker M. Improved predictions of secondary structures for RNA. Proc Natl Acad Sci. 1989;86:7706–7710. doi: 10.1073/pnas.86.20.7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kable ML, Seiwert SD, Heidmann S, Stuart K. RNA editing: A mechanism for gRNA-specified uridylate insertion into precursor mRNA. Science. 1996;273:1189–1195. doi: 10.1126/science.273.5279.1189. [DOI] [PubMed] [Google Scholar]
- Kao C, Zheng M, Rudisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA. 1999;5:1268–1272. doi: 10.1017/s1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol A, Carbon P. A guide for probing native small nuclear RNA and ribonucleoprotein structures. Methods Enzymol. 1989;180:212–227. doi: 10.1016/0076-6879(89)80103-x. [DOI] [PubMed] [Google Scholar]
- Lebruska LL, Maher LJ., III Selection and characterization of an RNA decoy for transcription factor NF-κB. Biochemistry. 1999;38:3168–3174. doi: 10.1021/bi982515x. [DOI] [PubMed] [Google Scholar]
- Madison-Antenucci S, Sabatini RS, Pollard VW, Hajduk SL. Kinetoplastid RNA-editing-associated protein 1 (Reap-1)—a novel editing complex protein with repetitive domains. EMBO J. 1998;17:6368–6376. doi: 10.1093/emboj/17.21.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: The 2′-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- Oppegard LM, Kabb AL, Connell GJ. Activation of guide RNA-directed editing of a cytochrome b mRNA. J Biol Chem. 2000;275:33911–33919. doi: 10.1074/jbc.M003002200. [DOI] [PubMed] [Google Scholar]
- Oppegard LM, Hillestad M, McCarthy RT, Pai RD, Connell GJ. Cis-acting elements stimulating kinetoplastid guide RNA-directed editing. J Biol Chem. 2003;278:51167–51175. doi: 10.1074/jbc.M307997200. [DOI] [PubMed] [Google Scholar]
- Pai RD, Oppegard LM, Connell GJ. Sequence and structural requirements for optimal guide RNA-directed insertional editing within Leishmania tarentolae. RNA. 2003;9:469–483. doi: 10.1261/rna.2175703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Gygi SP, Ernst NL, Igo RP, Palazzo SS, Schnaufer A, Weston DS, Carmean N, Salavati R, Aebersold R, et al. Association of two novel proteins, TbMP52 and TbMP48, with the Trypanosoma brucei RNA editing complex. Mol Cell Biol. 2001;21:380–389. doi: 10.1128/MCB.21.2.380-389.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Ernst NL, Domingo GJ, Fleck M, Salavati R, Stuart KD. Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA. 2006;12:1038–1049. doi: 10.1261/rna.45506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran M, Ellington AD. Selecting nucleic acids for biosensor applications. Comb Chem High Throughput Screen. 2002;5:263–270. doi: 10.2174/1386207023330246. [DOI] [PubMed] [Google Scholar]
- Rusche LN, Cruzreyes J, Piller KJ, Sollnerwebb B. Purification of a functional enzymatic editing complex from Trypanosoma brucei mitochondria. EMBO J. 1997;16:4069–4081. doi: 10.1093/emboj/16.13.4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaufer A, Panigrahi AK, Panicucci B, Igo RP, Jr, Wirtz E, Salavati R, Stuart K. An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science. 2001;291:2159–2162. doi: 10.1126/science.1058955. [DOI] [PubMed] [Google Scholar]
- Seiwert SD, Heidmann S, Stuart K. Direct visualization of uridylate deletion in vitro suggests a mechanism for kinetoplastid RNA editing. Cell. 1996;84:831–841. doi: 10.1016/s0092-8674(00)81062-4. [DOI] [PubMed] [Google Scholar]
- Srisawat C, Engelke DR. Streptavidin aptamers: Affinity tags for the study of RNAs and ribonucleoproteins. RNA. 2001;7:632–641. doi: 10.1017/s135583820100245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. Kinetoplastids: Related protozoan pathogens, different diseases. J Clin Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Wang B, Salavati R, Heidmann S, Stuart K. A hammerhead ribozyme substrate and reporter for in vitro kinetoplastid RNA editing. RNA. 2002;8:548–554. doi: 10.1017/s135583820202962x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]