

Abstract
We studied the peripheral motor axons of the two pyloric dilator (PD) neurons of the stomatogastric ganglion in the lobster, Homarus americanus. Intracellular recordings from the motor nerve showed both fast and slow voltage- and activity-dependent dynamics. During rhythmic bursts, the PD axons displayed changes in spike amplitude and duration. Pharmacological experiments and the voltage dependence of these phenomena suggest that inactivation of sodium and A-type potassium channels are responsible. In addition, the “resting” membrane potential was dependent on ongoing spike or burst activity, with more hyperpolarized values when activity was strong. Nerve stimulations, pharmacological block and current clamp experiments suggest that this is due to a functional antagonism between a slow after-hyperpolarization (sAHP) and inward rectification through hyperpolarization-activated current (I H). Dopamine application resulted in modest depolarization and “ectopic” peripheral spike initiation in the absence of centrally generated activity. This effect was blocked by CsCl and ZD7288, consistent with a role of I H. High frequency nerve stimulation inhibited peripheral spike initiation for several seconds, presumably due to the sAHP. Both during normal bursting activity and antidromic nerve stimulation, the conduction delay over the length of the peripheral nerve changed in a complex manner. This suggests that axonal membrane dynamics can have a substantial effect on the temporal fidelity of spike patterns propagated from a spike initiation site to a synaptic target, and that neuromodulators can influence the extent to which spike patterns are modified.
Introduction
Axon trunks are still commonly viewed as neuronal compartments whose sole function is to reliably propagate action potentials (spikes) from somatic and dendritic integration sites to electrotonically distant synaptic targets. The potential role of axon trunks in information processing and shaping of neuronal output is often underappreciated, despite the fact that extrasynaptic axonal regions can display dynamic properties that affect the reliability and temporal features of spike conduction (Debanne, 2004; Sidiropoulou et al., 2006). These properties largely result from rather complex intrinsic membrane properties that far exceed the commonly assumed minimal complement of voltage-gated ion channels required for spike propagation.
In fact, it is now well established that the diversity of voltage-gated ion channels that play a role in spike generation, spike shape, and control of activity can be substantial (Baranauskas, 2007; Bean, 2007). However, because of the difficulty of obtaining electrophysiological data from axons and the limited information about the spatial distribution of channels, it is often not clear whether their functional role lies primarily in spike initiation at the soma and proximal axon, or whether they also contribute to the dynamics of conduction and presynaptic transmitter release (Khaliq and Raman, 2006; Kole et al., 2007; Shu et al., 2007; Shah et al., 2008).
That said, the temporal patterns of spikes generated at soma and axon hillock can change during the conduction toward synaptic terminals. Such changes include frequency-dependent slowing or acceleration of conduction that result in changes of interspike intervals (Manor et al., 1991; Meeks and Mennerick, 2007; De Col et al., 2008), spike failures (Grossman et al., 1979; Miller and Rinzel, 1981; Debanne et al., 1997), and ectopic spike initiation (Kepler and Marder, 1993; Ma and LaMotte, 2007; Jiang et al., 2008).
Additional evidence that axon trunks have complex dynamic properties and are involved in shaping neuronal output comes from the finding that axonal excitability and spike conduction can be affected by a number of neuromodulators in a variety of vertebrate and invertebrate preparations. These modulators include serotonin (Meyrand et al., 1992; Mar and Drapeau, 1996; Beck et al., 2002; Lang et al., 2006; Scuri et al., 2007), dopamine (DA) (Bucher et al., 2003), Octopamine (Goaillard et al., 2004), GABA (Sakatani et al., 1993, 1994; Sun and Chiu, 1999; Verdier et al., 2003), and acetylcholine (Lang et al., 2003; Ding et al., 2004; Zhang et al., 2004; Kawai et al., 2007), and they affect both central and peripheral axons, in some cases even myelinated ones.
We have recently shown that the peripheral axon of the pyloric dilator (PD) neuron in the stomatogastric nervous system (STNS) of the lobster can initiate spikes in response to DA (Bucher et al., 2003). We now show that the PD axon displays complex membrane properties that affect spike shape and resting membrane potential in an activity-dependent manner and most likely the temporal fidelity of conducted bursts of spikes. In addition, we show that DA depolarizes the axon and present evidence that it acts on the hyperpolarization-activated inward current, I H.
Materials and Methods
Adult lobsters, Homarus americanus, were obtained from Commercial Lobster (Boston, MA) and kept in aerated filtered seawater tanks at 10−12°C. Lobsters were anesthetized on ice for at least 15 min before dissection. The STNS was removed from the stomach and placed into a transparent Sylgard-lined (Dow Corning) dish in saline. The saline composition was as follows (in mm): 479.12 NaCl, 12.74 KCl, 13.67 CaCl2, 10 MgSO4, 3.91 Na2SO4, and 10 HEPES, pH 7.4–7.5. To block potentially present Ca2+ currents, in some cases Ca2+ was mostly replaced with Mn2+ (1.3 CaCl2 and 11.62 MnCl2)
An outline of the ganglia and nerves that were kept intact is depicted in Figure 1 A (nomenclature after Maynard and Dando, 1974). They included the paired commissural ganglia, the esophageal ganglion, the stomatogastric ganglion (STG), and nerves connecting them. The PD neurons have their somata in the STG. They are unipolar and have a very complex central branching structure (Bucher et al., 2007). A single 10- to 20-μm-thick neurite projects into the core of the STG, gives off a number of secondary branches into the synaptic neuropil, and continues on as an axon that leaves the STG via the unpaired dvn. From there they project through the paired lateral ventricular nerves (lvn) which the dvn gives rise to, and the pyloric dilator nerves (pdn) which originate from the ventral lvn. The pdn innervates the ventral pyloric dilator muscles in the intact animal. The PD neurons are not bilateral homologues and each innervates muscles on both sides of the stomach. Therefore, not only the unpaired dvn, but also each lvn and pdn contains both PD axons.
Figure 1.

Intracellular PD axon recordings from peripheral nerve. A, Intracellular recordings of the PD soma and axon along with extracellular recording from the pdn during ongoing rhythmic pyloric activity. The schematic of the stomatogastric nervous system indicates the somatic and axonal recording sites. The soma recording shows attenuated spikes, whereas the axonal recording shows overshooting spikes. Spike amplitudes change over the course of the burst (white arrow) and ride on top of a baseline depolarization that starts abruptly with the first spike (black arrow). B, Simultaneous intracellular recordings from the soma and axon. The axon recording is from a site more proximal to the STG than used for the rest of this study. The bottom showing only the voltage range around the baseline of the axon recording reveals a slow depolarization that likely represents the attenuated remnant of the slow-wave oscillation seen in the soma recording (black arrows). C, Voltage attenuation from soma to axon. In the same experiment as shown in B, centrally generated activity was blocked and the soma penetrated with a second electrode. Steps of depolarizing and hyperpolarizing current injections show that voltage responses were substantially attenuated in the axon.
Electrophysiological recordings and data analysis.
For intracellular soma recordings and pharmacological application onto the central compartments of the PD neurons, the STG was desheathed. For intracellular axon recordings, a substantial part of the dvn was desheathed. In H. americanus, this nerve is covered with a thin outer and thick inner perineural sheath (Maynard and Dando, 1974; Bucher et al., 2007). The outer sheath was removed and the inner sheath slit open along the dvn, using the cut end of a pin made from tungsten wire (California Fine Wire). The sheath was then pinned to the side, exposing the bundle of axons.
During recordings, preparations were continuously superfused with cooled saline (12°C). Extracellular recordings of PD neuron spiking activity were obtained by placing a stainless steel wire electrode into a petroleum jelly well (diameter ≈1 mm) built around the distal part of the pdn. Signals were amplified and filtered using a differential AC amplifier (model 1700, A-M Systems). Nerve stimulation was achieved through the same electrode, using an isolated pulse stimulator (model 2100, A-M Systems).
Intracellular recordings were obtained from the PD axons in the dvn and/or the PD somata in the STG, using sharp glass microelectrodes filled with 0.6 m K2SO4 + 20 mm KCl, or 3 m KCl. Electrode resistance was between 20 and 40 MΩ. K2SO4 is used in STG soma recordings to avoid altering chloride conductances. In the PD axon, we did not detect a difference between the behavior of the membrane between recordings with either electrolyte, and subsequently mostly used KCl. For unknown reasons, using capacity overcompensation (“buzz”) to penetrate the axon membrane proved difficult and, when successful, often yielded rather depolarized potentials. Therefore, membrane penetration was achieved by tapping the micromanipulator (Leica) lightly with the back end of dissection forceps. Because of the relative difficulty to penetrate the axon membrane even with sharp electrodes, presumably because of the presence of glia cells, we refrained from attempting patch clamp recordings. Signals were amplified using Axoclamp 2A and 2B amplifiers (Molecular Devices). The PD axons were identified by the correspondence of their spike patterns with the extracellular pdn recording.
Although almost the entire PD axon is sensitive to application of DA, the most prevalent site of peripheral spike initiation in response to DA application is the lower dvn, close to the split into the 2 lvn (Bucher et al., 2003). We therefore wanted to obtain intracellular recordings from this site. The dvn contains some 20 large-diameter (∼10 μm) motor axons. These axons run as a cohesive bundle for most of the length of the dvn, but defasciculate near the dvn-lvn junction, which makes it difficult to obtain mechanically stable intracellular recordings at this site. Intracellular recordings from PD axons were therefore obtained slightly more proximal to the STG. The length of the dvn varies substantially across individuals. Therefore, recording sites varied between distances of 5 to 20 mm from the STG. Axon recordings showing interburst membrane potentials more depolarized than −55 mV were discarded and recordings showing spike amplitudes >3 SDs smaller than the mean were later excluded from analysis.
The large electrode resistances that we needed for axon recordings precluded switching rates in discontinuous current clamp that were sufficient to inject current and simultaneously accurately monitor fast transients like spikes, even when electrode tips were coated with Sylgard to reduce capacitance artifacts. Two-electrode recordings proved difficult to obtain because of axon size and limited mechanical stability. Nevertheless, in 21 experiments we successfully obtained simultaneous recordings with two electrodes, <1 mm apart from each other. Most of these recordings were used to characterize membrane responses to current injection in the absence of centrally generated activity.
Data were acquired using a micro 1401 digitizing board and Spike2 version 6 software (CED), and analyzed using programs written in the Spike2 script language. Plots were generated in Grapher (version 4, Golden Software) and Statview (version 5, SAS Institute), final figure mounting in Canvas 11 (ACD Systems). Statistical tests were performed in Statview and included linear regression analysis, paired t tests, and repeated-measurement ANOVA. Statistical significance is indicated in bar plots by asterisks (*p < 0.05; **p < 0.01, ***p < 0.001).
Pharmacological applications.
Tetrodotoxin (TTX) (100 nm, Sigma) in saline was bath-applied to block spiking activity when needed, and applied to a petroleum jelly well around the STG to block centrally generated bursting activity. In addition, the following blockers were bath-applied in different experiments: 5 mm CsCl (Acros Organics), or 50–100 μm ZD7288 (Tocris Bioscience) to block hyperpolarization-activated inward current (I H), 0.4–4 mm 4-aminopyridine (4-AP) to block A-type K+ currents, 0.5–2 mm CdCl2 (Sigma) to block Ca2+ currents, and 0.03–0.5 mm ouabain (Sigma) to block the electrogenic Na+/K+-pump.
DA (3-hydroxytyramine hydrochloride) (Sigma) was bath-applied at 1 nm or 1 μm. To avoid degradation, 1 mm stock solutions were always made fresh and kept in the dark on ice for no longer than 1 h. The concentrations were chosen because DA is not directly released into the peripheral nerves but present in the hemolymph and effective at hormonal concentrations (greater than nanomolar) (Bucher et al., 2003).
Results
Intracellular recordings from PD axons during ongoing rhythmic pyloric activity
Figure 1 A shows intracellular recordings from the PD soma in the STG and the PD axon in the dvn from two different experiments. The soma recording shows the typical slow-wave membrane potential oscillation underlying the rhythmic pyloric motor pattern (Harris-Warrick et al., 1992; Marder and Bucher, 2007). The spike initiation site in the STG is at some electrotonic distance to the mostly passive soma and fast membrane events like spikes are substantially attenuated in intracellular soma recordings. In contrast, PD axon recordings from the dvn displayed large spikes (Fig. 1 A). They also displayed a biphasic change in spike amplitude over the burst, with larger amplitudes at the beginning and at the end (Fig. 1 A, white arrow), and a substantial baseline depolarization during the burst. The latter did not rise from a slow-wave oscillation, but started abruptly with the first spike (black arrow).
PD axon recordings were obtained from sites electrotonically separated from the central part of the neuron
Before analyzing spike shapes and axonal membrane properties, we wanted to ensure that our axon recording sites were sufficiently distant from the STG to exclude a contribution of central cell compartments. Figure 1 B shows simultaneous recordings from the PD soma and the proximal axon. The first spike of the burst recorded in the axon is preceded by a small depolarization, presumably an attenuated remnant of the slow-wave depolarization seen in the soma recording (Fig. 1 B, arrows). We blocked impulse activity in the entire STNS and used two-electrode current clamp of the soma and simultaneous recording of the PD axon to estimate the length constant (n = 4). Figure 1 C shows the voltage responses in soma and axon to current injection into the soma. The voltage response in the axon is substantially attenuated. The mean length constant calculated from fits of exponential decay to the attenuation of both depolarizing and hyperpolarizing current steps was 1.50 mm (±0.32 SD), suggesting that dvn recording sites >5 mm from the STG are at an electrotonic distance of >3 length constants. Therefore, we are confident that in our experiments recording sites 5–20 mm from the STG were sufficiently electrotonically separated from central compartments of the neuron and our measurements do not reflect the intrinsic membrane properties of central branches.
Spike shape analysis
We analyzed spike shape during ongoing pyloric activity in 46 preparations. The mean baseline membrane potential in these preparations was −66.1 mV (±9.0 SD). The mean peak voltage of the largest spike in the burst showed considerable variability across preparations and ranged from −6.3 to +28.8 mV (+8.7 ± 7.8 mV, mean ± SD), but was overshooting in 40 of the 46 preparations (87%). 100 consecutive bursts from each experiment were analyzed. The mean pyloric cycle period was 0.99 s (±0.29 SD), the mean PD duty cycle was 0.35 (±0.05 SD), and the mean number of spikes per burst was 20.4 (±3.4 SD). Each measurement from the 100 bursts was sorted by index (spike number in the burst), and each data point represents the mean value for each spike index, plotted as a function of time after the first spike.
Reduction of spike amplitude was dependent on instantaneous spike frequency
The PD neurons in H. americanus are parabolic bursters, i.e., their instantaneous spike frequency increases toward the middle of the burst and decreases toward the end (Fig. 2 A), similar to the PD neurons in the California spiny lobster, Panulirus interruptus (Szucs et al., 2003). Across experiments, instantaneous spike frequency changed (mean ± SD) from 39.2 ± 10.3 Hz to 72.4 ± 16.1 Hz, with a mean difference between minimum and maximum instantaneous spike frequencies ranging from 17.2 to 58.4 Hz (33.2 ± 9.0 Hz). Changes in spike amplitude followed this pattern reciprocally (Fig. 2 A), with an average difference between maximum and minimum peak voltage of 4.7 mV (±3.0 SD), ranging from 0.9 to 16.0 mV. In five experiments, the axon did not display a biphasic change in spike amplitude. These data were excluded from quantification.
Figure 2.

Activity-dependent changes in spike peak voltage. A, Peak voltage and instantaneous spike frequency as a function of time over the course of a burst. Data are from 100 bursts in a single experiment. Single data points are superimposed with mean values sorted by spike index (±SD in x and y). spk, Spike. B, Peak voltage plotted as a function of instantaneous spike frequency, from the same data as in A. Mean values show slight differences for the first (right arrow) and second (left arrow) halves of the burst. C, Across experiments, the maximum difference in spike peak voltage was not dependent on the interburst membrane potential (Vm). D, Across experiments, the maximum difference in spike peak voltage was dependent on the maximum instantaneous spike frequency. IF, Instantaneous spike frequency; adj., adjusted; max, maximum.
The change in peak voltage was correlated with instantaneous spike frequency (Fig. 2 B). In all 41 experiments analyzed, linear regressions were statistically significant (adjusted R 2 values between 0.63 and 0.97, all p values <0.0001). In some experiments, the dependence of peak voltage on instantaneous frequency was slightly different for spikes during increasing or decreasing frequencies in the first and second half of the burst (Fig. 2 B, arrows), suggesting an additional dependence on spike history.
Across experiments, the difference between maximum and minimum peak voltages was not dependent on interburst membrane potential (R 2, 0.10, p = 0.34) (Fig. 2 C), but correlated with the maximum instantaneous spike frequency, with larger differences occurring at higher maximum spike frequencies (Fig. 2 D) (adjusted R 2: 0.12, p < 0.05).
Baseline depolarization during the burst was due to summation and enhanced by an increase in spike duration
Due to summation of spikes at the high instantaneous spike frequencies, a baseline depolarization of the PD axon membrane occurred during the burst. Figure 3 A shows a plot of the amplitudes of voltage minima between spikes over the burst from one experiment. For this analysis, the trough voltage between spike peaks was subtracted from the interburst membrane potential (Fig. 3 A, inset). Across experiments, the maximum amplitudes of this baseline depolarization ranged from 1.9 to 15.0 mV (6.9 ± 3.0 mV, mean ± SD).
Figure 3.

Baseline depolarization over the course of the burst. A, Trough amplitude as a function of time over the course of a burst. The inset indicates that trough amplitude was obtained as the difference between voltage minima between spikes and the resting membrane potential. Data are from 100 bursts in a single experiment. Single data points are superimposed with mean values sorted by spike index (±SD in x and y). spk, Spike. B, Trough amplitude plotted as a function of instantaneous spike frequency, from the same data as in A. Average values show slight differences for the first (lower arrow) and second half of the burst (upper arrow). C, D, Across experiments, the maximum trough amplitude was dependent on the maximum instantaneous spike frequency and the interburst membrane potential. amp, Amplitude; IF, instantaneous spike frequency; Vm, membrane potential; adj., adjusted; max, maximum.
The trough amplitudes were correlated with instantaneous spike frequency (Fig. 3 B). In all 46 experiments analyzed, linear regressions were statistically significant (adjusted R 2 values between 0.52 and 0.96, all p values <0.0001). In all experiments, the dependence of trough amplitude on instantaneous frequency was different for spikes during increasing or decreasing frequencies in the first and second half of the burst (Fig. 3 B, arrows), suggesting an additional dependence on spike history.
Across experiments, the maximum trough amplitude was dependent on both the maximum spike frequency (Fig. 3 C) (adjusted R 2, 0.34, p < 0.0001) and the interburst membrane potential (Fig. 3 D) (adjusted R 2, 0.37, p < 0.0001).
The spike repolarization time increased over the course of the burst. Figure 4 A shows multiple sweeps of all spikes in a single burst from a PD axon recording. Figure 4 B shows waveform averages of the first and last spike from each of the 100 bursts from the same experiment. Figure 4 C shows a plot of the spike duration at 1/6 of the amplitude of the first spike as a function of time after the first spike in one experiment. Across experiments, this duration ranged from 4.0 to 8.3 ms (5.9 ± 1.1 ms, mean ± SD), and the maximum spike duration in the burst ranged from 5.7 to 11.4 ms (8.5 ± 1.5 ms, mean ± SD). The average increase in spike duration over the burst was 2.6 ms (±1.1 SD).
Figure 4.

Increase in spike duration over the course of the burst. A, Multiple sweeps of all spikes in a single burst, aligned at the peak. B, Averaged traces of the first and last spike from 100 bursts. C, Spike duration at 1/6 of the amplitude of the first spike from 100 bursts in a single experiment. Single data points are superimposed with mean values sorted by spike index (±SD in x and y). D, Across experiments, the change in spike duration was not dependent on the maximum instantaneous spike frequency. E, Across experiments, the change in spike duration was dependent on the interburst membrane potential (Vm). F, Across experiments, the baseline depolarization under the burst maximum trough amplitude) was dependent on the increase in spike duration. adj., Adjusted.
The increase in spike duration was not dependent on the maximum spike frequency across experiments (Fig. 4 D) (adjusted R 2, 0.02; p = 0.17), but was greater at more hyperpolarized interburst membrane potentials (Fig. 4 E) (adjusted R 2, 0.35; p < 0.0001).
Because summation should depend on spike duration, we tested whether the increase in duration had an effect on baseline depolarization during the burst. Across experiments, the maximum trough amplitude was correlated with the increase in spike duration, with larger baseline depolarizations in experiments with a greater increase in spike duration (Fig. 4 F) (adjusted R 2, 0.75; p < 0.0001).
Changes in spike amplitude and duration were dependent on interburst membrane potential
Both the decrease of spike amplitude at higher frequencies and the increase in spike duration are consistent with channel inactivation. Sodium channel inactivation affects spike amplitude during repetitive firing (Grossman et al., 1979; Brody and Yue, 2000), and K+ channel inactivation, particularly of A- or D-type channels, leads to broadening of spikes (Geiger and Jonas, 2000; Kole et al., 2007; Shu et al., 2007). Changes in spike shape should therefore be dependent on the interburst membrane potential. Across experiments, we found a correlation between interburst membrane potential and spike broadening (Fig. 4 E) but not spike amplitude (Fig. 2 C). We therefore directly manipulated the membrane potential by injecting current through a second electrode (n = 4). Figure 5 A shows single bursts from the same recording at different membrane potentials, and Figure 5 B shows the corresponding multiple sweeps of all spikes in each burst. The change in spike amplitude was more severe at more depolarized membrane potentials. Spike duration was longer at more depolarized potentials, but did not increase as much as at hyperpolarized potentials. Both findings are consistent with larger portions of inactivated channels at more depolarized potentials.
Figure 5.

Changes in spike shape are likely due to channel inactivation. A, Bursts from a PD axon recording at three different interburst membrane potentials set by current injection through a second electrode. The change in spike amplitude is more pronounced at more depolarized potentials. B, Multiple sweeps from the bursts shown in A. The change in spike duration is more pronounced at more hyperpolarized potentials. C, The 1st and 100th spike from a PD axon recording in response to 20 Hz stimulation of the pdn, in control saline and 0.4 mm 4-AP. In 4-AP, duration was increased but did not change as much between 1st and 100th spike. Vm, Membrane potential. D, The increase of spike duration as a function of spike index in control saline (black) and 4-AP (gray). dur, Duration; amp, amplitude.
We tested whether the increase in duration could be due to the presence and inactivation of A-type K+ channels. In P. interruptus, the PD axons are immunoreactive for a shaker K+ channel subunit (Baro et al., 2000) but axonal A-type K+ currents have not been characterized. 4 mm 4-AP reliably blocks A-type K+ currents in the central compartments of STG neurons, but there are two technical caveats. In PD neurons, 4-AP transiently induces a leak current (Kloppenburg et al., 1999; Johnson et al., 2003) and 4 mm 4-AP also blocks up to 20% of delayed rectifier currents in a voltage-dependent manner (Gruhn et al., 2005). In our hands, 4 mm 4-AP in some experiments depolarized the PD axon by several mV when centrally generated activity was blocked. We therefore used 0.4 mm, which had no effect on membrane potential (mean ± SD) (control: −56.6 ± 5.1 mV, 4-AP: 55.1 ± 3.6 mV; n = 5; paired t test, p = 0.39). We stimulated the pdn to elicit 100 spikes at a frequency of 20 Hz. Figure 5 C shows that in the presence of 0.4 mm 4-AP, the duration of the first spike was larger than in control (7.9 ± 2.4 ms vs 5.6 ± 1.1 ms, mean ± SD; paired t test, p < 0.05) but the 100th spike did not increase in duration to the same degree. Figure 5 D shows that the mean increase in spike duration across all experiments as a function of spike index was larger in control than in 4-AP, consistent with a role of A-type K+ channel inactivation. The activation of a Ca2+ current would be another possible mechanism but experiments in which Ca2+ in the saline was replaced with Mn2+ did not show an effect on spike duration (data not shown).
The resting membrane potential was dependent on activity
In addition to the short-term activity- and voltage-dependent effects on spike shape, we found slower activity-dependent mechanisms that affected the resting membrane potential. Therefore, in the following the term “resting membrane potential” will be used in parentheses. In the experiments in which spike shapes were analyzed, the interburst membrane potential was weakly but significantly correlated with the strength of ongoing burst activity (Fig. 6 A). Linear regressions showed that across experiments the interburst membrane potential was more hyperpolarized at higher burst frequencies (adjusted R 2: 0.19; p < 0.01), higher spike frequencies within the burst (adjusted R 2: 0.12; p < 0.05), and higher overall spike frequencies (number of spikes/cycle period; adjusted R 2: 0.14; p < 0.01).
Figure 6.

The interburst membrane potential is dependent on the strength of bursting activity. A, Across experiments, the interburst membrane potential (Vm) was dependent on burst frequency, spike frequency within the burst, and overall spike frequency (spikes/cycle). B, PD axon recording showing depolarization in response to blocking centrally generated activity. adj., Adjusted. C, Change in “resting” membrane potential across experiments. Individual experiments are shown in gray, and mean data points (±SEM) in black. The difference between means was significant (paired t test; p < 0.0001; n = 35). D, PD axon recording with centrally generated activity blocked, showing hyperpolarization in response to stimulation of the pdn mimicking pyloric bursting activity. E, Change in “resting” membrane potential across experiments. Individual experiments are shown in gray, and mean data points (±SEM) in black. The difference between means was significant (paired t test; p < 0.0001; n = 17). F, PD axon recordings showing the response to stimulating the pdn with 10 s trains at 5, 10, 20, and 40 Hz. G, Hyperpolarization in response to stimulation was dependent on frequency (repeated-measures ANOVA, p < 0.0001; Fisher's PLSD post hoc, p values between 0.004 and <0.0001). stim, Stimulation.
In 35 experiments, we blocked centrally generated bursting activity. In all experiments, the membrane potential depolarized when spiking activity became weaker and ceased (Fig. 6 B). This effect was slow and a new steady state potential was only reached after 268–865 s (460 ± 147 s, mean ± SD). Across animals, the membrane potential changed from −64.8 ± 5.5 mV to −57.6 ± 4.5 mV SD (mean ± SD) (paired t test, p < 0.0001), depolarizing by 2.4–17.0 mV (Fig. 6 C, 7.3 ± 3.1 mV). In 10 of 35 experiments, the depolarization caused low frequency irregular firing or bursting, which is consistent with earlier findings from extracellular recordings of the isolated PD axon (Bucher et al., 2003).
In 17 experiments, we stimulated the pdn after central activity had ceased and a new steady state membrane potential had been reached. We mimicked realistic bursting by stimulating with trains of 19 pulses in a parabolic interval structure, with a period of 1 s, and 350 ms train duration. During a 5 min stimulus protocol, the interburst membrane potential hyperpolarized over the course of several minutes (Fig. 6 D). The average “resting” membrane potential changed from −58.6 ± 3.8 mV to −63.4 ± 4.1 mV (mean ± SD) (paired t test, p < 0.0001), hyperpolarizing by −2.0 to −13.0 mV (Fig. 6 E).
We also tested the dependency of interburst membrane potential on strength of ongoing spiking activity. We stimulated the pdn with single 10 s trains at tonic frequencies of 5, 10, 20, and 40 Hz. Figure 6 F shows that the hyperpolarization during the stimulus train increased with increasing stimulation frequencies. In addition, the axon membrane displayed a substantial sAHP that was also dependent on stimulation frequency. Across 24 experiments, the mean total changes in baseline membrane potential were –0.4 mV for 5 Hz, −0.9 mV for 10 Hz, −1.4 mV for 20 Hz, and −2.4 mV for 40 Hz (Fig. 6 G). Repeated-measures ANOVA (p < 0.0001) and subsequent Fisher's PLSD post hoc tests showed significant differences between measurements (p values between 0.004 and <0.0001).
We used 10 s/40 Hz pdn stimulations to test whether the hyperpolarization during stimulation and the following after-hyperpolarization were affected by ouabain and/or block of Ca2+ currents. Ouabain is an effective blocker of the Na+/K+-pump in the STNS (Hermann, 1981) and in our hands eventually lead to the breakdown of axonal membrane potential when we stimulated the nerve repeatedly. However, it did not noticeably block the sAHP (n = 3, data not shown). CdCl2 effectively blocks Ca2+-currents in STG neurons, and therefore subsequently calcium-dependent K+-currents (Golowasch and Marder, 1992). We used CdCl2 concentrations ranging from 0.5 to 2 mm (n = 4), and also blocked Ca2+ currents by replacing Ca2+ in the saline with Mn2+ (n = 3). Both treatments did not effectively block or substantially reduce the sAHP (Data not shown).
Intrinsic axonal membrane properties
In 21 experiments, we penetrated the axon membrane with two electrodes simultaneously, < 1 mm apart. We used these experiments to characterize intrinsic membrane properties in the absence of centrally generated activity.
The PD axon displayed substantial depolarizing sag potentials in response to hyperpolarizing current injections (Fig. 7 A, left trace, black arrow). These sag potentials were blocked by 5 mm CsCl (n = 7, Fig. 7 A, right trace) or 100 μm ZD7288 (n = 2, data not shown). We conclude the presence of a hyperpolarization-activated inward current (I H) in the PD axon. The axon membrane also displayed rebound depolarization following hyperpolarizing current injection (Fig. 7 A, white arrow). In all experiments in which I H was blocked (n = 9) this rebound firing persisted and often was only slightly weaker. In our experiments, rebound depolarization persisted when Ca2+ was replaced with Mn2+ (n = 2), and in the presence of TTX (n = 4), excluding a significant contribution of low threshold Ca2+ currents (Aizenman and Linden, 1999; Mitra and Miller, 2007) or TTX-sensitive persistent Na+ currents (Wu et al., 2005).
Figure 7.

Intrinsic membrane properties of the PD axon, tested in two-electrode current clamp in the absence of centrally generated activity. A, Depolarizing sag potential (black arrow) and rebound depolarization (white arrow) in response to hyperpolarizing current injection. The sag potential substantially reduced by CsCl, but the rebound to some degree persists. B, Spike frequency adaptation and sAHP in response to depolarizing current injections. The top panel shows the instantaneous spike frequency for each spike. Black arrows indicate the reduction in spike amplitude. The inset shows the sAHP (white arrow) from the voltage and time range indicated in the response to the third square pulse (dashed box).
In response to depolarizing current injections, the axon displayed spike frequency adaptation (SFA, n = 15, Fig. 7 B). SFA can be due to Ca2+-activated K+ currents, Na+ channel inactivation, and activation of the Na+/K+-pump (Powers et al., 1999). Both replacing Ca2+ with Mn2+ (n = 2) and bath application of ouabain (n = 2) did not block SFA (data not shown). However, SFA was accompanied by a substantial reduction in spike amplitude which was enhanced with larger current injections (Fig. 7 B, black arrows). This is consistent with a role of Na+ channel inactivation. We also consistently observed an sAHP (Fig. 7 B, inset), comparable to the one in response to nerve stimulations described in Figure 6, F and G.
We also obtained additional evidence for the presence of an A-type K+ current. One of the effects of A-currents is a delayed onset of spiking in response to depolarization. In 2 experiments, we blocked I H with 5 mm CsCl, and then held the membrane potential at −60 mV in current clamp, stepped it to a more hyperpolarized potential for 1 s to inactivate channels (to maximally −90 mV), and then to approximately −40 mV to elicit spikes. Figure 8 A shows that in control saline the delay to the first spike was larger with larger hyperpolarizing pre-steps, and that this effect was blocked by 4 mm 4-AP. Figure 8 B shows the delay to the first spike as a function of prestep voltage for one experiment. The delay effect was clearly blocked by 4-AP and reappeared after a 30 min wash. 4 mm 4-AP slightly depolarized the axon and the amount of current to hold the membrane potential at −60 mV had to be changed, consistent with the observation that 4 mm 4-AP induces a leak current in PD neurons (Kloppenburg et al., 1999; Johnson et al., 2003).
Figure 8.

Sensitivity of the voltage-dependent rebound delay to 4-AP. A, Two-electrode current clamp records of the PD axon's responses to depolarizing steps after hyperpolarizing presteps of different amplitude. The resting membrane potential was held at −60 mV, stepped to various hyperpolarized potentials up to −90 mV for 1 s, and then to −40 mV. In control saline, more hyperpolarized presteps lead to larger delays in the onset of spiking. In 4-AP, this delay is substantially reduced. B, Plots of delay over the prestep potential for the same experiment shown in A, for control saline, 4-AP, and after 30 min wash.
DA elicited peripheral spike initiation was due to modest depolarization and dependent on the presence of I H
We have shown previously that DA elicits peripheral spike initiation in the PD axon (Bucher et al., 2003), but these findings were based on intracellular soma recordings and extracellular nerve recordings and gave no insight into the underlying mechanism. Figure 9 A shows an intracellular PD axon recording in which bath application of 1 μm DA caused a modest depolarization that was sufficient to elicit spikes. Across experiments, both in 1 nm (n = 5) and 1 μm (n = 6) DA, we never observed more than a few millivolts of depolarization (Fig. 9 B), on average, 0.9 ± 1.5 mV and 6.8 ± 4.5 mV for 1 nm and 1 μm, respectively. Together with the finding that the small depolarization resulting from block of centrally generated activity (Fig. 6 B) are often sufficient to elicit spikes, our results suggest that the spike threshold of the axon is very close to the “resting” membrane potential of the quiescent axon. If modulation of voltage-gated ion channels underlies the DA effect, the channels involved should activate around “resting” membrane potential. The DA elicited depolarization persisted in the presence of TTX (n = 3) and peripheral spike initiation persisted in the presence of Cd2+ (n = 2) and when Ca2+ was replaced with Mn2+ (n = 2). However, both 5 mm CsCl (n = 2) and 50 μm ZD7288 (n = 3) blocked peripheral spike initiation (Fig. 9 C). It is therefore likely that DA elicits peripheral spike initiation predominantly by modulation of I H.
Figure 9.

Dopamine-induced depolarization of the PD axon. A, PD axon recording showing a slow depolarizing response to bath application of 1 μm dopamine. Centrally generated activity was blocked. B, “Resting” membrane potential (Vm) between control saline and dopamine for different experiments (open circles/gray, 1 nm; closed circles/black, 1 μm). C, PD axon recording showing that 1 μm dopamine elicited spikes on its own, but failed to do so in the presence of CsCl. After CsCl was washed, dopamine elicited spikes again.
Peripheral spike initiation was inhibited by the sAHP
The presence of I H and its modulation by DA together with the hyperpolarizing effect of spiking activity (Fig. 6) suggests that there exists a “tug-o-war” that determines the interburst or “resting” membrane potential. The sAHP is balanced by inward rectification through I H, and depolarization caused by DA activation of I H is counteracted by the sAHP. We have previously shown that DA-elicited peripheral spike initiation is inhibited by centrally generated bursts (Bucher et al., 2003). Therefore, we tested whether the sAHP was sufficient to exert such an effect. Figure 10 A shows an intracellular PD axon recording during a 10 s/40 Hz pdn stimulation in the presence of 1 μm DA. During stimulation, there is a pronounced hyperpolarization of the membrane potential. Following the stimulation, the axon was silent for several seconds as the membrane potential slowly depolarized again toward the spike threshold. Figure 10 B shows the mean duration of this “gap” in spiking for all stimulus frequencies and both 1 nm (n = 10) and 1 μm (n = 12) DA. The effects were quite variable in magnitude across experiments, but overall significant across different frequencies (repeated-measures ANOVA; 1 nm: p < 0.0058; 1 μm: p < 0.0001). For both concentrations, the 40 Hz stimulation had the largest effect (Fisher's PLSD post hoc tests, p values between 0.0163 and >0.0001).
Figure 10.

High frequency spiking suppresses dopamine elicited peripheral spike initiation. A, PD axon recording in the absence of centrally generated activity and the presence of 1 μm dopamine. Black boxes indicate timing of 10 s trains of pdn stimulations at different frequencies. stim, Stimulation. B, Quantification of the interval from the last stimulus to the first spike afterward for different stimulus frequencies in both dopamine concentrations.
The conduction delay between the central spike initiation site and the peripheral axon terminals changed over the course of the burst
The relatively complex membrane dynamics we describe here led us to ask how reliably temporal patterns can be conducted along the 4–5 cm of peripheral axon between the STG and the muscles. Figure 11 A shows an intracellular PD axon recording from the dvn and an extracellular recording of only this PD axon from the pdn. Figure 11 B shows sweeps of all intracellularly recorded spikes in the burst aligned at the peak, and the corresponding traces from the pdn recording with a constant offset from the first to the last spike in the burst. Over this distance of ∼4 cm, the conduction delay changed substantially over the course of the burst, decreasing and increasing in a complex pattern. Figure 11 C shows staggered multiple sweeps from an intracellular PD axon recording during a 40 Hz extracellular stimulation of the pdn. 100 stimuli are shown aligned at the pdn stimulus time. Delay initially decreases and then increases. Figure 11 D shows the change in delay for the first 2.5 s of 10 s pdn stimulations with 5, 10, 20, and 40 Hz as a function of time after the first stimulus (n = 28). In all cases, conduction delay changes in a biphasic way, decreasing first and then decreasing. The total change and the time course were dependent on stimulation frequency. Across experiments, plotting conduction delay as a function of spike peak voltage, spike duration, or trough voltage preceding the spike did not show consistent dependencies.
Figure 11.
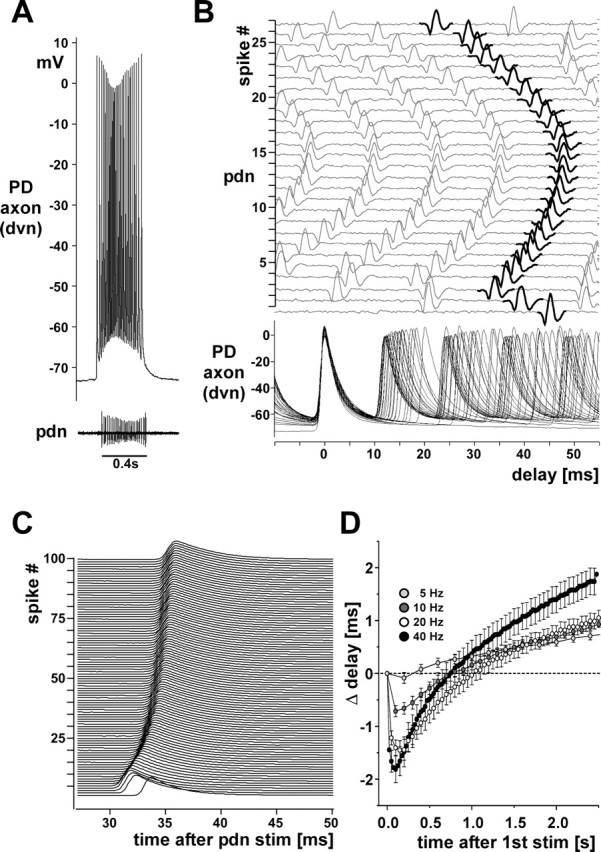
Changes in conduction delay. A, A single burst from a PD axon recording in an experiment in which the other PD neuron signal was missing on one of the pdns. B, Multiple sweeps of the recordings shown in A. Traces are aligned at the peaks of the intracellular spikes. Sweeps of the pdn recording are plotted with an offset and in order, with the extracellular spikes that correspond to the intracellular ones shown in black. Note the change of delay over the course of a single burst. C, Offset multiple sweeps from an intracellular PD axon recording during a 40 Hz extracellular stimulation of the pdn. One hundred stimuli are shown aligned at the pdn stimulus time. Delay initially decreases and then increases. D, Plot of the change in conduction delay over 2.5 s of pdn stimulations with 5, 10, 20, and 40 Hz (n = 28). stim, Stimulation.
Discussion
The peripheral motor axons of the PD neurons display voltage-, activity-, and modulator-dependent dynamics, including changes in spike shape and membrane potential, and DA-elicited peripheral spike initiation. These properties are largely due to a relatively rich complement of voltage-gated ion channels. Ephaptic interactions or changes in extracellular ionic composition due to activity in neighboring axons (Katz and Schmitt, 1940) are unlikely to play a significant role as the phenomena described persist when centrally generated activity is blocked and only the PD axons are stimulated. Axon trunks are still often thought to contain only the minimal number of channel types required for spike propagation (Hodgkin and Huxley, 1952; Connor, 1975). However, a range of different conductances can be present in distal nonsynaptic axonal regions, potentially lending computational power to the axon trunk (Debanne, 2004). Our results argue that the effect that axonal membrane properties and modulation have on spike conduction make axons conditional rather than reliable carriers of information.
Changes in spike shape
Even in axons that contain only fast Na+ and delayed rectifier K+ channels, channel inactivation and refractory effects limit spike conduction at higher frequencies (Miller and Rinzel, 1981). Na+ channel inactivation is implicated in slowed conduction and spike failures in a number of preparations (Chiu et al., 1979; Grossman et al., 1979; Brody and Yue, 2000; Meeks and Mennerick, 2004; De Col et al., 2008). The spike amplitude reduction at higher frequencies that we observed in the PD axons (Figs. 1 A, 2, 7 B) is consistent with Na+ channel inactivation, and it may also contribute to the SFA seen in response to depolarizing current injection (Fig. 7 B) (Powers et al., 1999).
Spike broadening can be due to K+ channel inactivation (Ma and Koester, 1996). In cortical axons, A-type and D-type K+ currents have such an effect, which has mostly been discussed with respect to consequences for presynaptic calcium influx (Geiger and Jonas, 2000; Kole et al., 2007; Shu et al., 2007). However, these channels are also expressed in nonsynaptic axonal membrane. In the PD axons, even centimeters away from the proximal spike initiation site and from distal synaptic terminals, 4-AP sensitive currents affect spike width. A-type K+ currents have also been implicated in the control of spike failures at axonal branch points (Debanne et al., 1997, 1999). We never observed spike failures along the PD axon trunk, which branches twice between our intracellular and extracellular recording sites (Fig. 1 A). However, spike failures may still occur at the terminal arborizations over the muscles.
Slower changes in membrane potential
The PD axon “resting” membrane potential is affected in several ways. During bursts, the high spike frequency in conjunction with slowing repolarization times lead to summation. All spikes after the first are therefore generated from a membrane potential more depolarized than the interburst potential.
In addition, the interburst membrane potential itself is dependent on activity. Due to an sAHP, spike and burst activity lead to hyperpolarization. The sAHP is relatively small in response to single bursts or spike trains, but summates over rhythmic activity. The PD neurons burst continuously during the lifetime of an animal (Marder and Bucher, 2007). In consequence, the actual interburst membrane potential would be >5 mV more hyperpolarized than the “resting” membrane potential in a quiescent axon. Changes in burst frequency and intensity would gradually affect the interburst membrane potential, with consequences for spike shape and conduction velocity.
We did not succeed in identifying the cellular basis of the sAHP. Most commonly, the sAHP in axons results from activation of the electrogenic Na+/K+-pump (Bostock and Grafe, 1985; Beaumont et al., 2002; Kiernan et al., 2004; Scuri et al., 2007). However, ouabain did not block the sAHP in the PD axon. Ca2+-dependent K+ channels are another possibility (Sah and Faber, 2002). To our knowledge, only channels or currents underlying faster AHPs have been described in axons (Lev-Ram and Grinvald, 1987; Knaus et al., 1996). In our experiments, blocking Ca2+ currents did not block the sAHP. We did not test for Na2+-dependent K+ currents. Such currents have been found in axons (Koh et al., 1994; Bhattacharjee et al., 2002) but are unlikely to be slow enough to generate an sAHP.
We also found slow inward rectification in response to hyperpolarizing current injections. I H has been found in a number of axons (Baker et al., 1987; Eng et al., 1990; Beaumont and Zucker, 2000; Soleng et al., 2003; Boyes et al., 2007; Luo et al., 2007; Jiang et al., 2008). A role for I H in balancing the sAHP has been suggested before for peripheral axons (Birch et al., 1991; Grafe et al., 1997; Kiernan et al., 2004) and presynaptic terminals (Beaumont et al., 2002). In the PD axon, this “tug-of-war” between sAHP and inward rectification is likely to be critical for the observed dependence of interburst membrane potential on ongoing activity. Any change in this balance, for example by modulation of either sAHP or I H, would shift the interburst membrane potential in opposite directions.
In addition, changes in interburst membrane potentials would affect spike shape and spike shape dynamics, as we demonstrated with DC current injections (Fig. 5 A,B). The inactivation and activation curves for both Na+ and A-type K+ currents overlap around resting membrane potential (Connor et al., 1977; Harris-Warrick et al., 1998). Therefore, how many channels are available and how substantial the effect of inactivation is for spike shape over the course of a burst depends on the interburst potential.
Neuromodulation of axons
The dynamic properties of axons and their involvement in shaping neuronal output potentially make them a target for neuromodulation. Serotonin can increase spike reliability in mammalian and leech axons (Mar and Drapeau, 1996; Lang et al., 2006). GABA changes conduction velocity in spinal and optic nerve axons in the neonatal rat (Sakatani et al., 1993, 1994), and blocks spike conduction in branches of sensory axons (Verdier et al., 2003). Acetylcholine increases axonal excitability in some unmyelinated axons (Armett and Ritchie, 1960, 1961; Lang et al., 2003).
In the STNS of the crab, serotonin elicits peripheral spike initiation in the axon of a motor neuron (Meyrand et al., 1992), and octopamine causes spike initiation in the axon of a modulatory projection neuron (Goaillard et al., 2004). In H. americanus, the peripheral PD axon initiates spikes in response to low concentrations of DA (Bucher et al., 2003). DA has rich and diverse actions on virtually all identified voltage-dependent currents in the central compartments of STG neurons (Harris-Warrick et al., 1998; Kloppenburg et al., 1999; Peck et al., 2001, 2006; Johnson et al., 2003, 2005; Gruhn et al., 2005). In the PD axons, it predominantly seems to act by increasing I H (Fig. 9 C).
We excluded the possibility that TTX-sensitive persistent Na+ currents or low threshold Ca2+ currents play a crucial role. Peripheral axons can express persistent Na+ currents (Bostock and Rothwell, 1997; Baker, 2000; Tamura et al., 2006), and there are numerous accounts of axonal Ca2+ channels (Lev-Ram and Grinvald, 1987; Wächtler et al., 1998; Mayer et al., 1999; Beck et al., 2001; Brown et al., 2001; Jackson et al., 2001; Lohr et al., 2001; Tamse and Yamoah, 2002; Le et al., 2006; Zhang et al., 2006; Tippens et al., 2008). In the PD axons, such currents are not involved in the DA-induced depolarization or in the rebound firing properties, but we cannot exclude the possibility that they are present. We also cannot exclude the possibility that DA may have additional targets, particularly outward currents. It may enhance or decrease K+ currents, i.e., either partially counteract or support axonal depolarization through I H activation.
Functional implications
Due to the low spike threshold and the relatively modest DA-induced depolarization, the sAHP is sufficient to explain the inhibition of peripheral spike initiation by centrally generated bursting activity (Fig. 10). As previously suggested, peripheral spike initiation during the interburst intervals may only play a role at lower rhythm frequencies and serve to rescue baseline tension in the muscles (Bucher et al., 2003).
In addition, DA-induced depolarization and increased I H may affect the temporal fidelity of axonal spike conduction. Changes in spike amplitude, duration, and interburst membrane potential can all affect conduction velocity (Miller and Rinzel, 1981; Kepler and Marder, 1993; Debanne, 2004; De Col et al., 2008). Figure 11 shows that conduction velocity during repetitive spiking changes in a complex manner, probably both dependent on instantaneous frequency and preceding spike patterns. Because DA affects interburst membrane potential and therefore also spike shape, the degree to which the centrally generated pattern is changed during the conduction over several centimeters of peripheral nerves is probably dependent on the presence of DA. We did not find simple correlations between spike shape parameters and conduction velocity. Therefore, a detailed analysis and pharmacological dissection of the contributions of different ion channels and their effects on spike shape and interburst membrane potential to the temporal fidelity of axonal conduction will be needed (Ballo and Bucher, in preparation).
Ultimately it is not clear whether changes in conduction velocity matter for the pyloric dilator muscles. However, membrane dynamics resulting from a complex complement of ionic conductances interacting at different time scales are likely to be a common phenomenon in axons and will affect temporal fidelity in many systems.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS058825 to D.B. We thank Dr. Eve Marder for initial support, and Drs. Marie-Luise Göritz, Peter A. Anderson, Jean-Marc Goaillard, and Adam L. Taylor for helpful discussions.
References
- Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–1709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Armett CJ, Ritchie JM. The action of acetylcholine on conduction in mammalian non-myelinated fibres and its prevention by an anticholinesterase. J Physiol. 1960;152:141–158. doi: 10.1113/jphysiol.1960.sp006476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armett CJ, Ritchie JM. The action of acetylcholine and some related substances on conduction in mammalian non-myelinated nerve fibres. J Physiol. 1961;155:372–384. doi: 10.1113/jphysiol.1961.sp006634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Bostock H, Grafe P, Martius P. Function and distribution of three types of rectifying channel in rat spinal root myelinated axons. J Physiol. 1987;383:45–67. doi: 10.1113/jphysiol.1987.sp016395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MD. Axonal flip-flops and oscillators. Trends Neurosci. 2000;23:514–519. doi: 10.1016/s0166-2236(00)01645-3. [DOI] [PubMed] [Google Scholar]
- Baranauskas G. Ionic channel function in action potential generation: current perspective. Mol Neurobiol. 2007;35:129–150. doi: 10.1007/s12035-007-8001-0. [DOI] [PubMed] [Google Scholar]
- Baro DJ, Ayali A, French L, Scholz NL, Labenia J, Lanning CC, Graubard K, Harris-Warrick RM. Molecular underpinnings of motor pattern generation: differential targeting of shal and shaker in the pyloric motor system. J Neurosci. 2000;20:6619–6630. doi: 10.1523/JNEUROSCI.20-17-06619.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Zucker RS. Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ih channels. Nat Neurosci. 2000;3:133–141. doi: 10.1038/72072. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Zhong N, Froemke RC, Ball RW, Zucker RS. Temporal synaptic tagging by I(h) activation and actin: involvement in long-term facilitation and cAMP-induced synaptic enhancement. Neuron. 2002;33:601–613. doi: 10.1016/s0896-6273(02)00581-0. [DOI] [PubMed] [Google Scholar]
- Beck A, Lohr C, Deitmer JW. Calcium transients in subcompartments of the leech Retzius neuron as induced by single action potentials. J Neurobiol. 2001;48:1–18. doi: 10.1002/neu.1039. [DOI] [PubMed] [Google Scholar]
- Beck A, Lohr C, Berthold H, Deitmer JW. Calcium influx into dendrites of the leech Retzius neuron evoked by 5-hydroxytryptamine. Cell Calcium. 2002;31:137–149. doi: 10.1054/ceca.2001.0268. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system. J Comp Neurol. 2002;454:241–254. doi: 10.1002/cne.10439. [DOI] [PubMed] [Google Scholar]
- Birch BD, Kocsis JD, Di Gregorio F, Bhisitkul RB, Waxman SG. A voltage- and time-dependent rectification in rat dorsal spinal root axons. J Neurophysiol. 1991;66:719–728. doi: 10.1152/jn.1991.66.3.719. [DOI] [PubMed] [Google Scholar]
- Bostock H, Grafe P. Activity-dependent excitability changes in normal and demyelinated rat spinal root axons. J Physiol. 1985;365:239–257. doi: 10.1113/jphysiol.1985.sp015769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol. 1997;498:277–294. doi: 10.1113/jphysiol.1997.sp021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes J, Bolam JP, Shigemoto R, Stanford IM. Functional presynaptic HCN channels in the rat globus pallidus. Eur J Neurosci. 2007;25:2081–2092. doi: 10.1111/j.1460-9568.2007.05463.x. [DOI] [PubMed] [Google Scholar]
- Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. J Neurosci. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Westenbroek RE, Catterall WA, Ransom BR. Axonal L-type Ca2+ channels and anoxic injury in rat CNS white matter. J Neurophysiol. 2001;85:900–911. doi: 10.1152/jn.2001.85.2.900. [DOI] [PubMed] [Google Scholar]
- Bucher D, Thirumalai V, Marder E. Axonal dopamine receptors activate peripheral spike initiation in a stomatogastric motor neuron. J Neurosci. 2003;23:6866–6875. doi: 10.1523/JNEUROSCI.23-17-06866.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher D, Johnson CD, Marder E. Neuronal morphology and neuropil structure in the stomatogastric ganglion of the lobster, Homarus americanus . J Comp Neurol. 2007;501:185–205. doi: 10.1002/cne.21169. [DOI] [PubMed] [Google Scholar]
- Chiu SY, Ritchie JM, Rogart RB, Stagg D. A quantitative description of membrane currents in rabbit myelinated nerve. J Physiol. 1979;292:149–166. doi: 10.1113/jphysiol.1979.sp012843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA. Neural repetitive firing: a comparative study of membrane properties of crustacean walking leg axons. J Neurophysiol. 1975;38:922–932. doi: 10.1152/jn.1975.38.4.922. [DOI] [PubMed] [Google Scholar]
- Connor JA, Walter D, McKown R. Neural repetitive firing: modifications of the Hodgkin-Huxley axon suggested by experimental results from crustacean axons. Biophys J. 1977;18:81–102. doi: 10.1016/S0006-3495(77)85598-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D. Information processing in the axon. Nat Rev Neurosci. 2004;5:304–316. doi: 10.1038/nrn1397. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Action-potential propagation gated by an axonal I(A)-like K+ conductance in hippocampus. Nature. 1997;389:286–289. doi: 10.1038/38502. [DOI] [PubMed] [Google Scholar]
- Debanne D, Kopysova IL, Bras H, Ferrand N. Gating of action potential propagation by an axonal A-like potassium conductance in the hippocampus: a new type of non-synaptic plasticity. J Physiol Paris. 1999;93:285–296. doi: 10.1016/s0928-4257(00)80057-1. [DOI] [PubMed] [Google Scholar]
- De Col R, Messlinger K, Carr RW. Conduction velocity is regulated by sodium channel inactivation in unmyelinated axons innervating the rat cranial meninges. J Physiol. 2008;586:1089–1103. doi: 10.1113/jphysiol.2007.145383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding YS, Fowler JS, Logan J, Wang GJ, Telang F, Garza V, Biegon A, Pareto D, Rooney W, Shea C, Alexoff D, Volkow ND, Vocci F. 6-[18F]Fluoro-A-85380, a new PET tracer for the nicotinic acetylcholine receptor: studies in the human brain and in vivo demonstration of specific binding in white matter. Synapse. 2004;53:184–189. doi: 10.1002/syn.20051. [DOI] [PubMed] [Google Scholar]
- Eng DL, Gordon TR, Kocsis JD, Waxman SG. Current-clamp analysis of a time-dependent rectification in rat optic nerve. J Physiol. 1990;421:185–202. doi: 10.1113/jphysiol.1990.sp017940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Goaillard JM, Schulz DJ, Kilman VL, Marder E. Octopamine modulates the axons of modulatory projection neurons. J Neurosci. 2004;24:7063–7073. doi: 10.1523/JNEUROSCI.2078-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golowasch J, Marder E. Ionic currents of the lateral pyloric neuron of the stomatogastric ganglion of the crab. J Neurophysiol. 1992;67:318–331. doi: 10.1152/jn.1992.67.2.318. [DOI] [PubMed] [Google Scholar]
- Grafe P, Quasthoff S, Grosskreutz J, Alzheimer C. Function of the hyperpolarization-activated inward rectification in nonmyelinated peripheral rat and human axons. J Neurophysiol. 1997;77:421–426. doi: 10.1152/jn.1997.77.1.421. [DOI] [PubMed] [Google Scholar]
- Grossman Y, Parnas I, Spira ME. Differential conduction block in branches of a bifurcating axon. J Physiol. 1979;295:283–305. doi: 10.1113/jphysiol.1979.sp012969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruhn M, Guckenheimer J, Land B, Harris-Warrick RM. Dopamine modulation of two delayed rectifier potassium currents in a small neural network. J Neurophysiol. 2005;94:2888–2900. doi: 10.1152/jn.00434.2005. [DOI] [PubMed] [Google Scholar]
- Harris-Warrick R, Marder E, Selverston AI, Moulins M, editors. Cambridge, MA: MIT; 1992. Dynamic biological networks: the stomatogastric nervous system. [Google Scholar]
- Harris-Warrick RM, Johnson BR, Peck JH, Kloppenburg P, Ayali A, Skarbinski J. Distributed effects of dopamine modulation in the crustacean pyloric network. Ann N Y Acad Sci. 1998;860:155–167. doi: 10.1111/j.1749-6632.1998.tb09046.x. [DOI] [PubMed] [Google Scholar]
- Hermann A. Action of caffeine on pyloric motorneurons in the crustacean stomatogastric ganglion. Comp Biochem Physiol C. 1981;69:191–197. doi: 10.1016/0306-4492(81)90128-3. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson VM, Trout SJ, Brain KL, Cunnane TC. Characterization of action potential-evoked calcium transients in mouse postganglionic sympathetic axon bundles. J Physiol. 2001;537:3–16. doi: 10.1111/j.1469-7793.2001.0003k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YQ, Xing GG, Wang SL, Tu HY, Chi YN, Li J, Liu FY, Han JS, Wan Y. Axonal accumulation of hyperpolarization-activated cyclic nucleotide-gated cation channels contributes to mechanical allodynia after peripheral nerve injury in rat. Pain. 2008 doi: 10.1016/j.pain.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Johnson BR, Kloppenburg P, Harris-Warrick RM. Dopamine modulation of calcium currents in pyloric neurons of the lobster stomatogastric ganglion. J Neurophysiol. 2003;90:631–643. doi: 10.1152/jn.00037.2003. [DOI] [PubMed] [Google Scholar]
- Johnson BR, Schneider LR, Nadim F, Harris-Warrick RM. Dopamine modulation of phasing of activity in a rhythmic motor network: contribution of synaptic and intrinsic modulatory actions. J Neurophysiol. 2005;94:3101–3111. doi: 10.1152/jn.00440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Schmitt OH. Electric interaction between two adjacent nerve fibres. J Physiol. 1940;97:471–488. doi: 10.1113/jphysiol.1940.sp003823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H, Lazar R, Metherate R. Nicotinic control of axon excitability regulates thalamocortical transmission. Nat Neurosci. 2007;10:1168–1175. doi: 10.1038/nn1956. [DOI] [PubMed] [Google Scholar]
- Kepler TB, Marder E. Spike initiation and propagation on axons with slow inward currents. Biol Cybern. 1993;68:209–214. doi: 10.1007/BF00224853. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26:1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan MC, Lin CS, Burke D. Differences in activity-dependent hyperpolarization in human sensory and motor axons. J Physiol. 2004;558:341–349. doi: 10.1113/jphysiol.2004.063966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloppenburg P, Levini RM, Harris-Warrick RM. Dopamine modulates two potassium currents and inhibits the intrinsic firing properties of an identified motor neuron in a central pattern generator network. J Neurophysiol. 1999;81:29–38. doi: 10.1152/jn.1999.81.1.29. [DOI] [PubMed] [Google Scholar]
- Knaus HG, Schwarzer C, Koch RO, Eberhart A, Kaczorowski GJ, Glossmann H, Wunder F, Pongs O, Garcia ML, Sperk G. Distribution of high-conductance Ca(2+)-activated K+ channels in rat brain: targeting to axons and nerve terminals. J Neurosci. 1996;16:955–963. doi: 10.1523/JNEUROSCI.16-03-00955.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DS, Jonas P, Vogel W. Na(+)-activated K+ channels localized in the nodal region of myelinated axons of Xenopus . J Physiol. 1994;479:183–197. doi: 10.1113/jphysiol.1994.sp020287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- Lang PM, Burgstahler R, Sippel W, Irnich D, Schlotter-Weigel B, Grafe P. Characterization of neuronal nicotinic acetylcholine receptors in the membrane of unmyelinated human C-fiber axons by in vitro studies. J Neurophysiol. 2003;90:3295–3303. doi: 10.1152/jn.00512.2003. [DOI] [PubMed] [Google Scholar]
- Lang PM, Moalem-Taylor G, Tracey D, Bostock H, Grafe P. Activity-dependent modulation of axonal excitability in unmyelinated peripheral rat nerve fibers by the 5-HT(3) serotonin receptor. J Neurophysiol. 2006;96:2963–2971. doi: 10.1152/jn.00716.2006. [DOI] [PubMed] [Google Scholar]
- Le T, Verley DR, Goaillard JM, Messinger DI, Christie AE, Birmingham JT. Bistable behavior originating in the axon of a crustacean motor neuron. J Neurophysiol. 2006;95:1356–1368. doi: 10.1152/jn.00893.2005. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Grinvald A. Activity-dependent calcium transients in central nervous system myelinated axons revealed by the calcium indicator Fura-2. Biophys J. 1987;52:571–576. doi: 10.1016/S0006-3495(87)83246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr C, Beck A, Deitmer JW. Activity-dependent accumulation of Ca2+ in axon and dendrites of the leech Leydig neuron. Neuroreport. 2001;12:3649–3653. doi: 10.1097/00001756-200112040-00009. [DOI] [PubMed] [Google Scholar]
- Luo L, Chang L, Brown SM, Ao H, Lee DH, Higuera ES, Dubin AE, Chaplan SR. Role of peripheral hyperpolarization-activated cyclic nucleotide-modulated channel pacemaker channels in acute and chronic pain models in the rat. Neuroscience. 2007;144:1477–1485. doi: 10.1016/j.neuroscience.2006.10.048. [DOI] [PubMed] [Google Scholar]
- Ma C, LaMotte RH. Multiple sites for generation of ectopic spontaneous activity in neurons of the chronically compressed dorsal root ganglion. J Neurosci. 2007;27:14059–14068. doi: 10.1523/JNEUROSCI.3699-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Koester J. The role of K+ currents in frequency-dependent spike broadening in Aplysia R20 neurons: a dynamic-clamp analysis. J Neurosci. 1996;16:4089–4101. doi: 10.1523/JNEUROSCI.16-13-04089.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor Y, Koch C, Segev I. Effect of geometrical irregularities on propagation delay in axonal trees. Biophys J. 1991;60:1424–1437. doi: 10.1016/S0006-3495(91)82179-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar A, Drapeau P. Modulation of conduction block in leech mechanosensory neurons. J Neurosci. 1996;16:4335–4343. doi: 10.1523/JNEUROSCI.16-14-04335.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Bucher D. Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu Rev Physiol. 2007;69:291–316. doi: 10.1146/annurev.physiol.69.031905.161516. [DOI] [PubMed] [Google Scholar]
- Mayer C, Quasthoff S, Grafe P. Confocal imaging reveals activity-dependent intracellular Ca2+ transients in nociceptive human C fibres. Pain. 1999;81:317–322. doi: 10.1016/S0304-3959(99)00015-9. [DOI] [PubMed] [Google Scholar]
- Maynard DM, Dando MR. The structure of the stomatogastric neuromuscular system in Callinectes sapidus, Homarus americanus and Panulirus argus (Decapoda Crustacea) Philos Trans R Soc Lond B. 1974;268:161–220. doi: 10.1098/rstb.1974.0024. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Selective effects of potassium elevations on glutamate signaling and action potential conduction in hippocampus. J Neurosci. 2004;24:197–206. doi: 10.1523/JNEUROSCI.4845-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Action potential initiation and propagation in CA3 pyramidal axons. J Neurophysiol. 2007;97:3460–3472. doi: 10.1152/jn.01288.2006. [DOI] [PubMed] [Google Scholar]
- Meyrand P, Weimann JM, Marder E. Multiple axonal spike initiation zones in a motor neuron: serotonin activation. J Neurosci. 1992;12:2803–2812. doi: 10.1523/JNEUROSCI.12-07-02803.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RN, Rinzel J. The dependence of impulse propagation speed on firing frequency, dispersion, for the Hodgkin-Huxley model. Biophys J. 1981;34:227–259. doi: 10.1016/S0006-3495(81)84847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra P, Miller RF. Mechanism underlying rebound excitation in retinal ganglion cells. Vis Neurosci. 2007;24:709–731. doi: 10.1017/S0952523807070654. [DOI] [PubMed] [Google Scholar]
- Peck JH, Nakanishi ST, Yaple R, Harris-Warrick RM. Amine modulation of the transient potassium current in identified cells of the lobster stomatogastric ganglion. J Neurophysiol. 2001;86:2957–2965. doi: 10.1152/jn.2001.86.6.2957. [DOI] [PubMed] [Google Scholar]
- Peck JH, Gaier E, Stevens E, Repicky S, Harris-Warrick RM. Amine modulation of Ih in a small neural network. J Neurophysiol. 2006;96:2931–2940. doi: 10.1152/jn.00423.2005. [DOI] [PubMed] [Google Scholar]
- Powers RK, Sawczuk A, Musick JR, Binder MD. Multiple mechanisms of spike-frequency adaptation in motoneurones. J Physiol Paris. 1999;93:101–114. doi: 10.1016/s0928-4257(99)80141-7. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Sakatani K, Chesler M, Hassan AZ, Lee M, Young W. Non-synaptic modulation of dorsal column conduction by endogenous GABA in neonatal rat spinal cord. Brain Res. 1993;622:43–50. doi: 10.1016/0006-8993(93)90799-s. [DOI] [PubMed] [Google Scholar]
- Sakatani K, Hassan AZ, Chesler M. Effects of GABA on axonal conduction and extracellular potassium activity in the neonatal rat optic nerve. Exp Neurol. 1994;127:291–297. doi: 10.1006/exnr.1994.1105. [DOI] [PubMed] [Google Scholar]
- Scuri R, Lombardo P, Cataldo E, Ristori C, Brunelli M. Inhibition of Na+/K+ ATPase potentiates synaptic transmission in tactile sensory neurons of the leech. Eur J Neurosci. 2007;25:159–167. doi: 10.1111/j.1460-9568.2006.05257.x. [DOI] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Yu Y, Yang J, McCormick DA. Selective control of cortical axonal spikes by a slowly inactivating K+ current. Proc Natl Acad Sci U S A. 2007;104:11453–11458. doi: 10.1073/pnas.0702041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidiropoulou K, Pissadaki EK, Poirazi P. Inside the brain of a neuron. EMBO Rep. 2006;7:886–892. doi: 10.1038/sj.embor.7400789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleng AF, Chiu K, Raastad M. Unmyelinated axons in the rat hippocampus hyperpolarize and activate an H current when spike frequency exceeds 1 Hz. J Physiol. 2003;552:459–470. doi: 10.1113/jphysiol.2003.048058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun BB, Chiu SY. N-type calcium channels and their regulation by GABAB receptors in axons of neonatal rat optic nerve. J Neurosci. 1999;19:5185–5194. doi: 10.1523/JNEUROSCI.19-13-05185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucs A, Pinto RD, Rabinovich MI, Abarbanel HD, Selverston AI. Synaptic modulation of the interspike interval signatures of bursting pyloric neurons. J Neurophysiol. 2003;89:1363–1377. doi: 10.1152/jn.00732.2002. [DOI] [PubMed] [Google Scholar]
- Tamse CT, Yamoah EN. Elementary properties of axonal calcium currents in type B photoreceptors in Hermissenda crassicornis . J Neurosci. 2002;22:10533–10538. doi: 10.1523/JNEUROSCI.22-24-10533.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura N, Kuwabara S, Misawa S, Kanai K, Nakata M, Sawai S, Hattori T. Increased nodal persistent Na+ currents in human neuropathy and motor neuron disease estimated by latent addition. Clin Neurophysiol. 2006;117:2451–2458. doi: 10.1016/j.clinph.2006.07.309. [DOI] [PubMed] [Google Scholar]
- Tippens AL, Pare JF, Langwieser N, Moosmang S, Milner TA, Smith Y, Lee A. Ultrastructural evidence for pre- and postsynaptic localization of Cav1.2 L-type Ca2+ channels in the rat hippocampus. J Comp Neurol. 2008;506:569–583. doi: 10.1002/cne.21567. [DOI] [PubMed] [Google Scholar]
- Verdier D, Lund JP, Kolta A. GABAergic control of action potential propagation along axonal branches of mammalian sensory neurons. J Neurosci. 2003;23:2002–2007. doi: 10.1523/JNEUROSCI.23-06-02002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wächtler J, Mayer C, Grafe P. Activity-dependent intracellular Ca2+ transients in unmyelinated nerve fibres of the isolated adult rat vagus nerve. Pflugers Arch. 1998;435:678–686. doi: 10.1007/s004240050569. [DOI] [PubMed] [Google Scholar]
- Wu N, Enomoto A, Tanaka S, Hsiao CF, Nykamp DQ, Izhikevich E, Chandler SH. Persistent sodium currents in mesencephalic v neurons participate in burst generation and control of membrane excitability. J Neurophysiol. 2005;93:2710–2722. doi: 10.1152/jn.00636.2004. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Verbny Y, Malek SA, Stys PK, Chiu SY. Nicotinic acetylcholine receptors in mouse and rat optic nerves. J Neurophysiol. 2004;91:1025–1035. doi: 10.1152/jn.00769.2003. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Wilson JA, Williams J, Chiu SY. Action potentials induce uniform calcium influx in mammalian myelinated optic nerves. J Neurophysiol. 2006;96:695–709. doi: 10.1152/jn.00083.2006. [DOI] [PubMed] [Google Scholar]