

Abstract
Signals that control the fine balance between cell death and cell survival are altered in cells during tumorigenesis. Understanding the mechanisms by which this balance is perturbed, leading to excessive cell survival, is important for designing effective therapies. Proteins belonging to the BCL family are known to regulate death responses to apoptotic signals, especially those originating within cells. A subset of BCL family members capable of inhibiting cell death is known to contribute to tumorigenesis; however, it is not known whether all six anti-apoptotic BCL family members play a causal role in tumor development. Using a mouse model of MYC-driven leukemia we demonstrate that, in addition to the well characterized BCL2 and BCLxl (BCL2L1), the other four family members- BCLw (BCL2L2), BCLb (BCL2L10), BFL1 (BCL2A1), and MCL1- also cooperate with MYC to accelerate leukemogenesis. In addition, high levels of each family member are found in either solid human tumors or cell lines derived from human leukemias or lymphomas.
Keywords: BCL2, BCLxl, BCLw, MCL1, BFL1, BCLb
The ability of cells to undergo a regulated and programmed death is critical to the development and survival of multi-cellular organisms (Cory et al., 2003; Youle & Strasser, 2008). De-regulation of the processes that govern this mode of cell death, known as apoptosis, has been linked to developmental disorders and diseases, including some cancers (Horvitz, 1999). The intrinsic apoptotic pathway is regulated, in large part, by the mitochondrial outer membrane permeabilization (MOMP); when MOMP occurs, cytochrome c is released, and downstream cysteinyl aspartate-specific proteases, called caspases become activated (Riedl & Salvesen, 2007). Members of the BCL2 (B-cell Lymphoma, BCL) family have the ability to regulate MOMP, positively or negatively, and are divided into two classes, pro- or anti-apoptotic, based on whether they lead to MOMP (pro-apoptotic) or inhibit MOMP (anti-apoptotic).
Since the initial discovery of BCL2, 24 additional family members have been identified based on the presence of a small amino acid motif, known as the BCL homology domain (BH) and only six members perform anti-apoptotic functions (BCL2, BCLxl/BCL2L1, BCLw/BCL2L2, BFL1/BCL2A1, MCL1, and BCLb/BCL2L10) (Lanave et al., 2004). Although each of the six anti-apoptotic BCL family members has been shown, in vitro, to inhibit apoptosis caused by various stimuli, there is mounting evidence that these proteins also perform distinct functions. The strongest support for this came from the analysis of mice engineered to lack the expression of individual BCL genes. The phenotypes of BCL-null mice range from embryonic lethality (BCLxl and MCL1), to abnormal death of sperm cells (BCLw), to no identifiable phenotype (BCLb) (Youle & Strasser, 2008).
Multiple studies have recently examined the in vitro protein-protein interactions and relative anti-apoptotic abilities of all six anti-apoptotic BCL family members (Chen et al., 2005; Zhai et al., 2006). However, a comprehensive in vivo comparison of the oncogenic potential of the six BCL family members has not been reported. To date, only BCL2 and BCLxl have been shown to be bona fide oncogenes (Swanson et al., 2004; Vaux et al., 1988). However, recent findings suggest that some BCL family members (MCL1, BCLw) are over-expressed in human malignancies (Chen et al., 2007; Kawasaki et al., 2007; Wesarg et al., 2007). Therefore, we decided to use a mouse model of MYC-induced leukemogenesis, in which hematopoietic progentior cells are transduced with retroviral vectors, ex vivo, followed by re-introduction into lethally irradiated sygeneic recipients. In fact, multiple groups have used variations of this, in vivo, assay to demonstrate the cooperative oncogenic potential for numerous genes, including BCL2 and MYC (Hemann et al., 2005; Luo et al., 2005).
There are several dissimilarities among the anti-apoptotic BCL family members; for instance, the amino acid sequence identities, compared to BCL2, range from 43.8% to 22.1% (Figure 1a). In addition, analysis using the “SMART” algorithm indicates that, while all six proteins encode at least three BCL homology domains (BH1-3), only BCL2, BCLxl, and BCLw encode a fourth BH domain near the amino termini of the proteins (Schultz et al., 1998). Furthermore, five of the family members encode recognizable trans-membrane domains that would potentiate insertion into the outer mitochondrial membrane, whereas BFL1 lacks this motif. Finally, a phylogeny algorithm indicated that BCL2, BCLxl and BCLw comprise a group closer in relative evolutionary time, whereas BCLb, MCL1 and BFL1 appear to comprise a distinct evolutionary group that is less related (Figure 1b) (Larkin et al., 2007).
Figure 1.
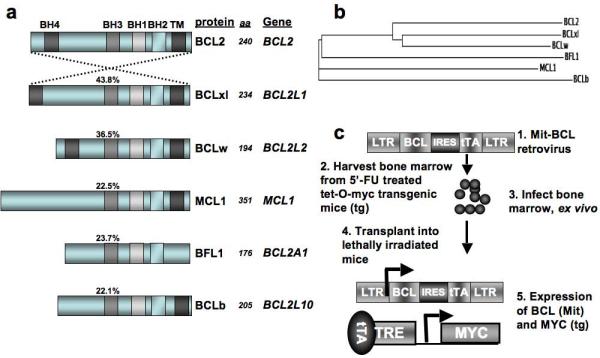
BCL proteins and experimental design. (a) To show that anti-apoptotic BCL proteins encode related, but distinct proteins, sequences were entered into the “ALIGN” program and the amino acid sequence identity of each full-length protein was compared to the sequence of BCL2. The protein domains that were identified using the “SMART” algorithm are indicated with shaded boxes of similar color. The protein designations (protein) used in this work are indicated next to the schematics, along with number of amino acids (aa) encoded by each protein. The gene symbol (gene) is also given as a reference. (b) The “CLUSTALW” algorithm generates a phylogeny tree and the relative evolutionary distances between human BCL proteins are indicated by the relative lengths of the lines. (c) Experimental design for testing the in vivo potential of BCL genes to cooperate with MYC in tumorigenesis.
Taking into consideration the levels of sequence divergence among the six BCL proteins and the observations made by others regarding sub-cellular localization and biochemical interactions, we decided to test whether each of the BCL genes could cooperate with MYC in leukemogenesis. Retroviruses used to infect bone marrow cells prepared from mice harboring a tetracycline trans-activator (tTA) dependent MYC allele (Tet-O-MYC), allowed for constitutive expression of the BCL gene from the provirus and tTA-dependent expression of the Tet-O-MYC transgene (Figure 1c). Following infection ex vivo, cells were transplanted into the tail vein of lethally irradiated syngeneic recipients, and disease occurrence was monitored by the presentation of clinical signs, including lethargy, ruffled fur, a distended abdomen and occasionally hind limb paralysis (Figure 2a). Upon presentation of disease, usually between twenty and ninety days following transplant, mice were euthanized for further analysis.
Figure 2.
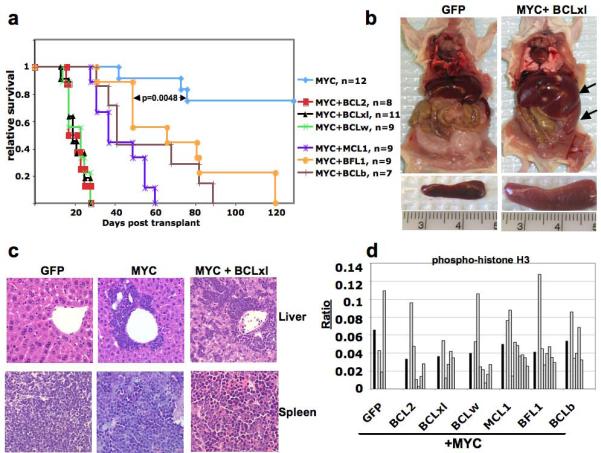
Induction of leukemia by cooperative action of MYC and BCL family members. (a) Kaplan-Meier survival curves are the compilation of two independent sets of bone marrow infections and transplantations performed as described in Figure 1c, and the number of mice for each cohort is indicated. MYC curve represents infections with a retrovirus encoding either tTA + GFP or tTA + luciferase. p-values were determined using student t-test for the values of the MYC + BFL1 cohort compared to the MYC cohort. (b) Regardless of the genotype of the leukemic cells mice had organomagly of spleen and liver (right panel, arrows), but did not display enlargement of lymph nodes or thymus. A mouse from the negative control cohort (mouse receiving Tet-O-MYC bone marrow cells infected with a retrovirus expressing only GFP) and a mouse with leukemia (mouse receiving Tet-O-MYC bone marrow cells infected with a retrovirus expressing BCLxl and tTA) are shown as representative examples. (c) Hematoxylin and eosin staining of liver and spleen from all leukemic mice, regardless of genotype, show infiltration of hematopoietic cells into liver and abnormal splenic cellularity. (d) For each paraffin embedded liver section, immuno-histochemistry was performed using an antibody that recognizes phosphorylated-histone H3, to identify cells in mitosis. Infiltrating leukemic cells from three random photographs were counted, and the total ratio of phospho-histone H3 positive cells for all tumors of a given genotype (dark bars) or the ratio of phospho-histone H3 positive cells for each individual tumor (open bars) was determined. Analysis of these ratios using a student t-test showed no statistically significant differences in the proliferation rates compared to MYC + GFP tumors.
Mice that received cells co-expressing MYC and any of the BCL genes showed accelerated tumorigenesis compared to mice that received cells that expressed either MYC and luciferase or MYC and GFP (Figure 2a). Although each of the BCL genes significantly cooperated with MYC to accelerate disease onset, it appeared that the three closest related genes (BCL2, BCLxl and BCLw) were the most potent, with all mice succumbing to disease within thirty days post-transplant. Regardless of the genotype of the leukemic cells, all mice displayed organomegaly of spleen and liver, which was caused by leukemic cells disrupting the architecture of the spleen and infiltrating into the liver (Figure 2b and Figure 2c). Leukemic cells initiated by either MYC alone or MYC plus BCL genes had uniform rates of proliferation, as shown by immunohistochemistry with two markers of proliferation (phospho-histone H3 or Ki67), indicating that expression of BCL proteins did not significantly increase cellular proliferation (Figure 2d and data not shown). In addition, multiple tumors expressing MYC + BCL genes that were transplanted into sub-lethally irradiated secondary recipient mice readily formed tumors that exactly mimicked the primary malignancies. Taken together, these data indicate that co-expression of MYC and any of the individual BCL family members increases the penetrance and decreases the latency of tumorigenesis, without significantly altering the proliferation rates, compared to expression of MYC alone.
Although all BCL family members were capable of cooperating with MYC in leukemogenesis, it was important to determine whether the leukemic cell phenotype was altered when different BCL family members were expressed. To address this issue, we examined cell surface markers, known to be expressed on cells of either the lymphoid or myeloid lineages, from various hematopoietic tissues- spleen, thymus, lymph nodes and marrow of the long bones- by flow cytometry. Irrespective of whether tumors were initiated by MYC alone or by MYC and BCL genes, we observed a profound increase in the number of cells that express the myeloid markers Gr-1 and Mac-1(CD11b) in the spleen and bone marrow (Figure 3a and b). However, lymphopoiesis was not overtly perturbed in the leukemic mice, as the thymus and lymph nodes contained normal ratios of resident cells and there were no increases in the numbers of lymphoid cells in the spleen (not shown and Supplemental 1). Since tumor cells reside primarily in the bone and spleen and express the Gr-1 and Mac-1 cell surface markers, the malignancies resemble acute myeloid leukemia (AML).
Figure 3.
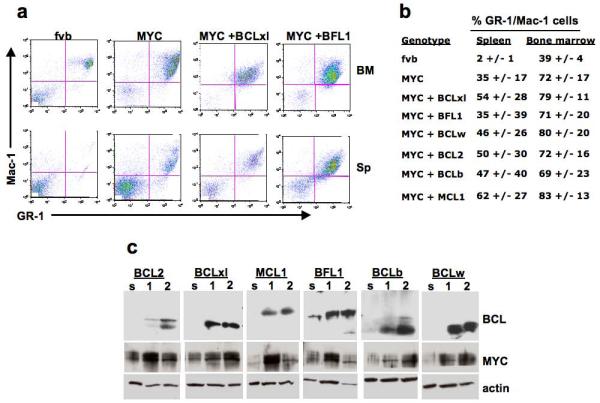
MYC-induced leukemias resemble acute myelogenous leukemia regardless of cooperating BCL gene. (a) Cells from all mice that required euthanization were analyzed by flow cytometry to determine the phenotypic nature of the disease. All mice examined had significant increases in the percentage of cells that stained for the myeloid cell surface markers GR-1 and Mac-1. Flow cytometry profiles from the spleen (sp) and bone marrow (BM) of cells from syngeneic FVB mouse and three AML induced by either MYC alone, MYC + BCLxl or MYC + BFL1 are shown as representative examples. (b) A table indicating the average of the percentage, and the range of the percentage, of GR1+/Mac1+ cells within the tissues of each tumor genotype. (c) Western blots of extracts of two representative tumors of each genotype (labeled 1 and 2) and normal splenocytes (s). All BCL antibodies were raised against human antigens and likely have preference for detecting the human BCL proteins encoded by the proviruses. An antibody that recognizes both the endogenous mouse Myc and transgenic human MYC proteins was used to determine the levels of MYC protein; antibodies against beta-actin were used as a loading control.
Next, we measured levels of MYC and BCL proteins in whole spleen samples by western blotting. Endogenous Myc was detected in cells of the normal spleen, with consistently increased levels of MYC protein observed in each of the tumor samples, consistent with induction of the transgene in the tumor cells (Figure 3c). In contrast, specific antibodies for each of the BCL proteins clearly show high levels of the respective BCL protein in the tumor samples. Only endogenous BFL1 was detected in whole spleen extracts from non-leukemic mice, consistent with previous data demonstrating expression of BFL1 in the hematopoietic compartment (Mandal et al., 2005).
To explore whether our observations using a MYC-driven mouse model of AML might be relevant to human malignancies, we measured expression of each of the BCL genes in a panel of solid human tumors and leukemia/lymphoma cell lines. Using the “TissueScan oncology survey panel” from Origene, we performed quantitative real time PCR analysis (qPCR) on cDNA acquired from 60 different tissues and tumor samples for all six BCL genes, plus ACTIN as a control (Figure 4a). The panel consists of cDNA derived from five tissues- 3 “normal” and 9 tumor samples for each tissue (breast, liver, lung, ovarian and prostrate)- deposited into individual wells. Following qPCR we observed elevated levels of BFL1 in eight of nine breast tumor samples (dark bars) and three of nine colon tumor samples, compared to the normal tissue controls (light bars). Increased BCL2 expression was also detected in seven of the nine breast cancer samples, and BCLxl was over-expressed in a number of ovarian tumors. BCLw was the only additional member that was highly expressed in more than two tumor samples from a particular tissue, being elevated in three lung cancer patients.
Figure 4.
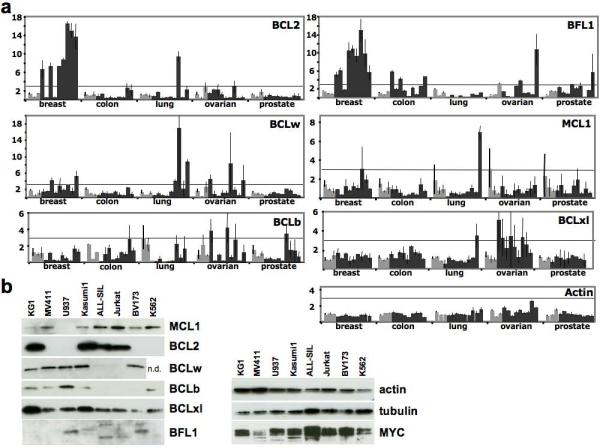
BCL family members are expressed in human solid tumor samples and in leukemia/lymphoma cell lines. (a) Expression of BCL genes in human solid tumor samples. TissueScan oncology survey cDNA panels were obtained from OriGene (Rockville, MD, USA) and quantitative PCR analysis was performed (Sybr Green, Applied Biosystems, Foster City, CA, USA) using two independent oligo pairs for each BCL gene. The average level of mRNA in normal tissue samples (light bars) was used for each tissue type to determine the fold-change in expression of each sample (dark bars). The average of the fold-change between the two pairs of gene specific oligos was determined and the standard error of the mean is depicted. The horizontal line indicating a three-fold increase in relative expression serves as a reference. (b) Antibodies specific for each BCL family member were used for western blot analysis of whole cell lysates (1% CHAPS, 150 mM NaCl, 10 mM Hepes) from multiple leukemia/lymphoma cell lines.. An antibody against MYC was used to determine the levels of MYC protein, antibodies against beta-actin and tubulin were used as a loading control.
As further support for the possible implications of our in vivo mouse studies, we sought to determine if BCL members and MYC are co-expressed in leukemia and lymphoma cell lines derived from human patients. All six BCL members were readily detectable in various human leukemia/lymphoma cell lines by western blotting with antibodies specific for each BCL protein. Importantly, MYC protein was also detected in each sample, indicating that the experiments conducted in this study, using MYC-induced murine AML, are likely pertinent to the study of human malignancies.
If BCL family members play a causal role in human neoplasms, it is important to determine whether inhibiting their anti-apoptotic effects will have therapeutic benefits. It has been known for more than ten years that over-expression of only the BH3 domain of certain pro-apoptotic BCL family members is sufficient to block the activity of BCLxl and BCL2 and can cause apoptosis in cultured tumor cells (Cosulich et al., 1997; Holinger et al., 1999). In addition, deinduction of an inducible BCL2 transgene, in the presence of a constitutive MYC allele, was sufficient to cause regression of lymphoid tumors in mice (Letai et al., 2004). This suggested that inhibiting the function of an anti-apoptotic gene (BCL2), even in the context of additional oncogenic lesions (MYC), might be sufficient to extend lives of patients with lymphoma over-expressing BCL2.
Such studies have stimulated research designed to target the anti-apoptotic machinery, and several small molecules that inhibit the anti-apoptotic BCL family members by binding within the BH3 hydrophobic pocket have been identified. One of the most promising first generation molecules, ABT-737, has a very high specificity for inhibiting BCL2, BCLxl and BCLw and has shown promise against many tumor types, including leukemias and solid tumors, as a single agent and in combination with other therapies (Kohl et al., 2007; Konopleva et al., 2006; Reed, 2006; Shoemaker et al., 2008). For instance, 8 out of 9 primary CD34+ human AML samples efficiently underwent apoptosis in the presence of ABT-737, whereas 3 out of 4 normal CD34+ stem cell samples were unaffected by the drug (Konopleva et al., 2006). Although the bulk of the work with this compound has been in cell culture and in animal models, this molecule, along with several others, is currently being tested in clinical trials to determine safety and efficacy in patients.
BCL genes may also have a role in both primary and acquired drug resistance. For example, increased expression of BCL2, BCLxl and BCLb has been observed in AML cell lines resistant to high levels of busulfan treatment, and increased copy numbers of genomic loci encoding BCLw, MCL1 and BCLb were identified in various cell lines that were treated with, and became resistant to, etoposide, campothecin or Ara-C, respectively (Valdez et al., 2008; Yasui et al., 2004). These data add emphasis to the findings we report here and support the need to understand the role of all BCL genes in cancer and to consider targeting multiple members of the BCL family in both primary and drug-resistant tumors.
Supplementary Material
Acknowledgements
The authors would like to thank members of the Varmus lab, especially G. Sanchez, A. Giannakou, and M.A. Melnick for expert handling of the mouse colony, including genotyping, and for performing immunohistochemistry on embedded tissue sections. Funding for this project was provided, in part, by NIH P01 2PO1CA094060-06A1 to H.E.V.
References
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–91. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- Cosulich SC, Worrall V, Hedge PJ, Green S, Clarke PR. Regulation of apoptosis by BH3 domains in a cell-free system. Curr Biol. 1997;7:913–20. doi: 10.1016/s0960-9822(06)00410-6. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–11. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holinger EP, Chittenden T, Lutz RJ. Bak BH3 peptides antagonize Bcl-xL function and induce apoptosis through cytochrome c-independent activation of caspases. J Biol Chem. 1999;274:13298–304. doi: 10.1074/jbc.274.19.13298. [DOI] [PubMed] [Google Scholar]
- Horvitz HR. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999;59:1701s–1706s. [PubMed] [Google Scholar]
- Kawasaki T, Yokoi S, Tsuda H, Izumi H, Kozaki K, Aida S, et al. BCL2L2 is a probable target for novel 14q11.2 amplification detected in a non-small cell lung cancer cell line. Cancer Sci. 2007;98:1070–7. doi: 10.1111/j.1349-7006.2007.00491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl TM, Hellinger C, Ahmed F, Buske C, Hiddemann W, Bohlander SK, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21:1763–72. doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Lanave C, Santamaria M, Saccone C. Comparative genomics: the evolutionary history of the Bcl-2 family. Gene. 2004;333:71–9. doi: 10.1016/j.gene.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–9. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Luo H, Li Q, O’Neal J, Kreisel F, Le Beau MM, Tomasson MH. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood. 2005;106:2452–61. doi: 10.1182/blood-2005-02-0734. [DOI] [PubMed] [Google Scholar]
- Mandal M, Borowski C, Palomero T, Ferrando AA, Oberdoerffer P, Meng F, et al. The BCL2A1 gene as a pre-T cell receptor-induced regulator of thymocyte survival. J Exp Med. 2005;201:603–14. doi: 10.1084/jem.20041924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nature clinical practice Oncology. 2006;3:388–98. doi: 10.1038/ncponc0538. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–13. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–64. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14:3268–77. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- Swanson PJ, Kuslak SL, Fang W, Tze L, Gaffney P, Selby S, et al. Fatal acute lymphoblastic leukemia in mice transgenic for B cell-restricted bcl-xL and c-myc. J Immunol. 2004;172:6684–91. doi: 10.4049/jimmunol.172.11.6684. [DOI] [PubMed] [Google Scholar]
- Valdez BC, Murray D, Ramdas L, de Lima M, Jones R, Kornblau S, et al. Altered gene expression in busulfan-resistant human myeloid leukemia. Leuk Res. 2008;32:1684–97. doi: 10.1016/j.leukres.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Wesarg E, Hoffarth S, Wiewrodt R, Kröll M, Biesterfeld S, Huber C, et al. Targeting BCL-2 family proteins to overcome drug resistance in non-small cell lung cancer. Int J Cancer. 2007;121:2387–94. doi: 10.1002/ijc.22977. [DOI] [PubMed] [Google Scholar]
- Yasui K, Mihara S, Zhao C, Okamoto H, Saito-Ohara F, Tomida A, et al. Alteration in copy numbers of genes as a mechanism for acquired drug resistance. Cancer Res. 2004;64:1403–10. doi: 10.1158/0008-5472.can-3263-2. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zhai D, Jin C, Satterthwait AC, Reed JC. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. 2006;13:1419–21. doi: 10.1038/sj.cdd.4401937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.