

Abstract
The dorsal striatum and the nucleus accumbens (NAc) shell of the ventral striatum have similar cellular components and are both richly innervated by dopamine neurons. Despite similarities that extend throughout the striatum, only the NAc shell has a conspicuous increase in basal dopamine upon the initial administration of psychostimulant drugs such as nicotine. As measured by microdialysis, the elevated dopamine in the NAc shell is considered an identifying functional characteristic of addictive drugs. To examine this general functional difference between nicotine's action on the dorsolateral striatum and NAc shell, we directly monitored dopamine release in rat striatal slices using fast-scan cyclic voltammetry. In addition, we separately monitored the in vivo unit firing activity of putative midbrain dopamine neurons from freely moving rats using chronic multiple tetrodes. Nicotine administration increased the firing frequency of dopamine neurons and specifically increased the number and the length of phasic burst firing. The frequency dependence for dopamine release in the dorsolateral striatum and NAc shell is fundamentally different, enabling mainly the NAc shell to capitalize on the nicotine-induced phasic burst firing by dopamine neurons. Although nicotine decreased low-frequency (tonic) dopamine release in both areas, the increased ratio of phasic bursts relative to tonic firing caused by nicotine boosted the basal dopamine concentration predominantly in the NAc shell. By favoring release from bursts while depressing release from tonic signals, nicotine spreads the range of dopamine signaling and effectively increases the signal-to-noise relationship along dopamine afferents.
Introduction
In the mammalian brain, the striatum is a large subcortical structure involved in motor coordination, cognitive functions, and disorders such as schizophrenia, Huntington's disease, Parkinson's disease, and drug addiction (Breiter et al., 1997; Schultz et al., 1997; Hornykiewicz, 1998; Lang and Lozano, 1998; Berke and Hyman, 2000; Cicchetti et al., 2000; Grace, 2000; Nicola et al., 2000; Zhou et al., 2001, 2002; Di Chiara, 2002; Wise, 2002). It is often divided into the dorsal and ventral portions that share general similarities. The vast majority of the striatal neurons are medium spiny GABAergic projection neurons, and there is dense cholinergic interneuron and dopaminergic afferent innervation (Björklund and Lindvall, 1984; Butcher and Woolf, 1984; Zhou et al., 2001).
Differences arise within the striatum from afferent and efferent connections. The dorsal striatum receives its main glutamatergic inputs from cortical areas, especially areas related to sensorimotor functions (Haber et al., 2000). Dense dopamine (DA) innervation arises primarily from the substantia nigra pars compacta (SNc) and to a lesser degree from the ventral tegmental area (VTA) (Björklund and Lindvall, 1984; Haber et al., 2000). The ventral striatum is composed of the nucleus accumbens (NAc) and portions of the olfactory tubercle, and is mainly limbic related. It receives extensive excitatory innervation from the prefrontal cortex, the hippocampus, and the amygdala (Pennartz et al., 1994; Haber et al., 2000; Heimer, 2000). The ventral striatum receives DA innervation primarily from the VTA and to a lesser degree from the SNc. The NAc is thought to serve as the limbic–motor or motivation–action interface, and these functions are part of the NAc's participation in reward-based learning and addiction (Mogenson et al., 1980; Haber et al., 2000).
Another distinguishing property is that addictive drugs (including nicotine) initially increase the basal DA concentration in the NAc shell as measured by microdialysis, but that increase is smaller or not detected in the dorsolateral striatum (Pontieri et al., 1996; Di Chiara, 1999; Pidoplichko et al., 2004). Nicotine administration excites DA neurons that innervate the whole dorsal to ventral extent of the striatum (Grenhoff et al., 1986; Pidoplichko et al., 1997; Schilström et al., 2003), suggesting that the DA signal should be comparable throughout the striatum. In addition, nicotinic mechanisms regulate the frequency dependence of DA release (Zhou et al., 2001; Cragg, 2003; Rice and Cragg, 2004; Zhang and Sulzer, 2004). There is rich dopaminergic innervation and nicotinic ACh receptor (nAChR) regulation of DA release throughout the dorsal to ventral extent of the striatum. Paradoxically, when there is general activation of DA neurons throughout the midbrain by the addictive drug nicotine, the DA increase measured by microdialysis resides predominantly in the NAc shell of the ventral striatum and not in the dorsolateral striatum. This study examines in vivo DA neuron firing and DA release in these specific targets to explain this paradoxical DA signaling.
Materials and Methods
In vivo unit recordings from midbrain DA neurons using tetrode electrodes.
Unit recordings were obtained from eight male Long–Evans rats (250–400 g). They were housed individually with food and water available ad libitum. Animals were kept on a 12/12 h light/dark schedule and tested in the light phase. Surgeries were conducted following the guidelines of National Institutes of Health and Baylor College of Medicine. Rats were injected (i.p.) with a xylazine, ketamine, and acepromazine mixture to achieve shallow anesthesia before being placed in a stereotaxic apparatus in which deep anesthesia was controlled using isoflurane inhalation. The body temperature was maintained with a thermostatically controlled heating blanket. The tetrode-microdrive containing 12 tetrodes was attached with dental cement to the skull by screws such that the tetrode wires passed through a 2.5 mm hole drilled in the skull (AP, −5.5 mm; ML, 1.1 mm from bregma to the center of the 2 mm cannula of the tetrode-microdrive). The tetrode-microdrive was grounded to one skull screw, and another steel wire was inserted into the neck muscles to monitor the electromyogram to help judge the sleep/wake state of the animal.
Each tetrode underwent a cleaning and gold plating process to reduce the final impedance to 200–300 kΩ. The tetrodes were slowly positioned into the midbrain DA area (Fig. 1A) (7–8.5 mm beneath the surface of the neocortex). Electrical signals were recorded by a multichannel Cheetah amplifier controlled by associated software (Neuralynx). Recordings were digitized gap-free at 26.5 kHz after variable high-pass filtering and low-pass filtering at 9 kHz, saved on computer disk, and analyzed off-line, where versatile digital filtering was applied for analysis and display. Spikes were identified and extracted in Matlab using Xtractor (program by M. Krause and W. Li, Baylor College of Medicine, Houston, TX). Individual unit isolation was achieved based on the relative amplitudes of their action potentials on the four channels of a tetrode in combination with spike waveform parameters (action potential duration, wave shape, and half-height length). This spike sorting procedure was accomplished using homemade software and software kindly provided by A. David Redish (University of Minnesota, Minneapolis, MN) (MClust) and Matt Wilson (Massachusetts Institute of Technology, Cambridge, MA) (X-clust).
Figure 1.

Nicotine increases the action potential firing rate of putative DA neurons in freely moving rats. A, The track of a recording tetrode (fluorescence from DiI) is shown within the ventral tegmental area with the DA neurons in blue (fluorescent antibody for tyrosine hydroxylase). The inset shows a waveform obtained with tetrodes. Calibration: 100 μV, 1 ms. B–D, Representative ISI histogram (B), autocorrelogram (C), and 2 s spike-recording epochs of a DA neuron (D), indicating that the DA neuron fires more bursts after nicotine administration than before [control (ctrl)]. Calibration: 50 μV, 200 ms. E, The normalized average firing rate of DA neurons in response to nicotine, quinpirole, and eticlopride.
Identification of DA neurons and data analysis.
A neuron was classified as dopaminergic if the following criteria were met: a low baseline firing rate (0.5–8 Hz) (Ungless et al., 2004; Anstrom and Woodward, 2005), a relatively long spike duration (>1.5 ms with our filter settings), a complex waveform, and no spikes within the first 2 ms of the interspike interval (ISI) histogram (Fig. 1B). The shortest ISIs were on average 7.6 ± 1.3 ms, but the absolute refractory time sets a limit of >2 ms (Wang, 1981). The absolute refractory time defines the closest two spikes can be in time even if the spikes are driven by external stimulation. In the midbrain the absolute refractory time is ∼2.6 ms for DA neurons and ∼1.1 ms for non-DA neurons (Wang, 1981), which we confirmed using antidromic stimulation (data not shown). In addition, the firing rate of putative DA neurons had to be inhibited by >70% by the D2-type agonist quinpirole (0.5 mg/kg, i.p.), and this inhibition had to be reversed by the D2-type antagonist eticlopride (0.5 mg/kg, i.p.) (Hyland et al., 2002; Robinson et al., 2004). It seems likely to us that these conservative requirements for identification will preferentially select a subset of DA neurons.
Postmortem histology confirmed that the electrodes were located among DA neurons of the midbrain (Figs. 1A, 2). In addition, recent studies indicate that lateral VTA neurons have classic DA neuron functional characteristics that make them easier to identify reliably (Ford et al., 2006; Lammel et al., 2008). Therefore, while obtaining nearby neurons, we focused on obtaining lateral VTA neurons, which have been shown to innervate the lateral NAc.
Figure 2.
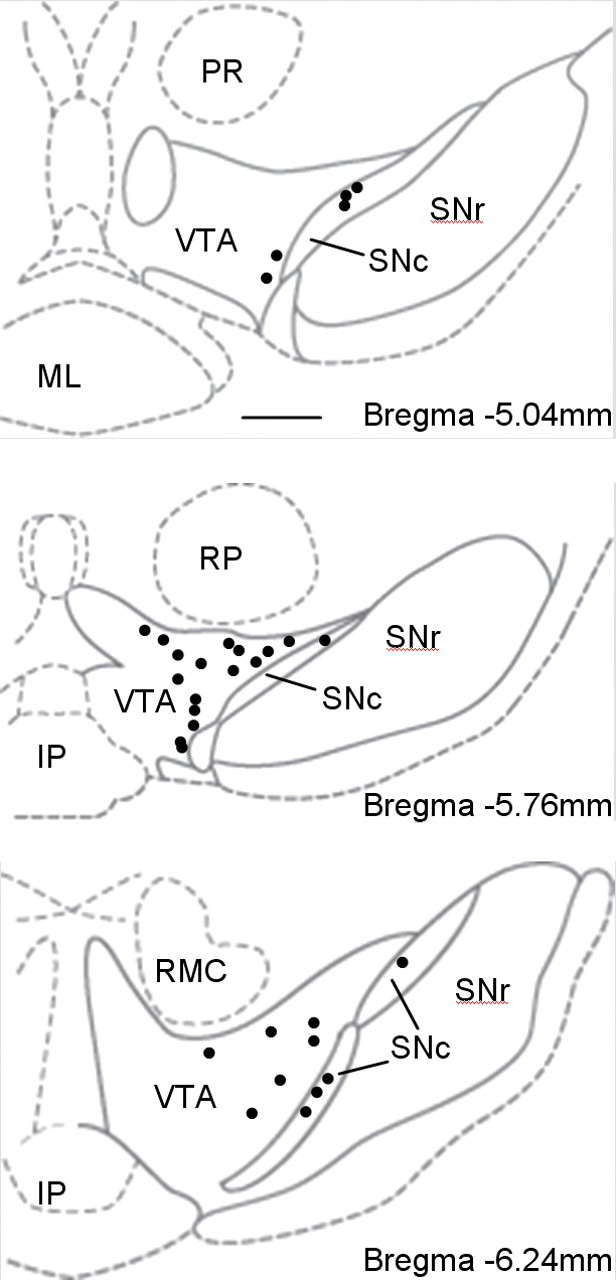
Anatomical locations of the putative DA neurons that were recorded in the VTA and the SNc. The labels indicate the substantia nigra reticulata (SNr), prerubral field (PR), mammillary nucleus (ML), interpeduncular nucleus (IP), parvicellular red nucleus (RP), and magnocellular red nucleus (RMC).
Based on doing several preliminary kinds of burst analysis, we settled on the most common traditional burst analysis (Grace and Bunney, 1984). Selection of this analysis was supported by our findings that the average intraburst frequency was not firing-rate dependent. That is, the average intraburst frequency was near 20 Hz during baseline or during nicotine or other drug administrations that altered the firing rate. The onset of a burst of action potentials was signified by two spikes with an interspike interval <80 ms, and the end of a burst was when the interspike interval became >160 ms (Grace and Bunney, 1984). As a control, a 20 min baseline was recorded before the nicotine injection (0.4–0.5 mg/kg, i.p., free-base equivalent). These recordings were used to calculate overall firing rate, bursts per second, spikes per burst, and intraburst frequency. Autocorrelogram and ISI histograms were used to identify the firing patterns (Fig. 1B,C), and p values <0.05 were considered statistically significant. To obtain average values for the graphs, the baseline spikes were counted for 10 min before nicotine treatment. After nicotine injection, we waited 2 min for nicotine to distribute before counting the spikes for 10 min.
Anatomical verification for in vivo recording sites.
One-percent solution of fluorescence dyes (DiI, DiD, or DiO, Invitrogen) was painted onto the surface of individual tetrode electrodes to mark the electrode tracks (Fig. 1A). After recording, most electrode tracks were also marked by a lesion (30 μA × 15 s of anodal current). The rat was overanesthetized and perfused through the left ventricle with 200 ml of saline followed by 300 ml of 10% neutral formalin. The intact rat brain was taken out and further fixed in 10% neutral formalin for 24 h at 4°C. Coronal sections (60 μm) were cut on a vibratome, rinsed six times in 0.01 m PBS. The slices were incubated with a primary rabbit polyclonal antibody to tyrosine hydroxylase (Millipore Bioscience Research Reagents) (1:500 dilution in 0.01 m PBS containing 0.3% Triton X-100) overnight at 4°C. After rinsing thoroughly, the slices were then reacted with the AMCA-conjugated secondary antibody (1:500; Jackson ImmunoResearch Laboratories) for 60 min at 37°C. After mounting, the slides were examined on an Olympus BX60F5 fluorescence microscope equipped with a digital camera. Based on postrecording histological examination (Fig. 2) of eight rats, 24 DA neurons were from the VTA and 8 from the SNc. For the purposes of the analysis presented, no substantial difference was observed between the firing patterns of VTA and SNc DA neurons in our samples (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Because the firing rate, bursts per second, spikes per burst, and percentage of spikes in bursts were not statistically different in the VTA and SNc, those data were combined for the final calculations.
Fast-scan cyclic voltammetry in rat striatal slices.
Horizontal slices (400 μm) were cut from Long–Evans rats of 300 ± 50 g body weight, held at room temperature for ≥1 h for equilibration, and studied at 34 ± 1°C in an artificial CSF containing (in mm): 125 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 26 NaH2PO4, 1.25 NaHCO3, and 10 glucose saturated with 95% O2 and 5% CO2 (Zhou et al., 2001). For the experiments, separate horizontal brain slices were cut to enable visual identification of the dorsolateral striatum (Fig. 3A) or NAc shell (Fig. 3B). The dashed enclosures (Fig. 3) set the limits of our recording positions, and those enclosures are well within the targets of the dorsal striatum or NAc shell. To avoid any uncertainty in identifying the NAc shell, only a portion of the NAc shell, medial to the anterior commissure, was studied, as indicated in Figure 3B.
Figure 3.

Photomicrographs of brain slices indicating the selected portion of the dorsal striatum or NAc shell studied using fast-scan cyclic voltammetry. The carbon-fiber recording microelectrodes were placed into the regions enclosed by the dashed lines. These regions are well within the areas of interest. A, A photomicrograph showing a horizontal brain slice containing the dorsal striatum. The area where the carbon-fiber electrode was placed to measure DA release is encircled by a dashed line indicating the lateral portion of the caudate/putamen within the dorsal striatum. B, A photomicrograph showing a more ventral horizontal slice containing the NAc core and shell. The NAc shell becomes larger in slices cut just ventral to this position, but the anterior commissure (ac) is easily seen in this slice. The ac was an important landmark for identifying the NAc shell, and the dashed line encircles where we placed the carbon-fiber electrode to record DA release from these slices.
Homemade carbon-fiber (P55S, Amoco Polymers) microelectrodes were used for fast-scan cyclic voltammetry (Zhou et al., 2001). An Axopatch 200B amplifier and pClamp 8 were used to acquire and analyze voltammetry data. Scans of the microelectrode potential (10 Hz) were from 0 mV to −400 mV to 1000 mV to −400 mV to 0 mV against an Ag/AgCl reference electrode at a rate of 300 mV/ms. Intrastriatal stimuli were delivered using bipolar tungsten electrodes. The two tips of the stimulating electrode were ∼100 μm away from each other. The tip of the carbon-fiber recording electrode was 200–300 μm away from the two tips of the stimulating electrode. Both the stimulating electrode and carbon-fiber recording electrode were placed well within the indicated regions (Fig. 3, dashed enclosures) of the dorsal striatum or NAc shell.
Each pulse of a single or burst stimulation was 0.5–1 ms in duration and 0.3–0.6 mA of constant current. As shown in supplemental Fig. S2 (available at www.jneurosci.org as supplemental material), after stimulation the DA signal completely recovered in 80 s. Therefore, we waited for a minimum of 120 s before applying the next stimulus to ensure complete recovery of DA release. Slices were exposed to nicotine at 0.1 μm, a concentration commonly experienced by smokers (Gourlay and Benowitz, 1997; Mansvelder et al., 2002; Karan et al., 2003), for 10 min before data were collected for nicotine-induced changes in DA release. To mimic DA firing patterns recorded in vivo before and after nicotine injection, we constructed stimulus patterns that were consistent with the in vivo average firing frequency, average number of bursts per second, average number of spikes per burst, and average intraburst frequency (bottom hash marks of Fig. 7 and supplemental Fig. S4, available at www.jneurosci.org as supplemental material). The stimulus train applied to the slices under control conditions mimicked the firing of DA neurons in vivo before nicotine, and the stimulus train with longer bursts mimicked the in vivo conditions in the presence of nicotine. The voltammetric DA signal was obtained by digital subtraction and calibrated against standards of 0.5–5 μm DA.
Figure 7.

The burst firing by DA neurons measured in vivo that is induced by nicotine causes a greater increase of DA release in the NAc shell than in the dorsal striatum (d. striatum). Stimulus patterns were designed to mimic the in vivo firing patterns of DA neurons measured from freely moving rats before and after nicotine injections (represented by vertical dashes inset below). A, B, Patterned stimulus trains based on the in vivo DA-unit recordings are shown below the evoked DA release in the absence (control) or presence of nicotine in the dorsal striatum (A) or the NAc shell (B). Calibration: 0.1 μm, 1 s. C, The relative DA signal (area under the curve) was unchanged by nicotine in the dorsal striatum but was increased in the NAc shell (n = 5, **p < 0.01).
The DA signals measured by voltammetry were all normalized to the area under the curve because an extremely wide range of stimulus trains and patterns were used in the study. When comparing a single-pulse stimulus with a stimulus train of 20 pulses at 20 Hz, the DA signal was spread out in time during the stimulus train. Also, when the stimulus trains were developed based on the in vivo unit recordings (e.g., Fig. 7 and supplemental Fig. S4, available at www.jneurosci.org as supplemental material), the stimulus trains were spread over seconds with varying frequencies. The most appropriate (and at times the only) way to normalize across experiments was to use the area under the complex curve. The relative DA signal induced by a burst was normalized to the signal induced by a single pulse. All data are means ± SEM. Differences between groups were assessed by paired t test or one-way ANOVA.
Results
Nicotine increases phasic burst action potential firing in freely moving rats
To quantify how nicotine changes the firing patterns of DA neurons, we placed tetrodes chronically into the midbrain DA area of freely moving rats (Wilson and McNaughton, 1994; Hyland et al., 2002). At the end of these experiments, the recording locations among tyrosine hydroxylase-positive (indicating DA) neurons were determined using stimulus-induced lesions and/or dye painted onto the tetrodes (Figs. 1A, 2). Putative DA neurons (see Materials and Methods) were required to have a low baseline firing rate (0.5–8 Hz) (Grenhoff et al., 1986; Ungless et al., 2004; Anstrom and Woodward, 2005), wide spike widths (>1.5 ms with our filter settings) (Hyland et al., 2002; Ungless et al., 2004), and an absolute refractory time that produced no spikes within the first 2 ms of the ISI (Wang, 1981). An ISI histogram (Fig. 1B), an autocorrelogram (Fig. 1C), and brief spike-recording epochs (Fig. 1D) from a representative DA neuron indicate more burst firing in nicotine (0.4–0.5 mg/kg, i.p.) than during the control (ctrl). As indicated by the summary data (Fig. 1E), nicotine increased the action potential firing rate. In addition, DA neurons express D2-type receptors, and we required that putative DA neurons were robustly inhibited (>70%) by the D2-type agonist quinpirole (0.5 mg/kg, i.p.) and activated by the D2-type antagonist eticlopride (0.5 mg/kg, i.p.) (Fig. 1E) (Grace and Bunney, 1985; Centonze et al., 2002; Hyland et al., 2002; Robinson et al., 2004).
As described in the Materials and Methods, the firing of VTA and SNc putative DA neurons identified with our conservative parameters showed similar firing characteristics (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Therefore, we combined the data from all the neurons to calculate the activity parameters. Nicotine (0.4–0.5 mg/kg, i.p.) administration increased the average DA unit firing frequency from 3.8 ± 0.3 Hz to 6.8 ± 0.7 Hz (n = 32, p < 0.01). More importantly, burst firing became more common after nicotine (Grenhoff et al., 1986). The number of bursts per second increased from 0.32 ± 0.04 to 0.47 ± 0.05 (p < 0.01). The number of spikes per burst increased from 5.1 ± 0.5 to 10.4 ± 2.1 (p < 0.01), and the proportion of spikes within bursts increased from 44.2 ± 4.9% to 56.5 ± 5.6% after nicotine (p < 0.01). There was no significant change in the average intraburst frequency: 17.8 ± 0.6 Hz in control and 18.3 ± 0.7 Hz after nicotine.
NAc shell responds more robustly to phasic burst stimulation than the dorsal striatum
Because nicotine increased burst firing by DA neurons, we used fast-scan cyclic voltammetry applied to in vitro brain slices to determine how burst firing influences DA release in the NAc shell and dorsolateral striatum. Conservative anatomical landmarks were used to place the carbon-fiber recording electrode in the dorsal striatum (Fig. 3A) or the NAc shell (Fig. 3B). We placed the carbon-fiber electrode and stimulating electrode well within the area enclosed by the dashed line, which delineates a conservative subset of the dorsal striatum (lateral caudate–putamen) (Fig. 3A) and the NAc shell (Fig. 3B). A bipolar simulating electrode was placed 200–300 μm from the carbon-fiber recording electrode.
Using fast-scan cyclic voltammetry, we examined the DA release evoked by 1 pulse or by trains of pulses. To illustrate the difference between the dorsal striatum and NAc shell, we used a stimulus frequency of 20 Hz because that is near to the consistent average intraburst frequency that we measured in vivo from freely moving rats. When the DA release produced by 1 pulse (1p) was compared with that released by 5 pulses at 20 Hz, we found greater facilitation in the NAc shell than in the dorsal striatum (Fig. 4A).
Figure 4.

Different DA release properties in the dorsal striatum (d. striatum) and NAc shell. A, Example measurements are shown of the DA signal evoked by 1p (black traces) or by 5p at 20 Hz (gray traces). In the dorsal striatum, the DA signal evoked by 1p is only slightly smaller than that evoked by a 5p train. In the NAc shell, the DA signal evoked by 1p is much smaller than that evoked by 5p. Calibration: 400 nm, 0.5 s in the dorsal striatum and 1 s in the NAc shell. B, Reducing the external concentration of calcium from 2.5 to 1 mm decreased the DA amplitude by 78.3 ± 4.5% (n = 3) to 21.7%. In low calcium, 5p at 20 Hz (gray trace) produced a larger increase in the DA signal than did 1p. Note that the calibration (100 nm, 0.5 s) refers to the smaller DA signal in low calcium. C, Example traces of paired-pulse stimulation at 50 ms intervals in the dorsal striatum and the NAc shell. Calibration: 0.5 μm, 0.5 s. D, Averaged paired-pulse ratios (p2/p1) in 2.5 mm (ctrl), 1.5 mm, or 1.0 mm external Ca2+ or in the nAChR inhibitor mecamylamine (Meca, 2 μm), all in the dorsal striatum (d.s., gray bars, n = 5–14), and NAc shell in 2.5 mm Ca2+ (ctrl, black bar, n = 7) compared with control in the dorsal striatum (*p < 0.05, **p < 0.01).
These results suggested that the probability of release was greater in the dorsal striatum to a single pulse, and thus there was less potential for facilitation. To test this possibility, we lowered the external calcium from 2.5 to 1 mm to decrease the probability of release in the dorsal striatum. As expected in that case, greater facilitation was then seen in the dorsal striatum in response to 5 pulses (Fig. 4B). The ratio of the DA signal, 5p at 20 Hz/1p, was significantly increased from the control value, 1.44 ± 0.15 (n = 7 in 2.5 mm Ca2+) to 2.02 ± 0.09 (n = 5, p < 0.05 in 1.0 mm Ca2+).
To test further our hypothesis that the probability of release was greater in the dorsal striatum, we measured DA release to paired-pulse stimulation (Fig. 4C), showing the response to the first pulse (p1) and separately to the second pulse 50 ms later (p2). As expected for an initial high probability of release, the p2/p1 was much smaller in the dorsal striatum (Fig. 4C,D). After lowering external Ca2+, to lower the probability of release to the first pulse (p1), the ratio becomes significantly larger (Fig. 4D). It has been shown previously that nAChRs increase the probability of release to a single pulse (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004). Blocking nAChRs with the nonselective inhibitor, mecamylamine (2.0 μm), lowered the p1, and the ratio of p2/p1 in the dorsal striatum became equal to that in the NAc. These results are consistent with the dorsal striatum having a higher probability of release to 1p (Zhang and Sulzer, 2004).
We examined this difference in the NAc shell and dorsal striatum over a wide range of pulse trains with and without nicotine (Fig. 5). Voltammetric measurements have shown that nicotine decreases tonically stimulated DA release and alters the frequency dependence of electrically evoked DA release in the striatum (Zhou et al., 2001; Cragg, 2003; Rice and Cragg, 2004; Zhang et al., 2004). Because the average intraburst firing frequency we recorded in vivo remained constant near 20 Hz, we applied stimulus bursts ranging in length from 1 to 20 pulses at 20 Hz in the presence or absence of nicotine (Fig. 5A,B). Increasing the stimulus burst length to 20 pulses increased the DA signal to 292 ± 23% in the dorsal striatum (n = 8) (Fig. 5A, open symbols), but there was a much larger increase to 1371 ± 161% in the NAc shell (n = 5) (Fig. 5B, open symbols). These results showed that afferent bursts boost DA release much more strongly in the NAc shell than in the dorsal striatum (Cragg, 2003).
Figure 5.

Fast-scan cyclic voltammetry indicates DA release and nicotine modulation differ in the dorsal striatum (d. striatum) and NAc shell. A, B, The DA release evoked by 20 Hz stimulus trains of differing length in the absence (open symbols, control) or presence of nicotine (0.1 μm, filled symbols) measured in the dorsal striatum (A) or the NAc shell (B). C, D, The DA release evoked by 5p at differing intraburst frequencies in the absence (open symbols, control) or presence (0.1 μm, filled symbols) of nicotine measured in the dorsal striatum (C) or the NAc shell (D). freq, Frequency.
We also measured nicotine's influence over the frequency dependence of DA release. In the presence of nicotine (0.1 μm) applied to the brain slice, the DA signal evoked by a single pulse decreased by 74 ± 6% (n = 8) in dorsal striatum (Fig. 5A,C, filled squares) and by 55 ± 5% (n = 5) in NAc shell (Fig. 5B,D, filled circles) (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004). When stimulus trains from 1 to 20 pulses long at 20 Hz were applied, nicotine caused a depression of the DA release that was greater in the dorsal striatum (Fig. 5, compare A, B). The frequency dependence of DA release was examined with a stimulus train of 5 pulses applied at frequencies ranging from 10 to 80 Hz (Fig. 5C,D, control, open symbols). When nicotine (100 nm) was applied to the bath, the DA release was suppressed at the lower frequencies but increased up to the control in the dorsal striatum (Fig. 5C, filled squares) and beyond the control in the NAc shell (Fig. 5D, filled circles) (Rice and Cragg, 2004). An important finding in the NAc shell is that 5 pulses at 20 Hz in nicotine increase the DA release to 325 ± 28% (n = 5, p < 0.01) of that released by a single pulse under control conditions (in the absence of nicotine).
It also is interesting to note that the maximum DA release was observed at the average intraburst firing frequency measured in vivo from freely moving rates (i.e., at 20 Hz) (Fig. 5C,D). To verify this finding, we measured DA release to pulse trains given in different orders to be sure that release was not altered by the stimulus history (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material), which is a factor we controlled in our methods (see supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Independent of the stimulus history, the DA signal (measured as the area under the curve) is smaller at 40 Hz and 80 Hz than it is at 20 Hz (Fig. 5C,D and supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). Finally, we verified that the oxidation and reduction peaks of the signal were indicative of DA and not some artifactual agent (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material). Thus, the results suggest that the maximum DA release tends to occur at the biologically relevant average intraburst frequency of the in vivo DA neurons (Exley et al., 2008).
The frequency dependence of DA signaling is not explained by DA reuptake by transporters
The DA signal measured with voltammetry in striatal tissue depends on both DA release and DA reuptake. The average density of DA transporters (DATs) is lower in the NAc shell than in the dorsal striatum (Coulter et al., 1996). To examine whether the difference in DAT density and efficacy is the underlying reason for the differences in the DA signal in the dorsal striatum and NAc shell, DAT activity was selectively blocked with GBR-12909 (GBR). After examining a dose range of 0.2–10 μm GBR (Fig. 6A), we determined that 2 μm GBR inhibited the majority of DAT activity and allowed stable recordings. With complete DAT inhibition, the voltammetric DA signal tended to decline over time. That result is as expected because the DA stores cannot be refilled when complete DAT inhibition prevents DA reuptake. Therefore, the voltammetry experiments depicted in Figure 4A were repeated with ∼80% inhibition of DAT activity. DAT inhibition prolonged the DA signal duration, but did not change the differences in burst-evoked DA facilitation between the NAc and dorsal striatal (Fig. 6B). As shown in the absence of DAT inhibition (Fig. 4A), the DA release in the dorsal striatum is not enhanced by afferent bursts nearly as strongly as is the DA release in the NAc shell (Fig. 6B).
Figure 6.

DATs do not account for the different relationships in DA signals in the dorsal striatum (d. striatum) and NAc shell. A, DA release in the dorsal striatum as a function of specific DAT inhibition by GBR-12909. The insets show DA release before (brief black traces) or with (larger gray traces) GBR at the labeled concentrations. Calibration: 2 μm, 10 s. B, In the presence of GBR (2 μm), DA release evoked by 1p (black) or by 5p at 20 Hz (gray) in the dorsal striatum and NAc shell. Calibration: 1 μm, 5 s. C, An example measurement is shown of the DA signal evoked by a single pulse before (black trace) or with (larger gray trace) DATs inhibited by 2 μm GBR-12909. Calibration: 2 μm, 10 s. D, The DA signal was measured for stimulus trains ranging from 1 to 20 pulses (at 20 Hz) in the presence of GBR (2 μm) normalized to the DA signal evoked by 1p. The dashed lines (points not shown for clarity) represent the response of the same slices under control conditions before the GBR was added (normalized to 1p under control conditions). As the stimulus train length increases, the DA signal in the dorsal striatum (filled square, n = 5) does not increase as much as in the NAc shell (filled circle, n = 5). When comparing the dorsal striatum to the NAc shell, inhibition of the DATs has little influence over the shape of the stimulus-number dependence for the DA signal as indicated by the dashed and solid lines. E, The DA signal was measured for stimulus trains of 5p ranging from 10 to 80 Hz in the presence of GBR (2 μm) normalized to the DA signal evoked by 1p. The dashed lines (points not shown for clarity) represent the response of the same slices under control conditions before the GBR was added (normalized to 1p under control conditions). As the stimulus frequency (freq) increases, the DA signal in the dorsal striatum (filled square, n = 9) does not increase as much as in the NAc shell (filled circle, n = 7). When comparing the dorsal striatum to the NAc shell, inhibition of the DATs does not account for the overall shape difference of the stimulus-frequency dependence for the DA signal.
After bath perfusion of GBR (2 μm, 30 min), the DA signal evoked by a single pulse increased dramatically (Fig. 6C). However, GBR did not alter the difference in the DA signaling relationships between the dorsal striatum and NAc shell (Fig. 6D,E). To examine the effect of burst length, we increased the number of pulses from 1 to 20 while applying 20 Hz trains in the presence of GBR (Fig. 6D). In Figure 6D, the filled symbols and solid lines were obtained in GBR (2 μm), and the dashed gray lines (controls) were obtained from the same slices before applying GBR. The difference between in the NAc shell (circles) and dorsal striatum (squares) in the presence of GBR (Fig. 6D, filled symbols) was similar to the control case (dashed lines, data obtained without GBR).
To examine the frequency dependence of the DA signal, we varied the frequency of a 5-pulse train from 10 to 80 Hz (Fig. 6E, solid lines in GBR, dashed lines in the absence of GBR). Qualitatively the difference between the NAc shell and dorsal striatum were retained in GBR. These results suggest that the difference in DAT density in the dorsal striatum and NAc shell does not primarily underlie the differences in DA signaling between the dorsal striatum and NAc shell. Rather, the differences in frequency dependence are more likely to arise from intrinsic differences in DA release properties in the striatal subregions.
DA neuron firing patterns induced by nicotine cause greater DA release in the NAc shell
We constructed stimulation trains based on the average firing patterns of DA neurons measured in vivo from free-moving rats before and after nicotine administration. Those two stimulus trains are shown as insets at the bottom of Figure 7, A and B. Based on the in vivo recordings, the intraburst frequency was held constant at 20 Hz, and the burst length was 5p in control and 10p in the presence of nicotine. The other spikes in the train set the overall firing frequencies to match the average firing rates of 4 Hz in control and 7 Hz in the presence of nicotine. Using carbon-fiber voltammetry, we measured the DA release evoked by those stimulus trains applied to slices from the dorsolateral striatum (Fig. 7A) or from the NAc shell (Fig. 7B). In the dorsal striatum, changes in the DA neuron firing caused by nicotine did not alter DA release significantly (92 ± 9%, n = 6, p > 0.05 when compared with the control area under the curve) (Fig. 7A,C). In the NAc shell, however, changes in the DA neuron firing caused by nicotine dramatically increased DA release (221 ± 20%, n = 5, p < 0.01 when compared with the control) (Fig. 7B,C). Although the biologically relevant cases are represented in Figure 7, for completeness all stimulus train combinations and nicotine treatments are shown in supplemental Figure S4 (available at www.jneurosci.org as supplemental material).
Discussion
In summary, the results show that DA axon terminals in the dorsolateral striatum and NAc shell respond differently to incoming action potentials (i.e., stimulus trains), consistent with the NAc shell having a lower basal probability of release than the dorsolateral striatum (Cragg, 2003; Rice and Cragg, 2004; Zhang and Sulzer, 2004). In freely moving rats, nicotine increases the phasic burst firing of the putative DA neurons (from the subset we identified). The NAc shell is much more responsive than the dorsolateral striatum to phasic bursts by the DA neurons. Consequently, microdialysis measurements detect a nicotine-induced DA rise mainly in the NAc shell, not the dorsolateral striatum, and this DA signal is likely important for the initiation of the addiction process (Balfour et al., 2000; Di Chiara, 2002; Dani and Harris, 2005). In addition, nicotine decreases DA release at low frequencies (e.g., single pulses) in the target areas of the dorsal striatum and NAc (Zhou et al., 2001; Rice and Cragg, 2004; Zhang et al., 2004). Because nicotine decreases tonic DA release much more effectively than release evoked by phasic bursts, nicotine stretches the range of mesostriatal DA signaling: low-frequency tonic signals are diminished while phasic-burst signals are favored. In that way, nicotine enhances the signal-to-noise relationship of DA transmission. This nicotine-induced change in the DA signaling may underlie the high prevalence of smoking seen in psychiatric patients, who are hypothesized to self-medicate to “normalize” their DA signaling properties (Dani and Harris, 2005; Kumari and Postma, 2005; Benowitz, 2008).
An interesting correlation was observed in the results. The maximum DA signal was observed at the relatively invariant average intraburst firing frequency measured in vivo from freely moving rats. The data suggest the intriguing possibility that phasic burst firing by the rat DA neurons occurs at the optimal frequency to give maximum DA signaling. This maximum in signaling was particularly evident when measuring the area under the DA concentration curve, but it was observed to a lesser extent in the DA peak amplitude too (Fig. 5D and supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). Higher-frequency stimulus trains would be expected to give more synchronized presynaptic Ca2+ signals to evoke release. However, at the optimal frequency of 20 Hz, a 5p stimulus train will be spread out more in time than a train given at 80 Hz, contributing to a more prolonged DA signal at 20 Hz.
Influence of nicotine over dopamine neurons in the midbrain
Synaptic events initiated by nicotine in the VTA/SNc underlie the DA signaling associated with tobacco use. Our present understanding suggests the following scenario. Nicotine obtained from tobacco initially activates β2-containing (β2*) nAChRs on the midbrain DA neurons (Pidoplichko et al., 1997; Picciotto et al., 1998; Jones et al., 2001). For a few minutes these DA neurons are directly depolarized and fire action potentials before the β2* nAChRs desensitize. On the same time scale, the nicotine slightly activates α7* nAChRs located on the presynaptic terminals of glutamatergic afferents, thereby increasing the excitatory drive onto the DA neurons (Mansvelder and McGehee, 2000; Schilström et al., 2003; Pidoplichko et al., 2004). The coincidence between enhanced presynaptic glutamatergic transmission and firing activity of the postsynaptic DA neurons induces synaptic potentiation (Mansvelder and McGehee, 2000, 2002; Mansvelder et al., 2002). Along with other synaptic events (Dani et al., 2001; Mansvelder and McGehee, 2002; Pidoplichko et al., 2004), this potentiated glutamatergic drive causes DA neurons to fire more in a burst or phasic mode (Grenhoff et al., 1986; Schilström et al., 2003). Nicotine administration increases the occurrence and the length of burst firing. This firing mode is expected to help mediate the phasic behavioral response to salient environmental stimuli as well as addictive drugs (Schultz et al., 1997; Maskos et al., 2005).
Nicotine differentially influences the dorsal striatum and nucleus accumbens shell
The frequency dependence and nicotinic modulation of DA release differ in the dorsolateral striatum and NAc shell (Chergui et al., 1994; Cragg et al., 2002; Cragg, 2003). There are anatomical and neurochemical differences that likely contribute to the functional inequality (Sesack, 2002). DA terminals in the NAc shell and dorsolateral striatum originate mainly from VTA and SNc, respectively (Haber et al., 2000). In the dorsolateral striatum, the DA varicosities contain almost exclusively small clear vesicles. In the NAc shell, DA varicosities contain mainly small clear vesicles, but there also are significant numbers of dense core vesicles and flattened clear vesicles. In the monkey, DA varicosities (or profiles) are ∼0.33 μm2 in the NAc shell compared with 0.14 μm2 in the dorsal striatum (Ikemoto et al., 1996). In addition, VTA DA neurons projecting mainly to the NAc shell often contain a calcium-binding protein, calbindin, whereas SNc DA neurons projecting to the dorsal striatum are often calbindin negative (Haber et al., 1995). These kinds of structural variations and differences in calcium buffering have been shown to regulate neurotransmitter release (Dittman and Regehr, 1998; Dittman et al., 2000; Thomson, 2000), and these factors likely contribute to the differences in DA release observed within the striatum.
Previous measurements in brain slices had indicated that there is a frequency-dependent function relating DA neuron activity and DA release (Cragg, 2003; Rice and Cragg, 2004; Zhang and Sulzer, 2004). Those results suggested that presynaptic nAChRs act to enhance DA release evoked at low frequency (Zhou et al., 2001). The ongoing activity of striatal cholinergic interneurons (Bennett and Wilson, 1999) excites presynaptic nAChRs on the DA terminals (Jones et al., 2001), producing an increase of intraterminal calcium that enhances DA release (Grady et al., 1997; Wonnacott et al., 2000). When nicotine was added to striatal brain slices, surprisingly, the presynaptic nAChRs were mainly desensitized, and low-frequency, tonic DA release was inhibited (Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004), but phasic DA release was regulated differently, such that phasic bursts induced large DA signals (Cragg, 2003; Rice and Cragg, 2004).
Our examination of nicotine's influence on the frequency dependence of DA release is consistent with the following interpretation. In the absence of other activity, a single action potential arriving at DA terminals in the dorsolateral striatum is more likely to cause release than one arriving at DA terminals in the NAc shell. The dorsal striatum seems to have a higher probability of release in response to a single stimulus. However, when nAChRs are inhibited either by an antagonist or by desensitization with nicotine (Zhou et al., 2001), the DA release to a single stimulus is inhibited more in the dorsal striatum than the NAc shell. This finding is consistent with presynaptic nAChRs contributing to the intraterminal calcium rise that causes DA release (Rathouz and Berg, 1994; Grady et al., 1997; Wonnacott et al., 2000; Zhou et al., 2001). When those nAChRs are inhibited, the boost to intraterminal calcium is lost, and those terminals then have a lowered probability of release. The DA terminals in the dorsal striatum appear to gain a significant proportion of their high release probability from the action of presynaptic nAChRs. Another difference is that high-frequency bursts enhance DA release in the NAc shell more than in the dorsal striatum. That result is explained, in part, by the finding that the basal probability of release is already high in the dorsal striatum (Zhang and Sulzer, 2004). Therefore, burst stimulation cannot boost the release probability much higher.
Overall, the data indicate that the frequency dependence of DA release is different in the dorsolateral striatum and NAc shell. That release difference arises from a number of factors, including intrinsic structural and calcium buffering differences as well as nicotinic mechanisms. Nicotine acts upon those DA release differences such that microdialysis measurements initially detect a nicotine-induced DA increase mainly in the NAc shell.
Footnotes
This work was supported by grants from the National Institute on Drug Abuse, the National Institute of Mental Health, the National Institute of Neurological Disorders and Stroke, and the National Alliance for Research on Schizophrenia and Depression. We thank K. Louie for helpful support with the unit recordings and M. A. Wilson for the opportunity to learn about the tetrode technique in his laboratory.
References
- Anstrom KK, Woodward DJ. Restraint increases dopaminergic burst firing in awake rats. Neuropsychopharmacology. 2005;30:1832–1840. doi: 10.1038/sj.npp.1300730. [DOI] [PubMed] [Google Scholar]
- Balfour DJ, Wright AE, Benwell ME, Birrell CE. The putative role of extra-synaptic mesolimbic dopamine in the neurobiology of nicotine dependence. Behav Brain Res. 2000;113:73–83. doi: 10.1016/s0166-4328(00)00202-3. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ. Spontaneous activity of neostriatal cholinergic interneurons in vitro. J Neurosci. 1999;19:5586–5596. doi: 10.1523/JNEUROSCI.19-13-05586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL. Clinical pharmacology of nicotine: implications for understanding, preventing, and treating tobacco addiction. Clin Pharmacol Ther. 2008;83:531–541. doi: 10.1038/clpt.2008.3. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Björklund A, Lindvall O. Dopamine-containing systems in the CNS. In: Björklund A, Hökfelt T, editors. Classical transmitters in the CNS, Part I. Amsterdam: Elsevier; 1984. pp. 55–122. [Google Scholar]
- Breiter HC, Gollub RL, Weisskoff RM, Kennedy DN, Makris N, Berke JD, Goodman JM, Kantor HL, Gastfriend DR, Riorden JP, Mathew RT, Rosen BR, Hyman SE. Acute effects of cocaine on human brain activity and emotion. Neuron. 1997;19:591–611. doi: 10.1016/s0896-6273(00)80374-8. [DOI] [PubMed] [Google Scholar]
- Butcher LL, Woolf NJ. Histochemical distribution of acetylcholinesterase in the central nervous system: clues to the localization of cholinergic neurons. In: Björklund A, Hökfelt T, Kuhar MJ, editors. Classical transmitters and transmitter receptors in the CNS. Part II. Amsterdam: Elsevier; 1984. pp. 1–50. [Google Scholar]
- Centonze D, Usiello A, Gubellini P, Pisani A, Borrelli E, Bernardi G, Calabresi P. Dopamine D2 receptor-mediated inhibition of dopaminergic neurons in mice lacking D2L receptors. Neuropsychopharmacology. 2002;27:723–726. doi: 10.1016/S0893-133X(02)00367-6. [DOI] [PubMed] [Google Scholar]
- Chergui K, Suaud-Chagny MF, Gonon F. Nonlinear relationship between impulse flow, dopamine release and dopamine elimination in the rat brain in vivo. Neuroscience. 1994;62:641–645. doi: 10.1016/0306-4522(94)90465-0. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington's disease. Brain Res Brain Res Rev. 2000;34:80–101. doi: 10.1016/s0165-0173(00)00039-4. [DOI] [PubMed] [Google Scholar]
- Coulter CL, Happe HK, Murrin LC. Postnatal development of the dopamine transporter: a quantitative autoradiographic study. Brain Res Dev Brain Res. 1996;92:172–181. doi: 10.1016/0165-3806(96)00004-1. [DOI] [PubMed] [Google Scholar]
- Cragg SJ. Variable dopamine release probability and short-term plasticity between functional domains of the primate striatum. J Neurosci. 2003;23:4378–4385. doi: 10.1523/JNEUROSCI.23-10-04378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ, Hille CJ, Greenfield SA. Functional domains in dorsal striatum of the nonhuman primate are defined by the dynamic behavior of dopamine. J Neurosci. 2002;22:5705–5712. doi: 10.1523/JNEUROSCI.22-13-05705.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–1470. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;31:349–352. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- Di Chiara G. Drug addiction as dopamine-dependent associative learning disorder. Eur J Pharmacol. 1999;375:13–30. doi: 10.1016/s0014-2999(99)00372-6. [DOI] [PubMed] [Google Scholar]
- Di Chiara G. Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav Brain Res. 2002;137:75–114. doi: 10.1016/s0166-4328(02)00286-3. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Kreitzer AC, Regehr WG. Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci. 2000;20:1374–1385. doi: 10.1523/JNEUROSCI.20-04-01374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, McIntosh JM, Cragg SJ. Alpha6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology. 2008;33:2158–2166. doi: 10.1038/sj.npp.1301617. [DOI] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlay SG, Benowitz NL. Arteriovenous differences in plasma concentration of nicotine and catecholamines and related cardiovascular effects after smoking, nicotine nasal spray, and intravenous nicotine. Clin Pharmacol Ther. 1997;62:453–463. doi: 10.1016/S0009-9236(97)90124-7. [DOI] [PubMed] [Google Scholar]
- Grace AA. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev. 2000;31:330–341. doi: 10.1016/s0165-0173(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Low doses of apomorphine elicit two opposing influences on dopamine cell electrophysiology. Brain Res. 1985;333:285–298. doi: 10.1016/0006-8993(85)91582-3. [DOI] [PubMed] [Google Scholar]
- Grady SR, Grun EU, Marks MJ, Collins AC. Pharmacological comparison of transient and persistent [3H]dopamine release from mouse striatal synaptosomes and response to chronic L-nicotine treatment. J Pharmacol Exp Ther. 1997;282:32–43. [PubMed] [Google Scholar]
- Grenhoff J, Aston-Jones G, Svensson TH. Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand. 1986;128:351–358. doi: 10.1111/j.1748-1716.1986.tb07988.x. [DOI] [PubMed] [Google Scholar]
- Haber SN, Ryoo H, Cox C, Lu W. Subsets of midbrain dopaminergic neurons in monkeys are distinguished by different levels of mRNA for the dopamine transporter: comparison with the mRNA for the D2 receptor, tyrosine hydroxylase and calbindin immunoreactivity. J Comp Neurol. 1995;362:400–410. doi: 10.1002/cne.903620308. [DOI] [PubMed] [Google Scholar]
- Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20:2369–2382. doi: 10.1523/JNEUROSCI.20-06-02369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer L. Basal forebrain in the context of schizophrenia. Brain Res Brain Res Rev. 2000;31:205–235. doi: 10.1016/s0165-0173(99)00039-9. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Biochemical aspects of Parkinson's disease. Neurology. 1998;51:S2–S9. doi: 10.1212/wnl.51.2_suppl_2.s2. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience. 2002;114:475–492. doi: 10.1016/s0306-4522(02)00267-1. [DOI] [PubMed] [Google Scholar]
- Ikemoto K, Satoh K, Kitahama K, Geffard M, Maeda T. Electron-microscopic study of dopaminergic structures in the medial subdivision of the monkey nucleus accumbens. Exp Brain Res. 1996;111:41–50. doi: 10.1007/BF00229554. [DOI] [PubMed] [Google Scholar]
- Jones IW, Bolam JP, Wonnacott S. Presynaptic localisation of the nicotinic acetylcholine receptor beta2 subunit immunoreactivity in rat nigrostriatal dopaminergic neurones. J Comp Neurol. 2001;439:235–247. doi: 10.1002/cne.1345. [DOI] [PubMed] [Google Scholar]
- Karan L, Dani JA, Benowitz N. The pharmacology of nicotine and tobacco. In: Graham AW, Schultz TK, Mayo-Smith MF, Ries RK, Wilford BB, editors. Principles of addiction medicine. Chevy Chase, MD: American Society of Addiction Medicine; 2003. pp. 225–248. [Google Scholar]
- Kumari V, Postma P. Nicotine use in schizophrenia: the self medication hypotheses. Neurosci Biobehav Rev. 2005;29:1021–1034. doi: 10.1016/j.neubiorev.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–773. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson's disease. Second of two parts. N Engl J Med. 1998;339:1130–1143. doi: 10.1056/NEJM199810153391607. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloëz-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ, Jones DL, Yim CY. From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol. 1980;14:69–97. doi: 10.1016/0301-0082(80)90018-0. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Groenewegen HJ, Lopes da Silva FH. The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioural, electrophysiological and anatomical data. Prog Neurobiol. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, Noguchi J, Areola OO, Liang Y, Peterson J, Zhang T, Dani JA. Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn Mem. 2004;11:60–69. doi: 10.1101/lm.70004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, Orzi F, Di Chiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature. 1996;382:255–257. doi: 10.1038/382255a0. [DOI] [PubMed] [Google Scholar]
- Rathouz MM, Berg DK. Synaptic-type acetylcholine receptors raise intracellular calcium levels in neurons by two mechanisms. J Neurosci. 1994;14:6935–6945. doi: 10.1523/JNEUROSCI.14-11-06935.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Robinson S, Smith DM, Mizumori SJ, Palmiter RD. Firing properties of dopamine neurons in freely moving dopamine-deficient mice: effects of dopamine receptor activation and anesthesia. Proc Natl Acad Sci U S A. 2004;101:13329–13334. doi: 10.1073/pnas.0405084101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilström B, Rawal N, Mameli-Engvall M, Nomikos GG, Svensson TH. Dual effects of nicotine on dopamine neurons mediated by different nicotinic receptor subtypes. Int J Neuropsychopharmacol. 2003;6:1–11. doi: 10.1017/S1461145702003188. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Sesack SR. Handbook of experimental pharmacology: dopamine in the CNS. Heidelberg: Springer; 2002. Synaptology of dopamine neurons. [Google Scholar]
- Thomson AM. Neurotransmission: chemical and electrical interneuron coupling. Curr Biol. 2000;10:R110–R112. doi: 10.1016/s0960-9822(00)00305-5. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Wang RY. Dopaminergic neurons in the rat ventral tegmental area. I. Identification and characterization. Brain Res Rev. 1981;3:123–140. [Google Scholar]
- Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265:676–679. doi: 10.1126/science.8036517. [DOI] [PubMed] [Google Scholar]
- Wise RA. Brain reward circuitry: insights from unsensed incentives. Neuron. 2002;36:229–240. doi: 10.1016/s0896-6273(02)00965-0. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Kaiser S, Mogg A, Soliakov L, Jones IW. Presynaptic nicotinic receptors modulating dopamine release in the rat striatum. Eur J Pharmacol. 2000;393:51–58. doi: 10.1016/s0014-2999(00)00005-4. [DOI] [PubMed] [Google Scholar]
- Zhang H, Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci. 2004;7:581–582. doi: 10.1038/nn1243. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhou FM, Dani JA. Cholinergic drugs for Alzheimer's disease enhance in vitro dopamine release. Mol Pharmacol. 2004;66:538–544. doi: 10.1124/mol.104.000299. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Wilson CJ, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol. 2002;53:590–605. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]