

Abstract
The stringent regulation of hematopoietic stem cell (HSC) quiescence versus cell cycle progression is essential for the preservation of a pool of long-term self-renewing cells and vital for sustaining an adequate supply of all blood lineages throughout life. Cell growth, the process that is mass increase, serves as a trigger for cell cycle progression and is regulated predominantly by mammalian target of rapamycin complex 1 (mTORC1) signaling. Emerging data from various mice models show deletion of several mTORC1 negative regulators, including PTEN, TSC1, PML and Fbxw7 result in similar HSC phenotypes characterized as HSC hyper-proliferation and subsequent exhaustion, and defective repopulating potential. Further pharmacological approaches show that PTEN, TSC1 and PML regulate HSC maintenance through mTORC1. mTORC1-mediated cell growth regulatory circuits thus plays a critical role in the regulation of HSC quiescence.
Keywords: hematopoietic stem cell, quiescence, cell growth, mTORC1, TSC
Hematopoietic stem cell quiescence, cell proliferation and cell growth
Stem cells are characterized by their differential capabilities and extensive self-renewal potential and play key roles in tissue maintenance and regeneration.1 Adult tissue stem cells exist in a variety of organs, including bone marrow, skin, gastrointestinal epithelium, and the central nervous system, among which hematopoietic stem cell (HSC), the stem cells functioning to sustain all blood effector lineages throughout life, has served as the paradigm for understanding of stem cell biology.2
HSCs exist in a relatively quiescent state in the bone marrow microenvironment, and experience an estimated single cell division every 2-4 weeks in the mouse. HSCs can be activated to rapidly enter cell cycle to regulate hematopoiesis as physiological demands dictate.2 The maintenance of HSC reserves therefore demands strict control over HSC quiescence and proliferation in the context of various intrinsic and extrinsic cues.3 Imbalances in these processes can lead to various hematological disorders: insufficient HSC proliferation would impair hematopoiesis and cause bone marrow failure, while hyper-proliferation of HSC might lead to exhaustion of HSC reserves and overproduction of various blood lineages, which could result in the development of hematological malignancy. Consistent with the essential role of quiescence in the regulation of HSC homeostasis, many key regulators of cell cycle progression including p27Kip1, p21Cip1, Bmi1, p18INK4c, p16INK4a, c-Myc have been shown to be integral in the regulation of self-renewal and multi-lineage differentiation of adult HSCs.4, 5
While cell proliferation leads to an increase in cell number, cell growth refers to increased cell mass brought about primarily by the synthesis of macromolecules such as protein and lipid. Cell growth is a prerequisite for cell cycle progression in that cells must grow to certain mass before they can commit to the cell division cycle and divide. Thus, sustained cell proliferation must be coupled to appropriate cell growth.6 While considerable information exists on the key roles of cell cycle components in the regulation of HSC biology, much less is known about the essentiality and functions of cell growth control regulators in HSC homeostasis.
mTORC1, the central mediator of cell growth
The mammalian target of rapamycin (mTOR) is an evolutionarily conserved serine/threonine kinase of the phosphatidylinositol kinase-related kinase (PIKK) subfamily. mTOR forms two distinct multi-protein complexes, rapamycin-sensitive mTOR complex 1 (mTORC1) and rapamycin-insensitive mTOR complex 2 (mTORC2).7 mTORC1 consists of mTOR, Raptor, PRAS40, and mLST8, and functions as the key regulator of protein synthesis and cell growth. mTORC1 controls mRNA translation, ribosome synthesis, metabolism-related gene expression, and autophagy via phosphorylation of a variety of downstream targets, among which are two key phosphorylation substrates S6 Kinase and 4E-BP1. mTORC2 is comprised of mTOR, Rictor, Sin1, and mLST8, and mainly functions to regulate cell survival/proliferation and actin cytoskeleton organization through phosphorylation of Akt. 7
One key upstream regulator of mTORC1 is TSC1-TSC2 complex (Figure 1). TSC1 and TSC2 are tumor suppressors mutated in Tuberous Sclerosis Complex Syndrome (TSC), a rare condition affecting ∼1 in 6,000 individuals and manifesting as hamartoma formation in a wide range of tissues.8 TSC1 and TSC2 form a stable complex and function as the GTPase activating protein (GAP) of the small GTPase, Rheb. Rheb cycles between GTP-bound active form and GDP-bound inactive form, and can potently activate mTORC1 when existing in active form. TSC1-TSC2 complex inhibits mTORC1 activity and restrains cell growth by stimulating Rheb GTP hydrolysis.8
Figrure 1. mTORC1-mediated cell growth machinery in the regulation of hematopoietic stem cell quiescence.
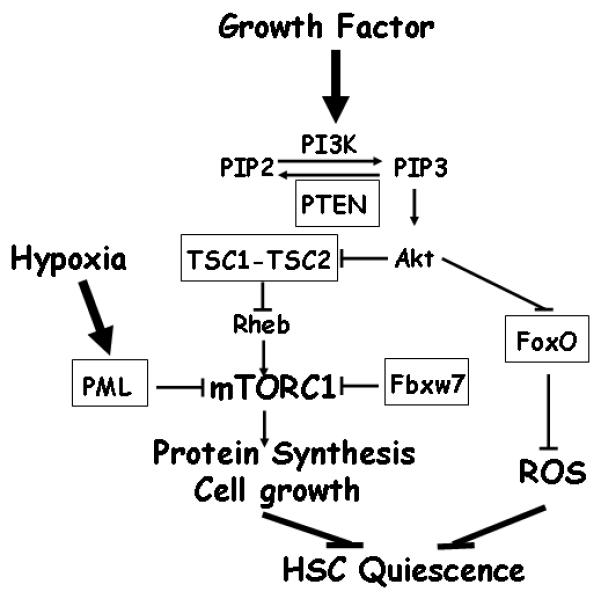
mTORC1 functions to integrates various upstream signaling pathways to regulate protein synthesis and cell growth. mTORC1 hyper-activation resulting from inactivation of PTEN, TSC1, PML and potentially Fbxw7 (all highlighted in box), drives HSC from quiescent state and leads to subsequent HSC exhaustion. Akt promotes mTORC1 activation and inhibits FoxO transcriptional factors. FoxOs (highlighted in box) function to maintain HSC quiescence through suppression of reactive oxygen species (ROS). For simplicity and for the focus of this review, several components of mTORC1 pathway, such as AMPK, REDD1, are not shown in this figure.
mTORC1 integrates various upstream signaling, including growth factor, energy stress and hypoxia, to regulate cell growth mainly through TSC1-TSC2 complex.9, 10 Growth factor stimulation results in PI3K-Akt activation, and activated Akt promotes mTORC1 signaling through Akt-mediated phosphorylation of both TSC2 and PRAS40, in which Akt-mediated phoshorylation of TSC2 relieves the inhibitory effect of TSC1-TSC2 complex on mTORC1 activation and cell growth. The PTEN tumor suppressor functions to antagonize PI3K, thus extinguishing AKT activation (Figure 1).11 Energy stress, on the other hand, leads to the activation of AMP-activated protein kinase (AMPK), which suppresses mTORC1 signaling via AMPK-mediated phosphorylation of both TSC2 and Raptor. Here AMPK-mediated phosphorylation of TSC2 promotes the inhibitory function of TSC1-TSC2 complex on mTORC1 activation and AMPK-mediated Raptor phosphorylation suppresses mTORC1 activation by Raptor.12-14 Furthermore, hypoxia inhibits mTORC1 activity through induction of REDD1 and REDD1-mediated dissociation of growth factor-stimulated TSC2/14-3-3 complex formation.15 Recently, it has been shown that PML also contributes to hypoxia suppression of mTORC1 activation through sequestration of mTOR into PML nuclear bodies upon hypoxia (Figure 1).16 Finally, amino acid signals activate mTORC1 through a TSC1-TSC2-independent, but Rag GTPases-dependent mechanism.17, 18
mTORC1-mediated cell growth signaling in the regulation of HSC maintenance
Recent genetic studies in murine models have established that several components of the mTORC1 signaling pathway play important roles in the regulation of HSC maintenance. Studies by Yilmaz et al 19 and Zhang et al 20 demonstrated that Mx-Cre-directed somatic deletion of tumor suppressor Pten in the hematopoietic system drives HSCs from a quiescence state into rapid cycling, resulting in HSC depletion. Pten deficient HSCs fail to sustain long-term hematopoietic reconstitution in the lethally irradiated recipient mice. Pten mutant mice also developed myeloproliferative disorder (MPD), which further progresses to frank leukemia. These studies thus established that PTEN plays differential roles in the maintenance of normal HSCs and leukaemic stem cells, thus providing a potential conceptual framework for the development of effective yet tolerable cancer drugs. Notably, treatment of mTORC1 inhibitor rapamycin reverses HSC depletion and leukemia phenotype in Pten deficient mice, implicating the critical role of mTORC1 signaling in the HSC depletion and leukemia phenotype observed in Pten deficient mice.19
Recent studies from our group 21 and Chen et al 22 showed that somatic deletion of TSC1 in HSC compartment leads to marked increased HSC proliferation, but subsequent progressive HSC depletion and defective long-term repopulating potential. Both studies further documented that rapamycin treatment can rescue HSC hyperproliferation and defective repopulating capability phenotypes observed in TSC1 deficient mice, strongly suggesting that TSC1, like PTEN, regulates HSC quiescence via mTORC1. Indeed, the phenotypes from TSC1 deficient mice described above share many similarities with those of Pten KO mice,19, 20 including increased proliferation leading to short-term expansion, but long-term depletion of HSCs, HSC mobilization to extramedullary sites, lineage development defects with blockade of lymphocyte development and expansion of myeloid development. It is noteworthy that, although our TSC1 KO mice develop MPD,21 the other study showed that TSC1 deletion leads to reduced myeloid development.22 These discrepancies are very likely due to different deletor strains used in these two studies (Rosa26CreERT2 used in our study vs Mx-Cre in the other study), which might result in different deletion patterns in hematopoietic microenvironment. Whether hematopoietic microenvironment plays any role in the regulation of myeloid proliferation resulting from TSC1 deficiency remains further investigation. Regardless of this, our TSC1 KO mice do not develop leukemia as shown in Pten KO mice, pointing to the existence of TSC-mTORC1 independent mechanisms to mediate PI3K-PTEN-Akt signaling function in HSCs. Along these lines, our recent studies23 and those of others24 show that the FoxO transcriptional factors, the other key downstream substrate of Akt, can also serve as important regulators of HSC homeostasis. Specifically, conditional deletion of FoxOs in adult murine hematopoietic system results in similar phenotype as observed in Pten deficient mice. Thus, these findings from TSC1, Pten and FoxO KO studies 19-24 collectively support the view that both TSC and FoxO pathways function as two key downstream effector arms of PI3K-PTEN-Akt signaling in the regulation of HSC maintenance (Figure 1). However, the inter-relationship of TSC and FoxO pathways in HSC biology is not understood.
Two other mTORC1 regulators also play similar roles in the regulation of HSC homeostasis. Pandolfi’s group demonstrated that PML deficient mice exhibit increase cell cycling, resulting in short-term increased, but long-term reduced HSC repopulating ability.25 Importantly, hyper-activation of mTORC1 is evident in PML KO HSCs, and rapamycin treatment rescues repopulation defect observed in PML KO mice.25 Mechanistically, they showed that PML can associate with mTOR and functions to inhibit mTORC1 signaling under hypoxia condition through sequestration of mTOR into PML nuclear bodies.16 In the other study from Matsuoka et al,26 deletion of tumor suppressor Fbxw7 in murine hematopoietic system led to similar phenotypes characterized as HSC depletion and impaired repopulating capacity due to active cell cycling. Fbxw7 is a subunit of SCF ubiquitin ligase complex and functions to target ubiquitination and degradation of a number of proteins.27 Interestingly, one such FBxw7 target is mTOR which is targeted by Fbxw7 for degradation, and mTORC1 was shown to be hyper-activated in the hematopoietic organs from Fbxw7 heterozygous mice.28 Since Fbxw7 also targets other component involved in the regulation of HSC homeostasis, such as c-Myc and Notch,27 it remains to be determined whether Fbxw7-mediated mTOR degradation plays any causal role in the regulation of HSC maintenance.
Conclusion and future perspectives
In summary, the confluence of data from recent genetic studies have demonstrated that deletion of one of several negative regulators of mTORC1, including PTEN,19, 20 TSC1,21, 22 PML,25 and Fbxw7,26 leads to strikingly similar phenotypes in the HSC compartment, namely active cell cycling driving HSCs from quiescence, leading to HSC exhaustion and defective long-term HSC repopulating potential. Importantly, pharmacological approaches convincingly pinpoint mTORC1 in mediating the effects from PTEN, TSC1 and PML.19, 21, 25 Together, these studies highlight the critical role of mTORC1-mediated cell growth signaling in the maintenance of HSC quiescence, and establish a causal role for mTORC1 hyper-activation in HSC active cell cycling and subsequent exhaustion (Figure 1).
Despite the emerging information supporting the important role of mTORC1 in HSC biology, many outstanding questions remain to be answered. Future studies will aim to dissect how mTORC1 integrate various upstream stress signals to regulate HSC maintenance, to identify the key downstream effectors of mTORC1 in mediating its functions in HSC biology, and to further explore the potential role of mTORC1 in other stem cell compartments. In addition, all the data so far regarding mTORC1 function in HSC biology have been derived from mTORC1 gain-of-function models resulting from deletion of mTORC1 negative regulators. mTORC1 loss-of-function mice models (such as Raptor conditional deletion mouse model) would be an important complementary approach for more in-depth understanding of mTORC1 functions in the regulation of HSC homeostasis. Finally, hyper-activation of mTORC1 results in the premature depletion of normal HSCs, but leads to the development of MPD and leukemia as shown in TSC1 and Pten KO mice models. Further exploring the differential role of mTORC1 in the regulation of leukaemic stem cells and HSCs would provide critical insight into HSC self-renewal mechanisms and may help illuminate specific therapeutic opportunities for leukaemia that spare normal stem cells.
Acknowledgements
This research was supported by National Cancer Institute grant R21CA135057 to RAD and BG. BG is the Research Fellow of the Leukemia & Lymphoma Society. RAD is the American Cancer Society Research Professor and is supported by the Robert A. and Renee E. Belfer Institute for Applied Cancer Science.
References
- 1.Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–5. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- 2.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–44. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9:115–28. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 5.Akala OO, Clarke MF. Hematopoietic stem cell self-renewal. Curr Opin Genet Dev. 2006;16:496–501. doi: 10.1016/j.gde.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Conlon I, Raff M. Size control in animal development. Cell. 1999;96:235–44. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 7.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14 Spec No. 2:R251–8. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 9.Reiling JH, Sabatini DM. Stress and mTORture signaling. Oncogene. 2006;25:6373–83. doi: 10.1038/sj.onc.1209889. [DOI] [PubMed] [Google Scholar]
- 10.Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene. 2006;25:6347–60. doi: 10.1038/sj.onc.1209885. [DOI] [PubMed] [Google Scholar]
- 11.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 13.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–51. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, et al. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779–85. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- 17.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–45. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–22. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 21.Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, et al. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008;105:19384–9. doi: 10.1073/pnas.0810584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008 doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–12. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–8. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008;22:986–91. doi: 10.1101/gad.1621808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 28.Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499–502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]