

Abstract
Many studies have suggested the involvement of wild-type (wt) p53 in the repair of DNA double-strand breaks (DSBs) via DNA end-joining (EJ) process. To investigate this possibility, we compared the capacity and fidelity of DNA EJ in RKO cells containing wt p53 and RKO cells containing no p53 (RKO cells with p53 knockdown). The p53 knockdown cells showed lower fidelity of DNA EJ compared to the control RKO cells. The DNA end-protection assay revealed the association of a protein complex including heterogeneous nuclear ribonucleoprotein G (hnRNP G) with the DNA ends in RKO cells containing wt p53, but not with the DNA ends in RKO cells with p53 knockdown. Depletion of endogenous hnRNP G notably diminished the fidelity of EJ in RKO cells expressing wt p53. Moreover, an ectopic expression of hnRNP G significantly enhanced the fidelity of DNA EJ and the protection of DNA ends in human cancer cells lacking hnRNP G protein or containing mutant hnRNP G. Finally, using recombinant hnRNP G proteins, we demonstrated the hnRNP G protein is able to bind to and protect DNA ends from degradation of nucleases. Our results suggest that wt p53 modulates DNA DSB repair by, in part, inducing hnRNP G, and the ability of hnRNP G to bind and protect DNA ends may contribute its ability to promote the fidelity of DNA EJ.
Keywords: hnRNP G, DSBs repair, DNA end-joining, p53
1. Introduction
Repair of DNA double-strand breaks (DSBs) involves ligation of DNA ends primarily through non-homologous end-joining (NHEJ) in mammalian cells [1]. The NHEJ pathway requires the Ku heterodimer and rejoins the DNA ends with an occasional loss of genetic information. Alternative DNA EJ pathways exist [2–4] in which the DSBs are joined with decreased fidelity and extensive deletion of the nucleotides at the DNA ends. The alternative mechanisms utilize small regions of microhomology at the DNA ends. Such erroneous DNA EJ may lead to accumulation of mutations and genetic instability [5–7].
The tumor suppressor p53 plays a critical role in DNA damage surveillance and maintenance of genetic integrity in mammalian cells [8]. Many studies have suggested a close association between p53 and DNA EJ. For example, in vitro p53 protein can bind to either double-stranded or single-stranded DNA ends [9–11]. The pleiotropic cellular activities of p53 include intrinsic 3′→5′ exonuclease activity, suggesting that p53 participates in the modulation of DNA EJ both directly and indirectly [12,13]. Presence of DSBs in cells can efficiently induce the biochemical activities of p53, resulting in cell cycle arrest, DNA repair, and/or apoptosis [14]. Also, treatment of cells with the chemical inhibitor of p53 (PFT-α) decreased the fidelity of DNA EJ activity in mouse fibroblasts, suggesting that p53 is necessary for the fidelity of DSBs repair [15].
We previously reported a significant diminution of precise DNA EJ activity in p53-defective human head and neck squamous cell carcinoma (HNSCC) cells compared with that of normal human oral keratinocytes (NHOK) [7]. When p53 in normal fibroblasts was abrogated by human papillomavirus (HPV) E6, the fidelity of DSBs repair was significantly decreased in vivo and in vitro [6].
Heterogenous nuclear ribonucleoprotein G (hnRNP G) was first identified as a nuclear protein with apparent molecular weight of 43 kDa [16,17]. It is composed of 391 amino acid (a.a.) residues encompassing the RNA binding domain (a.a. 10-88) at the amino terminus of the protein [17]. This structural feature suggests the physical association of hnRNP G with RNA, which may be required for its role in RNA processing and metabolism [17–19]. hnRNP G is detected primarily in the nuclei of mammalian cells and localized on lampbrush chromosomes of amphibian oocytes, supporting its association with nascent RNA transcripts as part of the transcriptional complexes [17]. hnRNP G alters pre-mRNA splicing pattern by antagonizing the effects of Tra2β, a splicing activator [20]. Therefore, the level of hnRNP G expression and its biological activity may affect global gene expression and maintenance of normal cellular homeostasis. Indeed, we reported that hnRNP G is a tumor suppressor against human head and neck cancer cells, but its mechanism of action remains to be investigated [21].
Here we show that hnRNP G promotes the fidelity of DSB repair by, in part, its ability to protect DNA ends.
2. Materials and methods
2.1. Cells and the culture conditions
Primary NHOK were prepared from separated epithelial tissue and serially subcultured in Keratinocyte Growth Medium (KGM, Cambrex) containing 0.15 mM Ca++ as described previously [22]. RKO, a human colorectal carcinoma cell line obtained from Dr. M. Brattain (Medical College of Ohio, Toledo, OH), was cultured in McCoy’s 5A (Invitrogen) medium supplemented with 10% FBS. Human head and neck squamous cell carcinoma cell lines SCC-4 and Tu-139 were obtained from the American Type Culture Collection (ATCC) and Drs. G. Clayman (University of Texas Medical Center, Houston, TX), respectively, and cultured in DMEM/Ham’s F12 supplemented with 10% FBS.
2.2. siRNA constructs, generation of virus, and infection of cells
The pSIREN-RetroQ vector (BD Biosciences), which drives the siRNAs expression under the control of human U6 RNA Pol III promoter, was utilized. The oligonucleotides containing the validated siRNA sequences of p53 [23] were obtained from Integrated DNA technologies. These oligonucleotides were annealed and inserted into the pSIREN-RetroQ vector downstream of the U6 promoter to construct pSIREN-RetroQ-p53. The resulting plasmid was transfected into the PA317 retrovirus packaging cell line, and the retrovirus expressing p53 siRNA was collected as described previously [24]. Retroviral vector expressing luciferase siRNA was also prepared as controls. RKO cells (5 × 105) were infected with the retroviral vectors in the presence of polybrene (5 μg/ml) at 37°C overnight and selected with 0.5 μg/ml puromycin (Invitrogen). The puromycin-resistant clones were subcultured and named Rp53KO (cells expressing p53 siRNAs) or RCTL (cells expressing luciferase siRNAs).
We constructed pLL3.7-RNPGi targeting hnRNP G. The detailed method of using pLL3.7 (Gift of Dr. Van Parijs, MIT, MA) to construct the lentivirus expressing siRNA is described elsewhere [25]. pLL3.7-RNPGi was constructed using double-stranded oligonucleotide cassette containing the hnRNP G target sequence (5′-GAGATATGAATGGA AAGTC -3′; +316 to +334) which was obtained from mouse hnRNP G gene (underlined nucleotide indicated a mismatch with human hnRNP G gene). The lentiviral vectors, LV-GFP and LV-RNPGi, were made from pLL3.7 (GFP alone) and pLL3.7-RNPGi, respectively.
2.3. Western blot analysis
Eighty-percent confluent cells were lysed. Protein concentration was determined using Protein Assay Reagent (BioRad). One hundred μg of protein was electrophoretically separated by SDS-polyacrylamide gel electrophoresis (PAGE). Western blot analysis was performed using antibodies against p53 (Oncogene Sciences), p21 (Oncogene Sciences), hnRNP G (G-17; Santa Cruz), and β-actin (Oncogene Sciences).
2.4. In vitro DNA EJ assay
In vitro DNA EJ activity was measured using the previously described method [6,7,26,27]. This in vitro assay utilizes whole cell lysates and pCR2.1 plasmid DNA (Invitrogen) linearized at the unique EcoRI or EcoRV site to analyze the end-joining of cohesive and blunt DNA ends, respectively. Briefly, cell lysates were incubated with the EcoRI or EcoRV-linearized plasmid at 37°C for 2 hours. After the end-joining reaction, PCR was performed with the EJ reaction using the M13 primers to amplify the rejoined DNA. PCR products were separated in agarose gel and visualized by staining with ethidium bromide. Erroneous non microhomology-mediated end-joining (NMEJ) can occur with few nucleotide alterations at the end-joined sites, undetectable by electrophoresis in agarose gels. This erroneous EJ can be detected by the following procedures: Individual 186 bp fragments were cloned in pcDNA3.1/V5-His TOPO plasmid (Invitrogen), transformed into Eschericia coli strain TOP10 (Invitrogen), and amplified by single colony PCR using the M13 primer set. The PCR product was then digested with EcoRI or EcoRV to determine whether the original restriction site is restored by end joining without nucleotide alteration. The PCR fragment resistant to the enzyme digestion was considered to be the product of erroneous DNA EJ activity [6,7,26].
2.5. In vivo DNA EJ assay
The pGL3 plasmid (Promega), in which expression of the luciferase gene is controlled by the cytomegalovirus promoter, was used to evaluate precise DNA EJ activity that precisely rejoins broken DNA ends in vivo [6,7,27]. The pGL3 plasmid was completely linearized by restriction endonuclease NarI (New England Biolabs), which cleaves within the luciferase-coding region and creates a DNA DSB with overhanging ends. The DNA was then transfected into cells with LipofectAMINE reagent (Invitrogen) following the procedures described by the supplier. The transfected cells were harvested and assayed for luciferase activity as described previously [6,7,27]. The reporter plasmid was digested to completion with NarI within the luciferase-coding region and only precise DNA EJ activity should restore the luciferase activity. The precise EJ activity was calculated from the luciferase activity of linearized pGL3 plasmids compared with that of the uncut plasmids. Each experiment was repeated three times.
2.6. Cellular exonuclease assay
The linearized DNA substrates used in the in vitro DNA EJ assay were incubated with cellular extracts for 10, 30, 60 or 120 minutes at 37°C in 30 μl reaction mixture containing 500 ng of DNA, 30 μg cell extract, and 1X exonuclease buffer (Promega). Controls included incubation without cell extract and incubation with 50U of exonuclease III (Promega). The reactions were stopped and deproteinized by adding 3.75 μl of 0.5 M EDTA pH 8.0, 2.25 μl of 2% SDS and 2.25 μl of 1 mg/ml Proteinase K followed by 10 minutes of incubation at 37°C. The deproteinized samples were separated by 0.8% agarose gel electrophoresis and visualized by ethidium bromide staining.
2.7. DNA end protection gel shift assay
To analyze DNA end protection, the EcoRV-linearized plasmid was 5′-end labeled with [γ-32P]ATP using T4 polynucleotide kinase (Invitrogen). The end-labeled DNA was incubated with the extracts for 30 min at 37°C in a 10 μl reaction mixture containing the indicated amount of extracts and 1X binding buffer. Ten unit of DNase I was added to the reaction and was incubated for 30 min at 37°C. After incubation, the reactions were run on a 3.5% acrylamide gel. Dried gels were analyzed and quantitated using PhosphorImager (Molecular Dynamics).
2.8. Identification of the proteins that co-migrate with the DNA end-binding complex
The DNA end-binding complex was excised from the gel and purified by electroelusion using the ElutaTube Protein Extraction Kit (Fermentas Inc.). Protein isolation and purification were performed according to the manufacturer’s guidelines. The isolated proteins were separated by SDS-PAGE and then stained with Coomassie blue. Identified band was excised and digested with 0.1 μg of trypsin per ml (Promega) overnight at 30°C. Tryptic peptide fragments were extracted with 60% acetonitrile and 0.1% trifluoroacetic acid using an automated Multiprobe II system (Packard Biosciences). The extracted peptides were dried under a vacuum, resuspended in a matrix, and subjected to matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) analysis using a Voyager DE Pro spectrometer (Voyager-DE STR, Applied Biosystems; UCLA Proteomics Core Facility). The resultant tryptic peptide molecular masses were entered into the Mascot Science search engine (www.matrixscience.com) for protein identification. The isolated proteins were separated by SDS-PAGE and subject to Western blot analysis using antibody against hnRNP G (Santa Cruz).
2.9. Cloning of hnRNP G cDNA, construction of retroviral vectors expressing hnRNP G, and infection of cells with the retroviruses
Wild-type and mutant K22R hnRNP G cDNAs were obtained from the total cDNA of NHOK and Tu-139, respectively. Detailed protocols for cloning, viral construction, and infection were described in previous publication [21].
2.10. Recombinant hnRNP G proteins and their activity to bind to DNA ends in vitro
Various GST fusion pGEX-2T (Amersham) vectors containing full-length wt, K22R or deletion mutants (Δ89-391) hnRNP G fragments were constructed. Each hnRNP G fragments was generated using PCR amplification. For cloning purpose, each sense primer was included a BamHI site and each antisense primer was included an EcoRI site. A termination codon (TGA) was created after the EcoRI site of antisense primers for the creation hnRNPG Δ89-391. The PCR product was digested with BamHI and EcoRI, and ligated into the pGEX-2T vector digested with the same enzymes. The primer sequences can be provided upon request. All fusion proteins were expressed in bacteria and affinity purified with glutathione-Sepharose (Pharmacia) according to manufacturer’s instruction.
To directly test hnRNP G’s binding to the ends of DNA, the in vitro end protection gel shift assay was performed by incubating only the purified hnRNP G recombinant proteins and the DNA substrates as described in Section 2.7. Briefly, one hundred ng of the DNA substrate labeled with 32P at the 5′ terminus was incubated with 300 ng of GST or GST-hnRNPG proteins for 30 min at 37°C. Then, DNase I was added to the reaction and incubated for 30 min at 37°C to digest unbound or unprotected DNA. After incubation, the reactions were run on 5.0 % acrylamide gel in 0.5X TBE. Dried gel was analyzed using STORM phosphorimager (Molecular Dynamics).
3. Results
3.1 Knockdown of endogenous p53 impairs the fidelity of DNA EJ activity in RKO cells
We previously showed that expression of HPV E6 in NHOK led to defective DNA EJ activity presumably by degradation of p53 [6]. In the recent study, we investigated the direct role of p53 in DNA EJ activity by knocking down the endogenous wt p53 by RNAi. RKO cells expressing wt p53 were infected with a retroviral vector expressing p53 siRNA, and the puromycin-resistant cell clones were selected. As a control, RKO cells were also infected with a retroviral vector expressing luciferase siRNAs. The drug-resistant cells were cloned and screened for changes in p53 expression by Western blot analysis. p53 expression in clones Rp53KO-1, Rp53KO-2, Rp53KO-9, and Rp53KO-19 was significantly inhibited (Fig. 1A). RCTL-1 and RCTL-2 cells infected with the control retrovirus showed normal level of p53 expressed compared to the parental RKO cells. The level of p2lWAF1/CIP1, a gene product whose expression is regulated by p53, was also downregulated in the p53 knockdown clones (Fig. 1A). We further defined p53 function by exposing both control (RKO and RCTL-1) and p53 knockdown RKO (Rp53KO-1 and -2) cells to DNA damaging agent, actinomycin D (data not shown). Genotoxic stress caused significant induction of p53 level in the control RKO cells but not in the p53 knockdown RKO cells. DNA damage also led to induction of the p53-dependent gene, p2lWAF1/CIP1. These data indicate that RKO has the functional p53, and that the p53 knockdown RKO cells are functionally p53-deficient. Not only is p53 expression abrogated, but induction of p53-dependent genes is also prevented. We also tested whether the p53 knockdown affected the levels of proteins involved in NHEJ such as Ku70, DNA-PK, XRCC4, DNA ligase IV, Mre11, NBS1, and Rad50. There were no significant differences in the levels of the tested proteins in the control and the p53 knockdown clones (data not shown). These results indicated that p53 siRNA inhibited the expression of p53 and that the knockdown of p53 did not alter the levels of NHEJ-related proteins tested in the study.
Fig. 1. Effect of p53 knockdown on the fidelity of DNA EJ.

(A) Cell lysates prepared from parental RKO, two control RKO clones (RCTL-1 and -2) infected with retroviruses expressing control siRNA, and four RKO clones (Rp53KO-1, -2, -9, and -19) infected with retroviruses expressing p53 siRNA. Western analysis was performed using anti-p53 and anti-p21. Anti-actin antibody was used as a loading control. (B) In vitro DNA EJ assay. Upper panel: The EcoRI-linearized plasmid was incubated with cellular extracts and amplified as described in Materials and Methods. Lower panel: The EcoRV-linearized plasmid was incubated with cellular extracts and amplified. (C) Effect of p53 knockdown on the frequency of erroneous DNA EJ activity in RKO cells. (D) In vivo DNA EJ assay. Effect of p53 knockdown on the relative error-free DNA EJ capacity in vivo. The relative error-free DNA EJ capacity was calculated by comparing luciferase activity expressed in cells transfected with NarI-digested plasmid with that of the uncut plasmid. The results were obtained from three independent transfection experiments. The error bars indicate ±S.D from three different assays.
In the next experiment, we investigated the effects of p53 knockdown on DNA EJ activity in RKO cells by the in vitro DNA EJ assay. With the pCR2.1 plasmid linearized with EcoRI digestion, all tested RKO clones revealed a PCR product of approximately 186 bp, giving the appearance that the EJ activity for the DNA substrate with 5′-overhang was not altered by p53 knockdown (Fig. 1B). In contrast, when the plasmid was linearized with EcoRV, creating blunt DNA ends, the extracts from the Rp53KO clones completely lacked the EJ activity, while the control cells (RKO parental cells and the RCTL clones) exhibited intact EJ activity (Fig. 1B). To determine whether there was any minor sequence alteration at the ligation site of the rejoined EcoRI-digested plasmid, the 186 bp PCR fragment was cloned into pcDNA3.1/V5-His TOPO plasmid in TOP10 E. coli strain and reamplified by single colony PCR. The 186 bp fragments amplified from the individual bacterial colonies were digested with EcoRI to determine whether the original EcoRI site was restored by DNA EJ. We found more frequent sequence alteration [NMEJ error; 6,7] in the plasmids rejoined with the extracts of the Rp53KO clones compared to the control cells (RKO parental cells and the RCTL clones) (Fig. 1C). The effect of p53 on the fidelity of DNA EJ activity was further confirmed using the linearized pGL3-Luc plasmids. The relative in vivo error-free DNA EJ activity was determined from the ratio of luciferase expression in the RKO clones transfected with the linearized plasmid and luciferase expression in the same clones transfected with the uncut circular plasmid (Fig. 1D). Approximately 11% of the linearized pGL3-Luc plasmids were correctly recircularized in the controls (RKO, RCTL-1 and -2), whereas the efficiency of error-free DNA EJ activity was substantially reduced in the p53 knockdown RKO clones (2.3–4.1%). These results indicate that abrogation of p53 leads to increased errors in DNA EJ reaction.
3.2. p53 knockdown RKO cellular extract showed elevated degradation of DNA substrate
It is possible that the erroneous EJ in the p53 knockdown cells resulted from cellular exonuclease activity causing nucleotide deletions. To check for this possibility, we compared the endogenous exonuclease activity of the cellular extracts prepared from control and p53 knockdown RKO clones (Fig. 2A, B, and C). The pCR2.1 plasmid DNA linearized with either EcoRI or EcoRV was incubated with the cell extracts for various time points and electrophoresed in 0.8% agarose gels (Fig. 2A). Whereas the control cellular extract (RCTL-2) showed a delay of 15 minutes and 30 minutes in degradation of overhang- and blunt-ended DNA substrates, respectively, the p53 knockdown extract (Rp53KO-2) demonstrated delays of only 5 minutes and 10 minutes, respectively, in the degradation of both DNA substrates (Fig. 2B and 2C). Afterward exonuclease digestion proceeded at a similar rate with control and knockdown cellular extracts. Thus, wt p53 activity increased the delay before exonuclease digestion occurred. These data indicated that the elevation of cellular exonuclease activity was mediated by p53 downregulation.
Fig. 2. Effect of p53 knockdown on cellular exonuclease activity.

(A) The EcoRV-linearized (upper) or EcoRI-linearized (lower) plasmids were incubated with 30 μg of extracts for indicated min. As controls, the DNA substrates were incubated without cell extract or with 50 U of exonuclease III. (B) Quantitative expression of exonuclease activity. The intensity determined by Scion imaging (Scion Corporation, Frederick, MD) of unincubated DNA substrate (no extract) was used as control (100%). (C) Quantitative expression of exonuclease activity. The EcoRI-linearized plasmids were incubated without cell extract or with 30 μg of extracts for indicated min. (D) Quantitative expression of exonuclease activity of NHOK.
To ascertain the validity of results obtained with RKO cells, endogenous exonuclease activity of NHOK was also measured by incubating the cellular extract with blunt-ended or overhang-ended DNA substrates. Like the control RKO cellular extract, the NHOK extract demonstrated a delay of 30 minutes in the degradation of both DNA substrates (Fig. 2D). Afterward exonuclease digestion proceeded at the same rate with overhang ended DNA and with blunt-ended DNA.
3.3. Identification of a novel DNA end-protection complex in RKO cells expressing wt p53 but not in the p53 knockdown RKO cells
The p53 knockdown RKO cellular extracts demonstrated elevated exonuclease activity and appeared to be defective in protecting the termini of a DNA substrate against nuclease digestion. Therefore, we speculated that the shortening of the delay for exonuclease activity in the p53 knockdown cellular extract could result from a loss of p53 molecules at the termini of the DNA substrate, thereby decreasing their protection from exonuclease. To test this possibility, we performed a modified gel shift assay (end-protection gel shift assay) with the DNA substrate labeled with 32P at the 5′ terminus (Fig. 3). After incubation of end-labeled, EcoRV-linearized plasmid with cellular extracts, the labeled DNA was digested with DNase I (Fig. 3A lanes 4 and 6). With control RCTL-2 cellular extract containing wt p53, a DNA end-binding complex was observed (Fig. 3A lane 6), indicating that the 5′ ends of the DNA substrate were protected from DNase I digestion by bound protein(s). In contrast, such a complex was not detected in the p53 knockdown cellular extract (Fig. 3A lane 4). The formation of the complex was enhanced by increasing the amount of control RCTL-2 cellular extract (Fig. 3B lanes 4, 5, and 6) but not detected with increasing the amount of the p53 knockdown cellular extract (data not shown). In addition, whereas the control cellular extract caused a large gel shift in the absence of DNase I treatment (Fig. 3A lane 5 and Fig. 3B lane 3), such a shift was not observed with the p53 knockdown extract (Fig. 3A lane 3). These results indicate that the 5′ ends of the linearized plasmid are protected from nuclease digestion by putative protein complex, and the DNA end-protection capacity is abolished by knockdown of wt p53.
Fig. 3. Effect of p53 knockdown on DNA end protection.

(A) End protection gel shift assay with the control RKO (RCTL-2) and the p53 knockdown RKO (Rp53KO-2) extracts. * indicates a novel DNA end-binding complex containing 5′-32P-ends of DNA substrates and protein(s). (B) Dose-dependent effect of RCTL-1 cellular extract on DNA ends protection. Lane 3, 5μg of the extract; Lane 4, 2μg of the extract; Lane 4, 5μg of the extract; Lane 4, 10μg of the extract.
3.4. Identification of hnRNP G protein in the DNA end protection complex
To identify the protein associated with DNA end-protection, the DNA and protein complex (Fig. 3, asterisk) from the modified gel shift experiment was isolated by electroelution. Cellular protein(s) was/were dissociated from the complex by boiling, separated by SDS-PAGE, and visualized by Coomassie blue staining. As shown in Figure 4A, a protein band (~40 kDa) was detected in the isolated DNA end-protection complex. To determine whether the p53 protein was present in the complex, the resolved protein(s) from the complex was/were subjected to Western blot analysis using anti-p53 antibody (Fig. 4B). The assay did not reveal the presence of p53 protein in the DNA end-protection complex, indicating that p53 mediates DNA end-protection through unknown factor(s).
Fig. 4. Identification of a ~40 kDa protein in the DNA end-binding complex.

(A) The DNA end-binding complex and whole lysate from the RKO control clone (RCTL-2) were resolved in an SDS-PAGE and then stained with Coomassie blue. (B) The DNA end-binding complex and whole lysate from the RKO control clone (RCTL-2) were resolved by SDS-PAGE and subjected to Western blotting using anti-p53.
To identify the protein that co-purified with the protected DNA ends, the ~40 kDa protein band was excised from the gel, digested with trypsin to obtain individual peptide fragments, and combined with matrices. Dried matrix-peptide mixtures were subjected to MALDI-TOF. Peptide masses obtained from a protein band at ~40 kDa matched the peptide fingerprint for hnRNP G. The identity of this protein as hnRNP G was confirmed by Western blot analysis using the DNA end-protection complex with specific antibodies against hnRNP G (Fig. 5A). We compared the level of hnRNP G in the control RKO cells (RCTL-1 and -2) and in NHOK with that in the p53 knockdown RKO clones (Rp53KO-1 and -2). Western blot analysis revealed that the level of hnRNP G was substantially decreased in the p53 knockdown RKO clones, compared to that of the control RKO clones (Fig. 5B). The above results raised the possibility that p53 maintains the fidelity of DNA EJ activity by DNA end-protection from cellular exonuclease through upregulation of hnRNP G expression.
Fig. 5. Identification of hnRNP G in the DNA end-binding complex.

(A) Upper panel: The DNA end-binding complex and whole lysates from RCTL-2 and Rp53KO-2 were subjected to Western using anti-hnRNP G. Lower panel: The complex and whole lysates were resolved in an SDS-PAGE and then stained with Coomassie blue. (B) Western analysis of hnRNP G in NHOK, two control RKO clones (RCTL-1 and -2) and two p53 knockdown RKO clones (Rp53KO-1, and -2). Membrane stained with Ponceau-S to serves as the internal control to account for the loading error.
3.5. Depletion of endogenous hnRNP G expression resulted in decrease fidelity of DNA EJ
To examine whether the p53-mediated EJ fidelity is specifically associated with hnRNP G, we knocked down the endogenous hnRNP G of RKO cells by infecting them with lentiviral vectors expressing hnRNP G siRNA or GFP alone. At two days post-infection, more than 90% of the RKO cells infected with LV-GFP or LV-RNPGi demonstrated green fluorescence (data not shown), indicating efficient infectivity. Also at two days post-infection, the endogenous hnRNP G expression level was decreased by 90% in RKO cells infected with LV-RNPGi if compared to the cells infected with LV-GFP (Fig. 6A). We next performed the in vivo DNA EJ assay using the RKO cells infected with LV-GFP or LV-RNPGi. Compared to the control RKO cells, the error-free DNA EJ activity in the hnRNP G-depleted RKO cells resulted in 3-fold reduction (Fig. 6B), the amount that is comparable to that in the p53 knockdown RKO cells (Fig. 1D).
Fig. 6. Inhibition of endogenous hnRNP G resulted in decreased fidelity of DNA EJ.

(A) RKO cells were infected with LV-RNPGi or LV-GFP, and, 48 hr after infection, hnRNP G knockdown was examined by RT-PCR and Western analysis. (B) Effect of hnRNP G depletion on the error-free DNA EJ capacity in vivo. RKO cells were transfected with the plasmids for 7 hr and incubated for additional 48 hr without the plasmids in order to obtain the maximum hnRNP G knockdown which was achieved at 48 hr post-infection. Then, the cells were subjected to the luciferase reporter assay. The relative error-free DNA EJ capacity was calculated by comparing luciferase activity expressed in cells transfected with NarI-digested plasmid with that of the uncut plasmid. The results were obtained from three independent transfection experiments. The error bars indicate ±S.D from three different assays.
3.6. wt hnRNP G enhanced the fidelity of DNA EJ in cancer cells lacking the expression of hnRNP G
We recently showed lack of wt hnRNP G expression in human head and neck squamous cell carcinoma (HNSCC) tissues and lines, which do not express wt p53 [21]. We also showed decreased fidelity of DNA EJ activity in these cancer cells compared with normal human epithelial cells, which expressed readily detectable level of hnRNP G [7,21]. To obtain a direct evidence of the role of hnRNP G in the fidelity of DNA EJ activity, we infected HNSCC cells lacking hnRNP G protein (human tongue carcinoma SCC-4 cells) or containing mutant K22R hnRNP G (human larynx carcinoma Tu-139 cells) with the retroviruses expressing wt or K22R mutant hnRNP G [21]. The infected cells successfully expressed the exogenous wt or mutant hnRNP G protein (Fig. 7A)
Fig. 7. Effect of hnRNP G on DNA EJ activity in cancer cells.

(A) Ectopic expression of wt hnRNP G or mutant hnRNP G (K22R) in two HNSCC cell lines (SCC-4 and Tu-139). Western blotting analysis was performed using the antibody against hnRNP G. Parent, non-infected cells; LX, cells infected with retrovirus expressing empty vector (LXSN); wtRG, cells infected with retrovirus expressing wt hnRNP G (LXSN-wtRG); K22R, cells infected with retrovirus expressing mutant hnRNP G (LXSN-K22R). (B) Effect of hnRNP G overexpression on the fidelity of DNA EJ in vivo. The relative error-free DNA EJ capacity was calculated as described in Materials and Methods. Each experiment was performed with a triplicate dish. The error bars indicate ±SD from three independent experiments. (C) Effect of hnRNP G overexpression on the fidelity of DNA EJ in vitro. The frequency of erroneous DNA EJ was calculated by counting the number of EcoRI or EcoRV-resistant PCR products as a percentage of the total number of tested PCR products. ND, not determined. (D) Effect of hnRNP G overexpression on cellular exonuclease activity. The EcoRV-linearized (upper) plasmids were incubated with 30 μg of extracts for indicated min.
To examine the effect of hnRNP G on DNA EJ activity, the in vivo DNA EJ assay was carried out using the control cells expressing empty vector and the cells overexpressing hnRNP G. An average of 6.2% of the linearized pGL3-Luc plasmids were correctly recircularized in the control SCC4-LX cells, whereas the error-free DNA EJ activity was substantially increased in the SCC4-wtRG cells expressing wt hnRNP G, but not in SCC4-K22R cells expressing mutant hnRNP G (Fig. 7B). Similar effect of hnRNP G was also found in the Tu-139 cell line. We further investigated the effect of hnRNP G expression on erroneous NMEJ activity in SCC-4 and Tu-139. In both cell lines, the restoration of wt hnRNP G notably lowered the frequency of erroneous DNA EJ (Fig. 7C).
We also compared the exonuclease activity of nuclear extracts prepared from SCC4-LX, SCC4-wtRG, and SCC4-K22R upon DNA substrates (Fig. 7D). The expression of wt hnRNP G significantly increased the amount of protected DNA substrate compared with those of LXSN and mutant K22R (Fig. 7D). These results indicate that hnRNP G enhances the fidelity of DNA EJ activity and the protection of DNA ends.
3.7. hnRNP G directly binds to DNA ends in vitro
To investigate whether hnRNP G directly binds to DNA ends, we performed the in vitro end protection gel shift assay by incubating only the purified hnRNP G recombinant proteins and the DNA substrates (Fig. 8). For this purpose, we first constructed and purified GST-tagged full-length wt hnRNP G and the K22R hnRNP G mutant and deleted hnRNP G (Δ89-391) recombinant proteins (data not shown). The most prominent structural feature of hnRNP G is the presence of RNA-binding domain (residues 10-88) at the amino terminus [17,18]. The physical interaction of hnRNP G with DNA ends was determined by the presence of shifted specific radioactive signal containing the 5′-32P-ends of DNA substrates. Shifted specific radioactive signals were observed from the reaction containing the full-length hnRNPG (Fig. 8, lane 4) or the hnRNPGΔ89-391 (Fig. 8, lane 6) protein, indicating that the 5′-32P-ends of DNA substrates were protected from DNase I digestion by bound wt hnRNP G or hnRNPGΔ89-391. We did not observe such signal when GST or GST-K22R was incubated with the labeled DNA substrates (lane 3 and 5). These results led us to conclude that (i) hnRNP G directly binds to DNA ends and (ii) the RNA-binding domain (residues 1-88) of hnRNP G is sufficient for the DNA end-binding activity of hnRNP G, and (iii) K22R point mutation causes the loss of DNA end-binding activity of wt hnRNP G.
Fig. 8. hnRNP G directly binds to DNA ends in vitro.
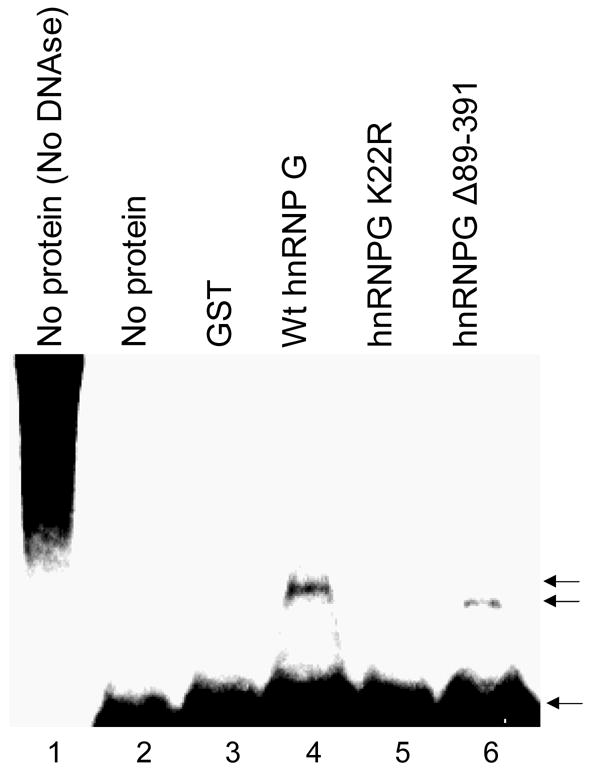
End protection gel shift assay was performed by incubating end-labeled DNA substrates and GST (lane 3), GST-tagged full-length wt hnRNP G (lane 4), the K22R hnRNP G mutant (lane 5) or deleted hnRNP G (Δ89-391; lane 6) recombinant proteins. The bottom arrow indicates the position of free DNA ends; the upper arrows indicate protected/bound DNA ends containing the recombinant hnRNP G proteins.
4. Discussion
DNA end-joining (EJ) represents the major DSBs repair mechanism in mammalian cells, but its fidelity remains to be elucidated. The fidelity represents the ability to rejoin the break accurately which affects genetic stability and the potential onset of oncogenesis. The present work is the first demonstration that hnRNP G protein is a novel p53 target gene product and plays an important role in the fidelity of DNA DSBs repair.
Our p53 siRNAs experiment demonstrated that cellular extracts derived from p53 knockdown RKO cells showed a higher error incidence and a significantly diminished precise in vivo DNA EJ activity if compared to extracts from RKO expressing wt p53. Together with our previous observation [6,7], our result showed an important role of p53 in the fidelity of DNA EJ. This is also consistent with a report that the p53 inhibitor pifithrin-α significantly reduced precise DNA ligation in murine fibroblasts [15]. However, the underlying mechanism or molecular determinant for the p53-mediated EJ fidelity has not been reported.
We detected a close relationship between the p53 status and the level of hnRNP G expression, i.e., a significant reduction of hnRNP G expression in both p53 knockdown cellular extracts and p53-defective cancer cells compared to that in wt p53 expressing cells [7,21]. We found transcriptional regulation of hnRNP G by p53 through a putative p53 binding site (−1,430 to −1,411) as well as a possible post-translational stabilization of hnRNP G by DNA damage, ubiquitin-dependent proteasomal pathway (unpublished our observation). We are currently investigating detail mechanisms underlying the transcription regulation of hnRNP G by p53 and a potential role for p53 in the post-translational stabilization of hnRNP G by DNA damage. Furthermore, the knockdown of endogenous hnRNP G in RKO cells expressing wt p53 resulted in a significant reduction in EJ fidelity, which is comparable to that in the p53 knockdown RKO cells (Fig. 1D and 6B). These data suggested that hnRNP G may be a target of the p53 tumor suppressor and a key molecular determinant in the p53-mediated EJ fidelity. This new role for the p53 protein in the regulation of hnRNP G is to be added to its role as a key regulator in genetic stability via regulation of its target genes [8].
The structural feature of hnRNP G indicated that like other hnRNPs, hnRNP G is directly associated with RNA and involved in RNA metabolism [17–19]. Moreover, protein domain-mapping studies showed the presence of RNP-consensus RNA binding domain (residues 10-88) at the amino-terminus of hnRNP G, including the codon 22 [17]. Others suggested that the RNA binding capability of hnRNPs is critical for its effects on gene regulation [24]. A recent study demonstrated that hnRNP G may modulate cellular splicing preferences, suggesting the deregulation of hnRNP G may impose profound effects on the alteration of cellular transcriptome [20]. Therefore, pleiotropic effects of hnRNP G on multiple biological pathways due to its ability to modulate RNA processing have been speculated, but its roles in DNA repair and carcinogenesis have not been investigated [28]. For instance, hnRNP G demonstrated tumor suppressive effect against human cancer cells [21], and here we report a novel effect of hnRNP G on the fidelity of DNA EJ activity by, in part, the ability of hnRNP G to bind and protect DNA ends. However, we would not rule out a possibility that other biological activities of hnRNP G, i.e., tumor suppression and EJ fidelity, are due to the global effect of hnRNP G in gene expression through the processing of mRNAs rather than through its direct effect on DNA EJ.
How could hnRNP G modulate the fidelity of DNA EJ? After exposure of free DNA end created from DSB, one cellular response would be to prevent exonuclease attack at DNA ends that arise as a consequence of DNA damage. Inhibition of cellular exonuclease activity and/or protection of DNA ends from exonuclease attack may be a critical initial step in the repairing damaged DNA and the maintenance of genomic stability by preventing deleterious degradation of DNA at inappropriate loci. Therefore, in this regard, our finding suggests an important role of hnRNP G for this process (Fig. 8). DNA end protection by hnRNP G prevents DNA ends from exonuclease attack, gives cells time to maintain intact DNA ends and eventually allow the DNA machinery to correct the damage without losing genetic information.
It is of interest to point out that the K22R hnRNP G mutant seems to be a very important mutant form that would help in elucidating the role of hnRNP G in EJ due to the following reasons: i) K22R point mutation is a naturally occurring mutant form that we obtained from Tu-139 cancer cells; ii) K22R resides in the RNA-binding domain that is critical for the RNA-splicing function of hnRNP G; and iii) K22R failed to enhance EJ fidelity and DNA end protection. Based on the above, we could speculate at least two non-mutually exclusive mechanisms: 1) hnRNP G has a global effect on the cellular levels of several proteins, which may indirectly affect the DNA EJ activity, and 2) the DNA end-binding and protection of hnRNP G may directly affect the fidelity of DNA EJ. The slower exonuclease activity in the hnRNP G overexpressing cells was attributable to an increased delay in the initiation of DNA degradation possibly caused by hnRNP G binding and protection of the DNA ends against exonuclease activity.
The pleiotropic cellular activities of p53 include intrinsic 3′→5′ exonuclease activity. Interestingly, the p53 knockdown RKO cells exhibited accelerated exonuclease activity with both 5′-overhang and blunt-ended DNA substrates when compared to the control RKO expressing wt p53. The slower exonuclease activity in the control RKO was attributable to an increased delay in the initiation of DNA degradation possibly caused by hnRNP G binding and protection of the DNA ends against exonuclease activity. Another possibility is that the intrinsic exonuclease activity of p53 itself may be involved since p53 must not be activated, e.g., phosphorylated, to possess this activity [29]. Alternatively, p53 inhibition of another cellular exonuclease activity could be involved in the increased exonuclease activity of p53 knockdown RKO cells since p53 transcriptionally inhibits cellular genes harboring exonuclease activity [30] or modulates exonuclease activity through physical interaction [31].
In summary, the above data revealed a novel function for the hnRNP G protein, i.e., a role in the error-free DSBs EJ pathway. Moreover, we have detected that the absence or significant reduction of hnRNP G protein in precancerous, malignant oral lesions, and cancer cells derived from head and neck SCC compared to normal human oral epithelia and cultured oral keratinocytes [21]. Thus, hnRNP G may play an important role in the maintenance of genetic integrity and possible carcinogenesis.
Acknowledgments
This work was supported in part by the grants DE14147 and DE15902 funded by the National Institute of Dental and Craniofacial Research (NIDCR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFFERENCES
- 1.Pierce AJ, Stark JM, Araujo FD, Moynahan ME, Berwick M, Jasin M. Double-strand breaks and tumorigenesis. Trends Cell Biol. 2001;11:S52–59. doi: 10.1016/s0962-8924(01)02149-3. [DOI] [PubMed] [Google Scholar]
- 2.Kabotyanski EB, Gomelsky L, Han J, Stamato TD, Roth DB. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 1998;6:5333–5342. doi: 10.1093/nar/26.23.5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liang F, Jasin M. Ku80-deficient cells exhibit excess degradation of extra chromosomal DNA. J Biol Chem. 1996;271:14405–14411. doi: 10.1074/jbc.271.24.14405. [DOI] [PubMed] [Google Scholar]
- 4.Roth DB. Restraining the V(D)J recombinase. Nature Rev Immunol. 2003;3:656–666. doi: 10.1038/nri1152. [DOI] [PubMed] [Google Scholar]
- 5.Bentley J, Diggle CP, Harnden P, Knowles MA, Kiltie AE. DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res. 2004;32:5249–5259. doi: 10.1093/nar/gkh842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin KH, Ahn JH, Kang MK, Lim PK, Yip FK, Baluda MA, Park NH. HPV-16 E6 oncoprotein impairs the fidelity of DNA end-joining via p53-dependent and -independent pathways. Int J Oncol. 2006;28:209–215. [PubMed] [Google Scholar]
- 7.Shin KH, Kang MK, Kim RH, Kameta A, Baluda MA, Park NH. Abnormal DNA end-joining activity in human head and neck cancer. Int J Mol Med. 2006;17:917–924. [PubMed] [Google Scholar]
- 8.Liu Y, Kulesz-Martin M. p53 protein at the hub of cellular DNA damage response pathways through sequence-specific and non-sequence-specific DNA binding. Carcinogenesis. 2001;22:851–860. doi: 10.1093/carcin/22.6.851. [DOI] [PubMed] [Google Scholar]
- 9.Bakalkin G, Selivanova G, Yakovleva T, Kiseleva E, Kashuba E, Magnusson K, Szekely L, Klein G, Terenius L, Wiman K. p53 binds single-stranded DNA ends through the C-terminal domain and internal DNA segments via the middle domain. Nucleic Acid Res. 1995;23:362–369. doi: 10.1093/nar/23.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S, Elenbaas B, Levine A, Griffith J. p53 and its 14 kDa C-terminal domain recognize primary DNA-damage in the form of insertion/deletion mismatches. Cell. 1995;81:1013–1020. doi: 10.1016/s0092-8674(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 11.Oberosler P, Hloch P, Ramsperger U, Stahl H. p53-catalyzed annealing of complementary single-stranded nucleic acids. EMBO J. 1993;12:2389–2396. doi: 10.1002/j.1460-2075.1993.tb05893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lilling G, Elena N, Sidi Y, Bakhanashvili M. p53-associated 3′-->5′ exonuclease activity in nuclear and cytoplasmic compartments of cells. Oncogene. 2003;22:233–245. doi: 10.1038/sj.onc.1206111. [DOI] [PubMed] [Google Scholar]
- 13.Mummenbrauer T, Janus F, Muller B, Wiesmuller L, Deppert W, Grosse F. p53 protein exhibits 3′-to-5′ exonuclease activity. Cell. 1996;85:1089–1099. doi: 10.1016/s0092-8674(00)81309-4. [DOI] [PubMed] [Google Scholar]
- 14.Nelson WG, Kastan MB. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol. 1994;14:1815–1823. doi: 10.1128/mcb.14.3.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin Y, Waldman BC, Waldman AS. Suppression of high-fidelity double-strand break repair in mammalian chromosomes by pifithrin-alpha, a chemical inhibitor of p53. DNA Repair. 2003;2:1–11. doi: 10.1016/s1568-7864(02)00183-0. [DOI] [PubMed] [Google Scholar]
- 16.Delbridge ML, Lingenfelter PA, Disteche CM, Graves JA. The candidate spermatogenesis gene RBMY has a homologue on the human X chromosome. Nat Genet. 1993;22:223–224. doi: 10.1038/10279. [DOI] [PubMed] [Google Scholar]
- 17.Soulard M, Della Valle V, Siomi MC, Pinol-Roma S, Codogno P, Bauvy C, Bellini M, Lacroix JC, Monod G, Dreyfuss G, Larsen CJ. HnRNP G: sequence and characterization of a glycosylated RNA-binding protein. Nucleic Acids Res. 1993;21:4210–4217. doi: 10.1093/nar/21.18.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dreyfuss G, Matunis MJ, Piuol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- 19.Soulard M, Barque JP, Della Valle V, Hernandez-Verdun D, Masson C, Danon F, Larsen CJ. A novel 43-kDa glycoprotein is detected in the nucleus of mammalian cells by autoantibodies from dogs with autoimmune disorders. Exp Cell Res. 1991;193:59–71. doi: 10.1016/0014-4827(91)90538-6. [DOI] [PubMed] [Google Scholar]
- 20.Nasim MT, Chernova TK, Chowdhury HM, Yue BG, Eperon IC. HnRNP G and Tra2beta: opposite effects on splicing matched by antagonism in RNA binding. Hum Mol Genet. 2003;12:1337–1348. doi: 10.1093/hmg/ddg136. [DOI] [PubMed] [Google Scholar]
- 21.Shin K-H, Kang MK, Kim RH, Christensen R, Park N-H. Heterogeneous nuclear ribonucleoprotein G (hnRNP G) demonstrates tumor suppressive activity against oral squamous cell carcinoma cells. Clin Cancer Res. 2006;12:3222–3228. doi: 10.1158/1078-0432.CCR-05-2656. [DOI] [PubMed] [Google Scholar]
- 22.Shin KH, Min BM, Cherrick HM, Park NH. Combined effects of human papillomavirus-18 and N-methyl-N′-nitro-N-nitrosoguanidine on the transformation of normal human oral keratinocytes. Mol Carcinog. 1994;9:76–86. doi: 10.1002/mc.2940090205. [DOI] [PubMed] [Google Scholar]
- 23.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 24.Alkan SA, Martincic K, Milcarek C. The hnRNPs F and H2 bind to similar sequences to influence gene expression. Biochem J. 2006;393:361–371. doi: 10.1042/BJ20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Zhang M, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 26.Kang MK, Shin KH, Yip FK, Park NH. Normal human oral keratinocytes demonstrate abnormal DNA end joining activity during replicative senescence. Mech Ageing Dev. 2005;126:475–479. doi: 10.1016/j.mad.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Shin KH, Kang MK, Dicterow E, Kameta A, Baluda MA, Park NH. Introduction of human telomerase reverse transcriptase to normal human fibroblasts enhances DNA repair capacity. Clin Cancer Res. 2004;10:2551–2560. doi: 10.1158/1078-0432.ccr-0669-3. [DOI] [PubMed] [Google Scholar]
- 28.Krecic AM, Swanson MS. hnRNP complexes: composition, structure, and function. Curr Opin Cell Biol. 1999;11:363–371. doi: 10.1016/S0955-0674(99)80051-9. [DOI] [PubMed] [Google Scholar]
- 29.Lilling G, Elena N, Sidi Y, Bakhanashvili M. p53-associated 3′-->5′ exonuclease activity in nuclear and cytoplasmic compartments of cells. Oncogene. 2003;22:233–245. doi: 10.1038/sj.onc.1206111. [DOI] [PubMed] [Google Scholar]
- 30.Li B, Lee MY. Transcriptional regulation of the human DNA polymerase delta catalytic subunit gene POLD1 by p53 tumor suppressor and Sp1. J Biol Chem. 2001;276:29729–29739. doi: 10.1074/jbc.M101167200. [DOI] [PubMed] [Google Scholar]
- 31.Brosh RM, Jr, Karmakar P, Sommers JA, Yang Q, Wang XW, Spillare EA, Harris CC, Bohr VA. p53 Modulates the exonuclease activity of Werner syndrome protein. J Biol Chem. 2001;276:35093–35102. doi: 10.1074/jbc.M103332200. [DOI] [PubMed] [Google Scholar]