

Abstract
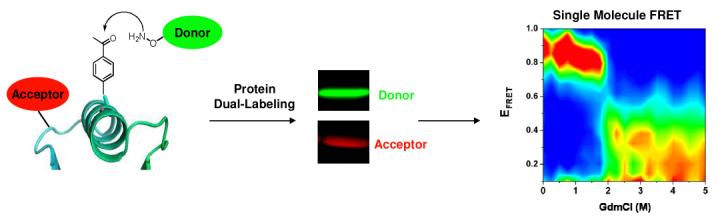 A general strategy for the site-specific dual-labeling of proteins for single-molecule fluorescence resonance energy transfer (smFRET) is presented. A genetically encoded unnatural ketone amino acid was labeled with a hydroxylamine-containing fluorophore with high yield (>95%) and specificity. This methodology was used to construct dual-labeled T4 lysozyme variants, allowing the study of T4 lysozyme folding at single-molecule resolution. The presented strategy is anticipated to expand the scope of single-molecule protein structure and function studies.
A general strategy for the site-specific dual-labeling of proteins for single-molecule fluorescence resonance energy transfer (smFRET) is presented. A genetically encoded unnatural ketone amino acid was labeled with a hydroxylamine-containing fluorophore with high yield (>95%) and specificity. This methodology was used to construct dual-labeled T4 lysozyme variants, allowing the study of T4 lysozyme folding at single-molecule resolution. The presented strategy is anticipated to expand the scope of single-molecule protein structure and function studies.
Single molecule fluorescence resonance energy transfer (smFRET) has emerged as a versatile tool for investigating biomolecular structure and dynamics.1-3 In contrast to ensemble measurements, direct measurements of conformational distributions and stochastic dynamics can be performed at single molecule resolution even in the context of complex structural landscapes and mixtures of molecular subpopulations. The strong distance dependence of energy transfer between donor (D) and acceptor (A) dyes makes smFRET especially useful for detailed studies of protein folding and conformational changes. However, a current inherent limitation is the need for site-specific labeling of the expressed protein with D and A dyes. Site-specific labeling is generally limited to reaction of cysteine residues with various electrophiles; however, random labeling of two cysteine residues produces a mixture of two species (D-A + A-D) and poses significant problems for subsequent single molecule studies. This uncertainty can be problematic in the analysis of conformationally heterogeneous populations, can contribute to errors in distance measurements, and could mimic the existence of two biologically significant species. Hence, considerable interest has been placed on the design of alternate site-specific protein labeling strategies, to significantly expand the number and scope of proteins that can be investigated using smFRET.1,4
An ideal labeling strategy for this purpose would fulfill at least three requirements. First, labeling should not be limited to a particular location on the protein surface. Thus, strategies such as N-terminal modifications and native chemical ligation are not optimal, since the site of labeling is generally restricted to protein termini.5,6 Second, the chemistry used for labeling should be high yielding because poor labeling efficiency degrades the quality of single molecule measurements or requires additional purification steps. Finally, it would be ideal for labeling reagents to be readily available from commercial sources. Herein we demonstrate that the orthogonal chemistry of a genetically encoded ketone handle together with a single cysteine residue permit efficient and versatile site-specific dual-labeling of proteins for smFRET studies.
We have previously demonstrated that a variety of unnatural amino acids (UAAs) can be introduced site-specifically into proteins in response to the amber (TAG) stop codon in E. coli through the use of an engineered orthogonal Methanococcus jannaschi derived tRNA/aminoacyl-tRNA synthetase pair.7 For this study, we selected a pair that incorporates p-acetylphenylalanine ((1), figure 1c), an unnatural ketone bearing amino acid, based on its orthogonal reactivity, and because several carbonyl reactive probes are commercially available.8,9 As a proof-of-principle, we labeled and studied a his-tagged cysteine-free variant of bacteriophage T4 lysozyme (T4L*) whose folding and physical characteristics have been examined extensively by other methods.10-12
Figure 1.
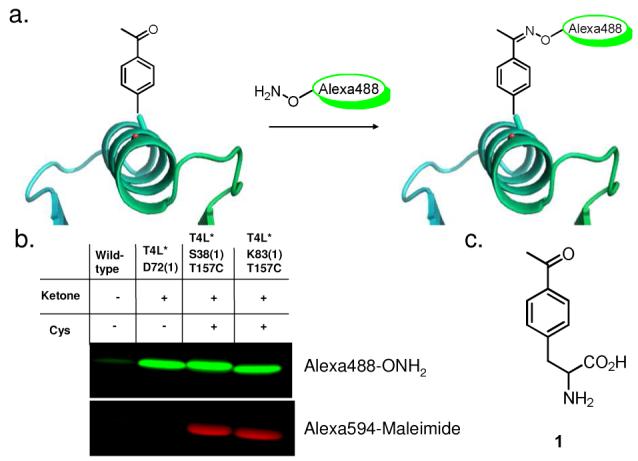
a) Oxime ligation with an unnatural ketone amino acid. b) Fluorescence image of site-specific labeled T4 lysozyme after SDS-PAGE separation. c) p-acetylphenylalanine (1)
To examine the selectivity of labeling, 1 was substituted for D72 of T4L* (T4L*D72(1)), and subsequently modified with commercially available hydrazide or alkoxyamino functionalized Alexa488 derivatives (Molecular Probes). In general, hydrazide containing dyes reacted less effectively than alkoxyamine derivatives with the ketone amino acid. Alkoxyamine derived Alexa488 provided on average >90% labeled protein under optimized conditions (Figures 1b, S1a and S1b) as measured by absorbance. Chemical modification was also confirmed by ESI mass spectrometry (figures S2a and S2b).
To achieve selective dual-labeling, two T4L* variants (T4L*S38(1)/T157C and T4L*K83(1)/T157C) were constructed, each containing a single cysteine for A dye labeling with Alexa594-maleimide and a single ketone (1) for D dye labeling with Alexa488-alkoxyamine. T4L is comprised of two domains13 with the A-labeled T157C located towards the end of the predominantly alpha-helical C-terminal domain. K83(1) lies in a small alpha helix at the start of the C-terminal domain and was D-labeled to provide smFRET information on C-terminal domain folding. S38(1) is instead located in a loop in the middle of the N-terminal domain, and when D-labeled and analyzed by smFRET, probes the general unfolding of both domains in concert. To confirm our orthogonal labeling strategy, wild-type T4L*, T4L*D72(1), T4L*S38(1)/T157C and T4L*K83(1)/T157C were labeled with either Alexa488 alkoxyamine or Alexa594-maleimide.
As shown in figure 1b, selective labeling was achieved with no cross-reactivity, suggesting that the combination of two orthogonal reactive handles allows for unique site-specific labeling with appropriately reactive probes. Dual labeling of both ketone/Cys mutants was confirmed by ESI (figure S2c and S2d). It should be noted that no subsequent purification other than the removal of excess dye was needed prior to single molecule experiments.
SmFRET experiments were performed on freely diffusing molecules in solution under equilibrium conditions as previously described.14 Briefly, laser excitation and confocal detection were used to detect FRET efficiencies (EFRET) from individual diffusing molecules, and these were then plotted in the form of a FRET histogram. This method permits rapid acquisition of FRET data on a significant population of single molecules, while minimizing potential perturbations from protein or dye interactions with a solid surface during immobilization. Experiments under native conditions yielded peaks in smFRET histograms with an observed EFRET of 0.86 and 0.68 for double-labeled T4L*K83(1)/T157C and T4L*S38(1)/T157C, respectively (figure 2A and figure S3). These EFRET values correspond to approximate distances of 34 Å and 41 Å (see supporting information for details on distance measurements), respectively, which are consistent with the T4L crystal structure (PDB: 2LZM).13
Figure 2.
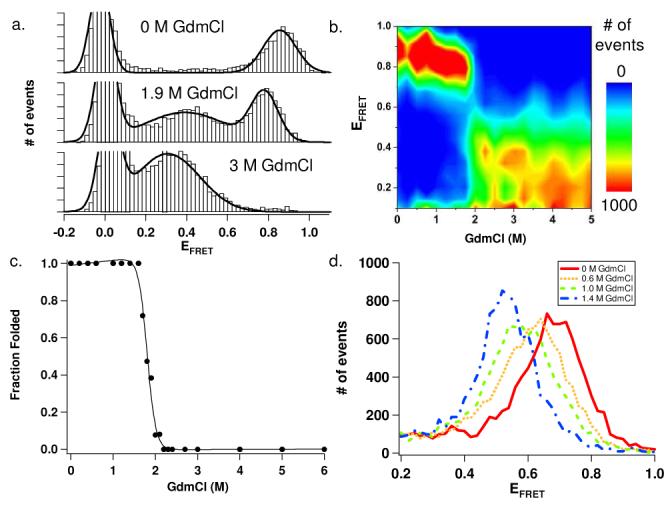
a) smFRET histograms of double-labeled T4L*K83(1)/T157C. b) contour plot of double-labeled T4L*K83(1)/T157C smFRET unfolding histograms. c) T4L*K83(1)/T157C single molecule unfolding curve d) Peak shift of folded T4L*S38(1)/T157C at low GdmCl concentrations.
We next monitored single molecule protein unfolding as a function of guanidinium chloride (GdmCl) concentration. Figure 2a and 2b show data from a typical denaturation series for double-labeled T4L*K83(1)/T157C. At low GdmCl concentrations, a single peak with a high EFRET (∼0.86, figure 2a top) is observed, consistent with a single folded state. At GdmCl concentrations above 1.7 M (figure 2a, middle and bottom), a decrease in the folded population peak-size is observed with the concomitant appearance of a population with a lower EFRET (∼0.4) corresponding to the formation of the denatured state. Consistent with a two state equilibrium unfolding transition for this protein, no additional populations are seen in the histograms. An unfolding curve was plotted using fractions of folded and unfolded protein populations calculated directly from Gaussian fits of the corresponding peaks in each histogram, which shows a two-state unfolding process with a midpoint transition around 1.9 M GdmCl (figures 2c and S5).
Single molecule unfolding of double-labeled T4L*S38(1)/T157C also shows a similar two-state transition (figure S5). In this case, however, we observed a significant decrease in the observed EFRET of the folded state prior to the cooperative unfolding transition. At 0 M GdmCl, a single broad peak is seen with an observed EFRET of 0.68, which decreases to an observed EFRET of 0.53 by 1.4 M GdmCl (figure 2d). Such a decrease in FRET may suggest that the T4L native state is expanding with increasing denaturant even prior to the unfolding transition. Control experiments indicate that these observed shifts are not the result of changes in dye photophysical properties (tables S1, S2, figure S4). Other studies have recorded similar smFRET shifts for denatured states of proteins, indicative of a continued expansion of the protein after denaturation;1,3,14-16 it is particularly interesting to observe such an effect for the T4L native state. Further experiments exploring the nature of this shift in the native state as well as other doubly-labeled T4L mutants will provide a more detailed picture of the T4L folding landscape.
In summary, we have successfully demonstrated that the in vivo incorporation of a ketone amino acid provides a general strategy for site-specific labeling of proteins with commercially available single-molecule dyes, bypassing a significant limitation in smFRET analysis of proteins. This technology allows proteins to be labeled in a truly site-specific manner because the location of a desired fluorophore need only be specified by the introduction of a unique TAG codon. Moreover, as in vivo unnatural amino acid incorporation methodology improves, we anticipate that multiple orthogonal reactive handles could be introduced, alleviating altogether the need for cysteine residues as reactive handles and allowing for multicolor smFRET or combined single-molecule fluorescence and manipulation protein studies. Finally, this chemistry is not limited to smFRET labeling and should be useful for attachment of other biophysical probes and directed protein immobilization.
Supplementary Material
Acknowledgements
We thank A. Ferreon, Y. Gambin, and E. Remba for their help in preparing this manuscript. This work is supported by the US Department of Energy, Division of Materials Sciences, under Award No. DE-FG03-00ER46051 (P.G.S), NIH grant GM066833 (A.A.D) and an AvH fellowship (E.A.L.)
Footnotes
Supporting Information Available: General procedures. Distance calculations. Supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Deniz AA, Mukhopadhyay S, Lemke EA. J. Royal Soc. Interface. 2008;5:15–45. doi: 10.1098/rsif.2007.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Annu. Rev. Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- 3.Schuler B, Eaton WA. Curr. Opin. Struct. Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jager M, Nir E, Weiss S. Protein Sci. 2006;15:640–646. doi: 10.1110/ps.051851506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheck RA, Dedeo MT, Iavarone AT, Francis MB. J. Amer. Chem. Soc. 2008;130:11762–11770. doi: 10.1021/ja802495w. [DOI] [PubMed] [Google Scholar]
- 6.Valiyaveetil FI, Sekedat M, MacKinnon R, Muir TW. Proc. Natl. Acad. Sci. USA. 2004;101:17045–17049. doi: 10.1073/pnas.0407820101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Xie J, Schultz PG. Annual Review of Biophysics and Biomolecular Structure. 2006;35:225–249. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Zhang Z, Brock A, Schultz PG. Proc. Natl. Acad. Sci. USA. 2003;100:56–61. doi: 10.1073/pnas.0234824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornish VW, Hahn KM, Schultz PG. J. Amer. Chem. Soc. 1996;118:8150–8151. [Google Scholar]
- 10.Cellitti J, Bernstein R, Marqusee S. Protein Science. 2007;16:852–862. doi: 10.1110/ps.062632807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llinas M, Gillespie B, Dahlquist FW, Marqusee S. Nature Structural Biology. 1999;6:1072–1078. doi: 10.1038/14956. [DOI] [PubMed] [Google Scholar]
- 12.Lu JR, Dahlquist FW. Biochemistry. 1992;31:4749–4756. doi: 10.1021/bi00135a002. [DOI] [PubMed] [Google Scholar]
- 13.Weaver LH, Matthews BW. J. Mol. Biol. 1987;193:189–199. doi: 10.1016/0022-2836(87)90636-x. [DOI] [PubMed] [Google Scholar]
- 14.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. Proc. Natl. Acad. Sci. USA. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzmenkina EV, Heyes CD, Nienhaus GU. Proc. Natl. Acad. Sci. USA. 2005;102:15471–15476. doi: 10.1073/pnas.0507728102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sherman E, Haran G. Proc. Natl. Acad. Sci. USA. 2006;103:11539–11543. doi: 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.