

Abstract
Objective
Necrotic cells evoke potent innate immune responses through unclear mechanisms. The mitochondrial fraction of the cell retains constituents of its bacterial ancestors, including N-formyl peptides, which are potentially immunogenic. Thus, we hypothesized that the mitochondrial fraction of the cell, particularly N-formyl peptides, contributes significantly to the activation of monocytes by necrotic cells.
Design
Human peripheral blood monocytes were incubated with necrotic cell fractions and mitochondrial proteins in order to investigate their potential for immune cell activation.
Setting
University medical center research laboratory.
Subjects
Healthy human adults served as blood donors.
Measurements and Main Results
Human blood monocyte activation was measured after treatment with cytosolic, nuclear and mitochondrial fractions of necrotic HepG2 cells or necrotic HepG2 cells depleted of N-formyl peptides [Rho(0) cells]. The specific role of the high affinity formyl peptide receptor (FPR) was then tested using specific pharmacological inhibitors and RNA-silencing. The capacity of mitochondrial N-formyl peptides to activate monocytes was confirmed using a synthetic peptide conforming to the N-terminus of mitochondrial NADH subunit 6. The results demonstrated that mitochondrial cell fractions most potently activated monocytes, and IL-8 was selectively released at low protein concentrations. Mitochondria from Rho(0) cells induced minimal monocyte IL-8 release, and specific pharmacological inhibitors and RNA-silencing confirmed that FPR contributes significantly to monocyte IL-8 responses to both necrotic cells and mitochondrial proteins. N-formyl peptides alone did not induce monocyte IL-8 release; whereas, the combination of mitochondrial N-formyl peptides and mitochondrial transcription factor A (TFAM) dramatically increased IL-8 release from monocytes. Likewise, HMGB1, the nuclear homologue of TFAM, did not induce monocyte IL-8 release unless combined with mitochondrial N-formyl peptides.
Conclusions
Interactions between mitochondrial N-formyl peptides and FPR in the presence of other mitochondrial antigens (e.g., TFAM) contributes significantly to the activation of monocytes by necrotic cells.
Keywords: Innate immunity, Inflammation, N-formyl proteins, TFAM, HMGB1, Necrosis
Introduction
Acute cell or tissue damage is known to promote inflammatory responses; however, the mechanisms have only recently been explored. Previous studies suggested that monocyte activation in response to cell necrosis is consequent to the liberation of a nuclear transcription factor, high mobility group box 1 (HMGB1), through the activation of receptors for advanced glycation end products (RAGE) (1). RAGE activation, in turn, initiates various pro-inflammatory events, including the recruitment of inflammatory cells and the production of pro-inflammatory cytokines (2, 3). However, new evidence indicates that HMGB1 is not essential for immune responses to necrotic cells in vivo (4), and other potentially immunogenic cell components, including DNA (5) and lipid membranes (6), should be considered in the context of inflammatory responses to damaged cells, such as occurs during trauma, pancreatitis, ischemia, severe systemic infections, and other acute illnesses. In this regard, a systematic analysis of the inflammatory properties of necrotic cell components has not been performed.
Acute, life-threatening illness is associated with characteristic changes in cell morphology and function with mitochondrial pathology being among the earliest manifestations (7, 8). Recent studies indicate that mitochondrial proteins are released under conditions associated with cell death (9) or tissue damage (10, 11), which has interesting implications for triggering immune responses.
Mitochondria are unique in that they are derived from bacterial ancestors and retain many of their features, including a circular genome, unique cell membrane lipids (e.g., cardiolipin), and N-formyl peptides, which are distinct byproducts of mitochondrial DNA expression in mammalian cells. Like their bacterial homologues, mitochondrial DNA-encoded N-formyl proteins serve as potent chemoattractants for neutrophils when released from degenerating mitochondria in the context of necrotic cell death (12, 13) and are recognized by high-affinity formyl peptide receptors (FPR) and formyl peptide-like 1 receptors on various immune cells (14, 15). Furthermore, mitochondrial DNA is rich in CpG dinucleotides, which are recognized by intracellular toll-like receptor 9 (TLR9) in specific immune cells, including monocytes (16). In mammals, the inner mitochondrial membrane is the only source of cardiolipin, which is potentially antigenic but does not directly induce innate immune responses (17). Finally, the immunostimulatory activity of mitochondrial transcription factor A (TFAM), the structural and functional homologue of HMGB1 (18), a potent pro-inflammatory mediator (2, 3), has not been considered. Based upon these observations, we hypothesized that the mitochondrial component of necrotic cells, particularly N-formyl peptides, mitochondrial DNA, and TFAM, would activate human peripheral blood monocytes.
Materials and Methods
Reagents and Cell Culture
Bacterial N-formyl-Met-Leu-Phe (fMLP) and purified cardiolipin were obtained from Sigma (St. Louis, MO). Human N-formyl peptide [fMMYALF, the N-terminal sequence of mitochondrial NADH dehydrogenase subunit 6 (ND6)] was synthesized by GenScript Corp. (Piscataway, NJ). Cyclosporin H (CsH), Boc-Phe-Leu-Phe-Leu-Phe (Boc-FLFLF) and pertussis toxin were acquired from Axxora, LLC (San Diego, CA), ChemPep, Inc. (Miami, FL) and Calbiochem (San Diego, CA) respectively. Most primary antibodies directed against specific proteins were obtained commercially: HMGB1 and CEACAM-1 (R&D Systems; Minneapolis, MN); cytochrome c oxidase, subunit II (COXII, MitoSciences, Inc.; Eugene, OR); calnexin and heat shock protein 60 (HSP60) (Abcam Inc.; Cambridge, MA), and ND1, ND6, β-actin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology, Inc.; Santa Cruz, CA). Anti-TFAM polyclonal antibody was made by Spring Valley Laboratories, Inc. (Woodbine, MD). The epitope used to create the antibody was KQRKYG, which was chosen because the corresponding peptide sequence was unique relative to that of HMGB1. Secondary antibodies were acquired from Cell Signaling Technology, Inc. (Danvers, MA). HepG2 and HEK-293 cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and cultured using DMEM/F-12 and MEM media (Invitrogen Corp.; Carlsbad, CA), respectively. Unless otherwise stated, all additional chemicals were obtained from Sigma using the best available grade.
Establishment of HepG2 Rho(0) Cell Lines
HepG2 cells depleted of mitochondrial DNA (mtDNA) and mtDNA-encoded proteins were generated using the protocol described by King et al (19). In brief, HepG2 cells were cultured in DMEM/F-12 medium supplemented by equal amounts (100 μg/ml) of uridine and pyruvate and treated with 100 ng/ml ethidium bromide for 6-8 weeks. Suppression of mtDNA-encoded genes and proteins was confirmed by evaluating mtDNA gene expression using real-time PCR, performing Western blot analysis using anti-COXII antibody, and examining cytochrome c oxidase activity using the Cytochrome c Oxidase Assay kit (Sigma) according to the manufacturer's instructions.
Subcellular Fractionation
HepG2 cell necrosis was induced by freeze/thaw, as described by Scaffidi et al (1), or by heating (44°C for 60 min) and was confirmed by the lactate dehydrogenase (LDH) assay (94.7 ± 4.9% vs. 86.2 ± 5.4% cell death). Nuclear, cytoplasmic and total mitochondrial protein fractions, along with submitochondrial particles, were prepared from HepG2 cells using a differential centrifugation approach as described by Nair et al (20) with minor modifications. Briefly, HepG2 cells were suspended in mitochondrial isolation buffer [230 mM mannitol, 70 mM sucrose, 3 mM HEPES (pH 7.4), 1 mM EGTA, 1 mM EDTA, 0.5 mg/ml fatty acid-free bovine serum albumin (BSA), 1 mM phenylmethylsulfonylfluoride (PMSF), 10 μl/ml protease inhibitor cocktail] in the presence or absence of 50 U/ml benzonase (Novagen; San Diego, CA), as indicated. Following freeze/thaw or heating, the necrotic HepG2 cell suspension was centrifuged at 300 × g for 10 min at 4°C to pellet unlysed cells and the nuclear fraction. The resultant supernatant was centrifuged again at 1500 × g for 10 min at 4°C to assure removal of nuclear contaminants. The supernatant was then centrifuged at 7000 × g for 15 min at 4°C to separate mitochondrial (pellet) from cytoplasmic (supernatant) fractions. The cytoplasmic fraction was centrifuged again at 10,000 × g for 15 min at 4°C to exclude any mitochondria. The mitochondrial pellet was re-suspended and lysed by three 5-minute freeze-thaw cycles using a dry ice/ethanol bath and a 37°C water bath to obtain soluble total mitochondrial proteins. These proteins were then centrifuged at 144,000 × g for 1 hr at 4°C to separate soluble intermembrane and matrix proteins from the mitochondrial inner and outer membranes. The mitochondrial membrane pellet was re-suspended and incubated in 2% CHAPS in Tris-buffered saline [25 mM Tris (pH 7.2), 0.15 M NaCl]. The final supernatant, containing soluble mitochondrial membrane proteins, was then exchanged to PBS buffer using a desalting column (Amersham Biosciences; Piscataway, NJ). Purity of the mitochondrial protein fractions was confirmed by Western blot using antibodies against markers specific for plasma membrane (CEACAM-1), endoplasmic reticulum (calnexin), and mitochondria (COXII, HSP60) (Figure 1). GAPDH served as a loading control.
Figure 1. Verification of Subcellular Fraction Purity.

(A) Representative Western blot of subcellular fractions (30 μg) showed a lack of cytosolic contamination by mitochondrial proteins (HSP60, COXII) and an absence of β-actin in the mitochondrial fractions. Furthermore, the mitochondrial matrix fraction was free of mitochondrial membrane contamination, as reflected by the absence of COXII. The COXII control was derived from human heart mitochondria (MitoSciences, Inc.). Protein loading was normalized to GAPDH. (B) Representative Western blot examining CEACAM-1 and calnexin immunoreactivity confirmed that the mitochondrial and cytosolic fractions (30 μg) were not contaminated by plasma membrane or endoplasmic reticulum, respectively.
Cytotoxicity of HepG2 Preparations
HepG2 cell death following freeze/thaw or heating was examined by analyzing the LDH concentration in the collected supernatant samples (Cytotoxicity Detection kit, Roche Applied Science; Indianapolis, IN). LDH content was measured spectrophotometrically at 490 nm according to the manufacturer's instructions and normalized to matching samples wherein cells were incubated for 60 min with 2% Triton X-100.
Plasmid Construction and Purification of Recombinant HMGB1 and TFAM
The coding region of human TFAM (GenBank Accession No. BC126366) and HMGB1 (GenBank Accession No. BC003378) were amplified by PCR using pT7-7-TFAM and pCMV-Sports6-HMGB1 (provided by Gerald Shadel, Ph.D.) as templates. The sense primers for TFAM (5′-CCGAATTCCCACCATGTCATCTGTCTTGGCAAGT-3′) and HMGB1 (5′-CCGAATTCCCACCATGGGCAAAGGAGATCCTAAG-3′) introduced a consensus Kozak translation initiation sequence and an EcoRI restriction site to facilitate cloning. The anti-sense primers of TFAM (5′-CCAAGCTTACACTCCTCAGCACCATATT-3′) and HMGB1 (5′-CCAAGCTTTTCATCATCATCATCTTCTTCT-3′) introduced a HindIII restriction site. The PCR product was digested with EcoRI and HindIII, cloned into a pcDNA3.1(-)/myc-His A vector (Invitrogen Corp.) in-frame with a C-terminal myc epitope and 6×histidine tags and then transformed into E. coli-competent cells (DH5α, Invitrogen Corp.). The sequences of TFAM and HMGB1 with the myc/His tag were confirmed by DNA sequencing. The pcDNA3.1-TFAM.myc.6×His and pcDNA3.1-HMGB1.myc.6×His plasmids were transfected into HEK-293 cells using calcium phosphate-DNA co-precipitation. The polyhistidine-tagged recombinant proteins were purified by Ni-NTA nickel-chelating resin (Invitrogen Corp.) according to the manufacturer's instructions.
The identity and function of the proteins were confirmed by Western blot using specific antibodies against TFAM and HMGB1, capillary-liquid chromatography-nanospray tandem mass spectrometry (Nano-LC/MS/MS) and the electrophoretic mobility shift assay (see below) (Figure 2). Proteins evaluated by mass spectrometry were scored based upon the number of matching peptide sequences (i.e., hits) and the probability that the match was a random event. This yielded a “mowse score” which when >51 indicated significant identity and/or extensive homology (p<0.05). Mass spectrometric analysis confirmed the identities of TFAM and HMGB1 with overall protein coverage corresponding to Mowse scores of 1005 (54% sequence coverage, 258 peptide matches) and 1031 (53% sequence coverage, 293 peptide matches) respectively.
Figure 2. Verification of Recombinant Human TFAM and HMGB1 Proteins.
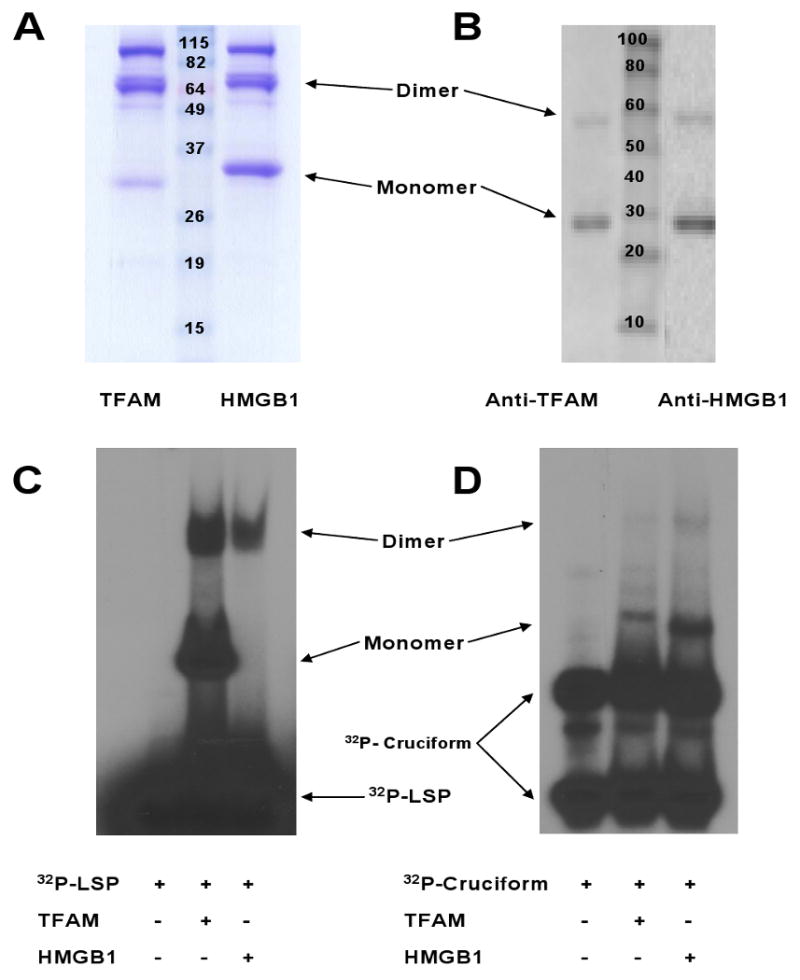
(A) Stained protein gel of TFAM and HMGB1 (3μg). TFAM.myc.6×His and HMGB1.myc.6×His monomers were ∼29 kD. (B) Western blot of proteins (2 μg) using antibodies directed against TFAM and HMGB1. (C-D) Protein gels of TFAM and HMGB1 (2 μg) following incubation with annealed oligonucleotide binding probes [32P-LSP (C) or 32P-cruciform (D) DNA].
Electrophoretic Mobility Shift Assay for TFAM and HMGB1
Synthetic cruciform DNA (four-way DNA junctions, 4WJs) for the HMGB1 gel shift assay was constructed according to published protocols (21, 22). Cruciform DNA was prepared by annealing the following four oligonucleotides synthesized (from 5′ to 3′) with 2 of them 32P-end-labeled: strand 1) CCCTATAACCCCTGCATTGAATTCCAGTCTGATAA; strand 2) GTAGTCGTGATAGGTGCAGGGGTTATAGGG; strand 3) AACAGTAGCTCTTATTCGAGCTCGCGCCCTATCACGACTA, and strand 4) TTTATCAGACTGGAATTCAAGCGCGAGCTCGAATAAGAGCTACTGT. Similarly, the LSP (light strand promoter of mitochondrial DNA) DNA duplex of 101 bp containing the TFAM-binding site was prepared by annealing oligonucleotides 1 (5′-TTAGTAGTATGGGAGTGGGAGGGGAAAATAATGTGTTAGTTGGGGGGTGACTGTTAAAAGTGCATACCGCCTAAAAGATAAAATTTGAAATCTGGTTAGGC-3′) and 2 (5′-GCCTAACCAGATTTCAAATTTTATCTTTTAGGCGGTATGCACTTTTAACAGTCACCCCCCAACAACACATTATTTTCCCCTCCCACTCCCATACTACTAA-3′). All oligonucleotides were highly purified by denaturing polyacrylamide gel electrophoresis and subsequently labeled at their 5′-termini by [γ-32P] ATP.
Endotoxin Analysis
The endotoxin level in each subcellular fraction protein, fMLP, fMMYALF and recombinant TFAM and HMGB1 proteins was determined using the chromogenic Limulus Amebocyte Lysate assay (LAL, Associates of Cape Cod; East Falmouth, MA). When needed, samples were passed through AffinityPak™ Detoxi-Gel™ columns (Pierce Biotechnology; Rockford, IL) according to the manufacturer's recommendations and re-tested by LAL to confirm that endotoxin levels were <5 pg/μg protein prior to later use for monocyte incubations.
Isolation, Culture and Stimulation of Human Monocytes
Normal peripheral blood monocytes were isolated from healthy human blood donors (n=10) according to approved university IRB guidelines (including signed written informed consent) using the MACS CD14 MicroBead positive selection kit (Miltenyi Biotec, Inc.; Auburn, CA) according to the manufacturer's instructions. Monocytes were cultured in 24-well plates at a concentration of 1 × 106/ml in X-VIVO™ 15 serum-free medium (Lonza Walkersville; Walkersville, MD) at 37°C in a 5% CO2-humidified atmosphere. Polymyxin B (10 μg/ml) was routinely added to the cells in each well (except the LPS-treated positive control well) to further block any possible endotoxin contamination. Desired concentrations of subcellular or mitochondrial fraction proteins were then added to the medium. All pre-incubations with FPR inhibitors (CsH, 3 μM; Boc-FLFLF, 10 μM) or the small G-protein inhibitor (pertussis toxin, 1 μg/ml) were made 30 min prior to protein fraction addition. Monocytes transfected to silence formyl peptide receptor (FPR1) RNA (see below) were incubated for 24 hr prior to additions. Further monocyte incubations involved adding N-formyl peptides (fMLP or fMMYALF at 100 ng/ml) with/without TFAM or HMGB1 (5 μg/ml). The cell supernatants were then collected at 3 and 6 hours post-treatment and stored at -80°C for later analyses. Cell pellets were also obtained and stored in a similar manner for later RNA isolation and analysis.
Cytokine Assays
Cell culture supernatants were analyzed for their IL-8 (R&D Systems), IL-6 and TNFα (eBioscience, Inc.; San Diego, CA) concentrations by ELISA according to the manufacturer's recommendations.
FPR siRNA and Transfection
Pre-designed human FPR1 siRNA was synthesized by Qiagen, Inc. (Valencia, CA) based upon GenBank Accession No. NM002029. The sequences were as follows: sense, r(GCAAGGCAUGUACAAAGAA)dTdT, and anti-sense, r(UUCUUUGUACAUGCCUU GC)dAdA. Negative non-silencing control siRNA (nc-siRNA, Qiagen, Inc.), labeled with Alexa Fluor 488 and having no known homology to mammalian genes, was used to control for nonspecific effects. FPR1 siRNA was transfected into monocytes using the Human Monocyte Nucleofector kit (Amaxa, Inc.; Gaithersburg, MD). In brief, 5 × 106 monocytes, re-suspended with 100 μl human monocyte nucleofector solution, were mixed with 3 μg FPR1 siRNA, transferred into an Amaxa-certified cuvette and transfected using Nucleofector Program Y-01. FPR gene expression was later evaluated in cell pellets by reverse transcription (RT)-PCR and real-time PCR. Transfection efficiency was determined in monocytes 24 hr post-transfection by analyzing Alexa 488 fluorescence. β-actin and CAP1 served as housekeeping gene controls.
RT-PCR and Real-Time PCR
Total RNA was isolated from HepG2 and Rho(0)-HepG2 cells as well as normal and FPR1 siRNA-transfected monocytes using the RNeasy Mini kit (Qiagen) according to the manufacturer's recommendations. The quality and concentration of RNA were analyzed using a ND-8000 spectrophotometer (NanoDrop Technologies, Inc.; Wilmington, DE) and a 2100 BioAnalyzer (Aligent Technologies, Inc.; Santa Clara, CA). The first strand cDNA was synthesized using 1.0 μg total RNA by MultiScribe reverse transcriptase (Applied Biosystems; Foster City, CA) with random hexamers in a 50 μl reaction volume. Semi-quantitative RT-PCR was performed in 50 μl containing 1 μl of diluted cDNA (1:10) from 50 μl cDNAs, 1.5 mM MgCl2, 0.2 mM dNTP, 0.5 μM of each primer and 1 U of Taq DNA polymerase (Invitrogen Corp.). The PCR conditions for FPR1, β-actin and CAP1 were 95°C for 2 min, followed by 25 cycles of 95°C for 30 sec, 56°C for 30 sec, 72°C for 30 sec and 72°C for 5 min. 10 μl of PCR product were separated by 1.5% agarose gel electrophoresis and stained with 0.05% ethidium bromide. The primers used in PCR are identified in Table 1 (Integrated DNA Technologies, Inc.; Coralville, IA).
Table 1.
Primer Sequences Used for Real-Time PCR.
| Gene | Amplicon (bp) | Primer Sequence | |
|---|---|---|---|
| ATP Synthase 6 | 72 | Sense | 5′-CCAATAGCCCTGGCCGTAC-3′ |
| Antisense | 5′-CGCTTCCAATTAGGTGCATGA-3′ | ||
| ATP Synthase 8 | 95 | Sense | 5′-CAACTAAAAATATTAAACACAA-3′ |
| Antisense | 5′-CGTTCATTTTGGTTCTCAGGG-3′ | ||
| β-actin | 268 | Sense | 5′-GCCAACCGCGAGAAGATGA-3′ |
| Antisense | 5′-TGGTGGTGAAGCTGTAGCC-3′ | ||
| CAP1 | 80 | Sense | 5′-ATTCCCTGGATTGTGAAATAGTC-3′ |
| Antisense | 5′-ATTAAAGTCACCGCCTTCTGTAG-3′ | ||
| COX I | 66 | Sense | 5′-TCCGCTACCATAATCATCGCT-3′ |
| Antisense | 5′-CCGTGGAGTGTGGCGAGT-3′ | ||
| COX II | 63 | Sense | 5′-TGCCCGCCATCATCCTA-3′ |
| Antisense | 5′-TCGTCTGTTATGTAAAGGATGCGT-3′ | ||
| COX III | 67 | Sense | 5′-CCAATGATGGCGCGATG-3′ |
| Antisense | 5′-CTTTTTGGACAGGTGGTGTGTG-3′ | ||
| COX IV | 151 | Sense | 5′-CCTCCTGGAGCAGCCTCTC-3′ |
| Antisense | 5′-TCAGCAAAGCTCTCCTTGAACTT-3′ | ||
| Cytochrome b | 60 | Sense | 5′-CCCCACCCCATCCAACAT-3′ |
| Antisense | 5′-TCAGGCAGGCGCCAAG-3′ | ||
| FPR1 | 147 | Sense | 5′-ATCAGGTGGTGGCCCTTA-3′ |
| Antisense | 5′-GGCCCATGAAGACATA-3′ | ||
| GAPDH | 123 | Sense | 5′-TGGTATCGTGGAAGGACTCA-3′ |
| Antisense | 5′-GCAGGGATGATGTTCTGGA-3′ | ||
| NADH 1 | 64 | Sense | 5′-CCCTAAAACCCGCCACATCT-3′ |
| Antisense | 5′-CGATGGTGAGAGCTAAGGTC-3′ | ||
| NADH 2 | 66 | Sense | 5′-CACCCTTAATTCCATCCACCC-3′ |
| Antisense | 5′-TGGGCAAAAAGCCGGTTAG-3′ | ||
| NADH 3 | 64 | Sense | 5′-AGAAAAATCCACCCCTTACGAGT-3′ |
| Antisense | 5′-TGGAGAAAGGGACGCGG-3′ | ||
| NADH 4 | 65 | Sense | 5′-ACCTTGGCTATCATCACCCG-3′ |
| Antisense | 5′-TAGGAAGTATGTGCCTGCGTTC-3′ | ||
| NADH 4L | 195 | Sense | 5′-ATCGCTCACACCTCATATCCTC-3′ |
| Antisense | 5′-GGCCATATGTGTTGGAGATTG-3′ | ||
| NADH 5 | 221 | Sense | 5′-ACATCTGTACCCACGCCTTC-3′ |
| Antisense | 5′-TATGTTTGCGGTTTCGATGA-3′ | ||
| NADH 6 | 52 | Sense | 5′-TGGTTGTCTTTGGATATACTACAGCG-3′ |
| Antisense | 5′-CCAAGACCTCAACCCCTGAC-3′ | ||
Real-time PCR was performed in 20 μl of a mixture containing 1 μl of a cDNA sample, 5 pmol of each primer needed (Table 1), and 10 μl of SYBR® green PCR master mix (Applied Biosystems). Amplification and detection were achieved with the 7900HT Fast Real-Time PCR System (Applied Biosystems) using the protocol of 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 sec and 60°C for 1 min. The expression of each gene was calculated based upon the cycle threshold number and normalized by β-actin /CAP1 expression.
Western Blotting
The presence of specific proteins in various subcellular and mitochondrial fractions was determined by Western blot analysis using standard techniques. Protein loading was normalized to GAPDH.
Measurement of Lipid and DNA Content in Mitochondrial Preparations
Mitochondrial DNA content was determined with the fluorescence-based Quant-iT™ PicoGreen® dsDNA Kit (Molecular Probes Inc., Eugene, OR), which has a sensitivity as low as 25 pg/ml DNA. Mitochondrial lipid content was determined by gas-liquid chromatography after samples had been prepared according to Folch et al (23). DNA and lipid contents were normalized to protein concentration.
Statistical Analyses
The data was derived from at least 5 independent experiments and was expressed as mean ± SEM, and statistical significance was based upon a value of p ≤ 0.05. SigmaPlot 10.0 and SYSTAT 12.0 software were used to plot the data and carry out the statistical analyses, respectively. Comparisons of IL-8 release following the treatment of mitochondrial protein under Rho(0) conditions and FPR expression after RNA silencing were carried out using the Wilcoxon Rank-Sum test. Additional comparisons of IL-8 release following the treatment of mitochondrial protein or cell lysates with or without pharmacological inhibitors or RNA silencing, necrotic cell supernatants, or N-formyl peptides +/- TFAM/HMGB1 were made using the Kruskal-Wallis test. Where appropriate, post hoc analyses between groups were performed using Dunn's test if the Kruskal-Wallis overall p-value is significant.
Results
Necrotic cells passively release mitochondrial proteins
HepG2 cells exposed to freeze/thaw, heating or detergent were shown to release mitochondrial proteins into the surrounding medium in the absence of cell manipulation (e.g., vortex or cell agitation) (Figure 3A). The mixture of mitochondrial and non-mitochondrial (e.g., HMGB1) proteins released from necrotic cells was shown to activate human monocytes (Figure 3B).
Figure 3. Necrotic Cells Release Mitochondrial Proteins and Promote Monocyte IL-8 Release.
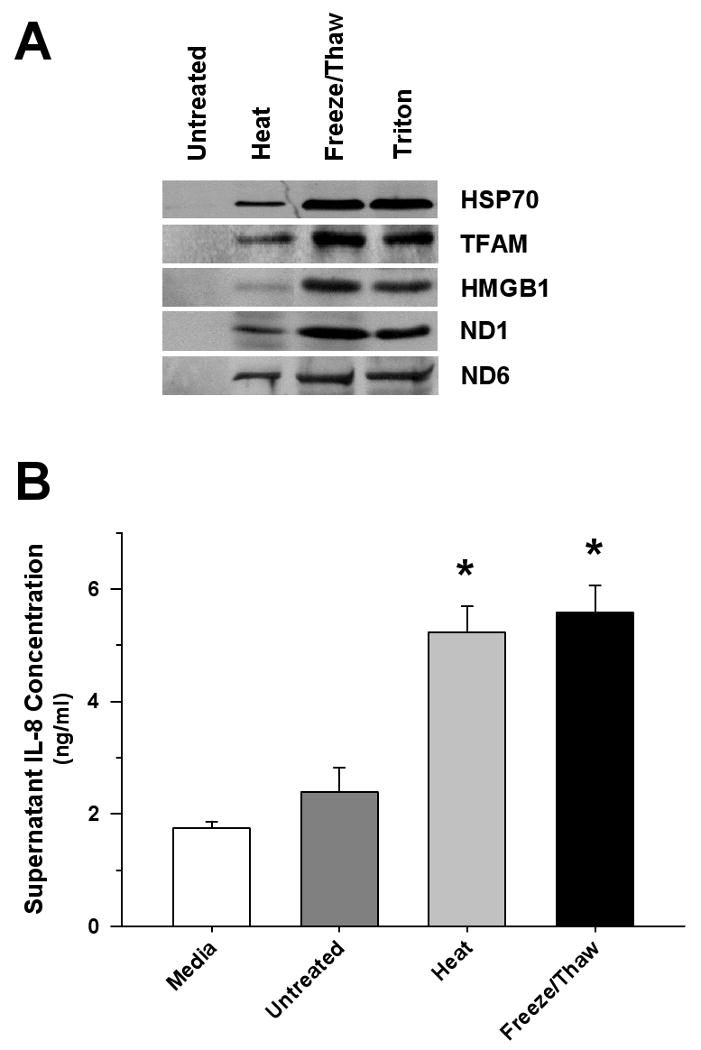
(A) Representative Western blot of cell supernatants derived from HepG2 cells exposed to heat, freeze/thaw or detergent (2% Triton X-100) demonstrating the release of mitochondrial (TFAM, ND1, ND6) and nuclear (HMGB1) proteins. (B) Significant monocyte IL-8 release was observed 6 hrs after treatment with equal supernatant volumes (50 μl) derived from freeze/thaw, or heat-treated HepG2 cells (∼107 cells/ml) compared to matching untreated preparations (*p<0.05, compared to untreated Control). The results were compared to spontaneous release of IL-8 from monocytes in culture medium.
Monocytes are primarily activated by the mitochondrial component of necrotic liver cells
Necrotic HepG2 cells were fractionated into nuclear, cytoplasmic, mitochondrial, and submitochondrial fractions and compared in terms of promoting monocyte activation, as reflected by IL-8 release. As shown in Figure 4A, the mitochondrial fraction of HepG2 cells, particularly the mitochondrial membrane, was the most potent inducer of IL-8 release by human peripheral blood monocytes. Nearly identical results were obtained when immune responses to monocyte treatment with matching heat-killed HepG2 cell components were determined (data not shown). Thus, all subsequent experiments were conducted using freeze/thaw cell preparations. Figure 4B shows similar dose-dependent induction of monocyte IL-8 release from mitochondrial preparations derived from either human liver or from HepG2 cells.
Figure 4. Monocyte Cytokine Responses to Treatment with Nuclear, Cytoplasmic and Mitochondrial Components of Necrotic Cells.

(A) Six hrs after exposure to increasing titrations of HepG2 subcellular fractions, dose-dependent monocyte IL-8 release was limited to the mitochondrial compartment, particularly the membrane fraction. (B) Monocyte IL-8 release was comparable when treated with mitochondria derived from HepG2 cells or human liver homogenates. LPS (10 ng/ml) was used as a positive control.
The observed pro-inflammatory effect of mitochondria was apparently mediated by one or more mitochondrial proteins, since depletion of mitochondrial CpG-containing DNA and mitochondrial lipids had no significant effect upon this response (Figure 5). Moreover, treatment of monocytes with purified cardiolipin (5 μg/ml) did not induce monocyte IL-8 release (917.0 ±153.3 vs. 879.5 ±53.2 pg/ml). With respect to the nature of the cytokine response, monocyte IL-8 release occurred at much lower concentrations of mitochondrial protein than were required to promote IL-6 or TNFα release (Figure 6). Thus, one or more of the proteins localized to mitochondrial membranes were recognized by monocyte receptors to trigger the release of IL-8.
Figure 5. Lack of Contribution of Mitochondrial DNA and Lipids to Monocyte Activation.

Monocyte IL-8 release 6 hrs after treatment with mitochondrial lysates was unchanged when compared with matching preparations treated with CHAPS and benzonase to remove lipids and DNA, respectively (*p<0.05, compared to untreated Control; †p<0.05, relative to Control and corresponding lower dose treatment). CHAPS-treated mitochondrial preparations had a 33.1 ±1.8% reduction in total lipid content compared to untreated mitochondria (p<0.01). DNA content was reduced by 83.9 ±2.8% (p<0.01) after benzonase treatment.
Figure 6. Dose-response Relationship between Mitochondrial Membrane Proteins and Monocyte Cytokine Release.

Human monocytes were treated with increasing titrations of mitochondrial membrane protein fractions derived from HepG2 cells. IL-8 and IL-6 release were measured 6 hrs after treatment; whereas, TNFα release was measured 3 hrs post-treatment. Significant dose-dependent IL-8 release was observed at much lower protein concentrations than those required for IL-6 release; TNFα release was not significantly increased at any concentration.
Depletion of mitochondrial N-formyl peptides attenuates monocyte IL-8 responses to mitochondrial proteins
HepG2 Rho(0) cells, which are selectively depleted of mitochondrial DNA (Table 2) and consequently N-formyl peptides (Figure 7A), exhibited the expected reduction in mitochondrial respiratory activity (Figure 7B). Compared to mitochondrial proteins derived from untreated HepG2 cells, monocyte IL-8 responses to mitochondrial fractions from Rho(0) cells were significantly attenuated (Figure 7C). Thus, N-formyl peptides appear to be integral to the activation of human monocytes by mitochondrial proteins.
Table 2.
Mitochondrial DNA (mtDNA)-Encoded Gene Expression by Real-Time PCR.
| Gene | Normal HepG2 Cell [(EtBr-)HepG2] | Ethidium Bromide-Treated HepG2 Cell [(EtBr+)HepG2] | |
|---|---|---|---|
| Relative Copy Number | Relative Copy Number | Fold Difference (Reduced) | |
| ATP synthase F0 6 | 66 | 0.023 | 2815 |
| Cytochrome c oxidase I | 181 | 0.047 | 3807 |
| Cytochrome c oxidase II | 99 | 0.024 | 4141 |
| Cytochrome c oxidase III | 73 | 0.015 | 5041 |
| Cytochrome c oxidase IVa | 1 | 0.046 | 20 |
| Cytochrome b | 49 | 0.011 | 4326 |
| NADH dehydrogenase 1 | 29 | 0.047 | 611 |
| NADH dehydrogenase 2 | 64 | 0.036 | 1753 |
| NADH dehydrogenase 3 | 26 | 0.006 | 4472 |
| NADH dehydrogenase 4 | 114 | 0.024 | 4813 |
| NADH dehydrogenase 4L | 94 | 0.025 | 3843 |
| NADH dehydrogenase 5 | 98 | 0.027 | 3593 |
| NADH dehydrogenase 6 | 58 | 0.012 | 4944 |
The expression of cytochrome c oxidase IV, a nuclear gene product, in normal HepG2 cells was used to normalize the expression of all other genes.
Figure 7. Treatment with Mitochondria Depleted of N-formyl Peptides Elicits Attenuated Monocyte IL-8 Release.
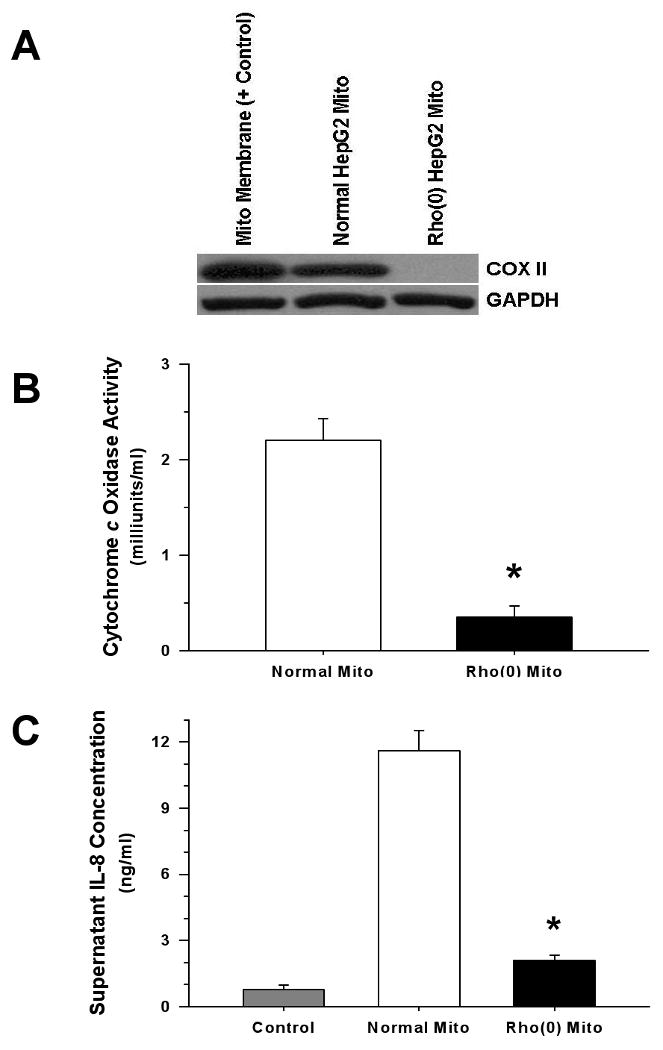
Rho(0) HepG2 cells exhibited reduced mitochondrial DNA-encoded cytochrome c oxidase subunit 2 (COXII) expression (A), and had diminished capacity for electron transport (B). Compared to proteins isolated from normal HepG2 mitochondria, mitochondrial proteins derived from Rho(0) HepG2 cells (500 ng/ml) elicited reduced monocyte IL-8 release (C), and the same relationship was observed when whole cell lysates from normal and Rho(0) cells (5 μg/ml) were compared (not shown) (*p <0.01, compared to Normal HepG2 mitochondrial protein).
Suppression of high-affinity formyl peptide receptor (FPR) activity attenuates IL-8 release by monocytes in response to mitochondrial proteins
To the extent that mitochondrial N-formyl peptides are responsible for the activation of monocytes, we reasoned that the observed IL-8 response was dependent upon interaction with FPR. CsH significantly attenuated the monocyte response to treatment with necrotic HepG2 cell lysates (Figure 8A), and both CsH and Boc-FLFLF strongly suppressed IL-8 release from monocytes exposed to mitochondrial proteins; whereas, reduction by pertussis toxin was not significant (Figure 8B). In keeping with these results, suppression of FPR expression by siRNA (Figure 9A) dramatically inhibited monocyte IL-8 release in response to mitochondrial proteins (Figure 9B).
Figure 8. FPR Inhibition Significantly Reduces Monocyte IL-8 Release Induced by Cell Lysates or Mitochondrial Proteins.

(A) The pharmacological inhibitor of FPR, CsH (3 μM), significantly decreased monocyte IL-8 release in response to treatment with necrotic HepG2 cell lysates (5 μg/ml) (*p<0.05, relative to Control; †p<0.05, compared to cell lysate alone). LPS (10 ng/ml) was used as the positive control. (B) Pretreatment with CsH (3 μM) or Boc-FLFLF (10 μM) attenuated monocyte IL-8 release in response to mitochondrial membrane proteins (50 ng/ml); whereas, the small G-protein inhibitor, pertussis toxin (1 μg/ml) had no significant effect (*p<0.05, relative to Control; †p<0.05, compared to mitochondrial membrane alone; ‡p<0.05, relative to Control and mitochondrial membrane alone).
Figure 9. FPR Plays a Significant Role in the Induction of Monocyte IL-8 Release by Mitochondrial Proteins.
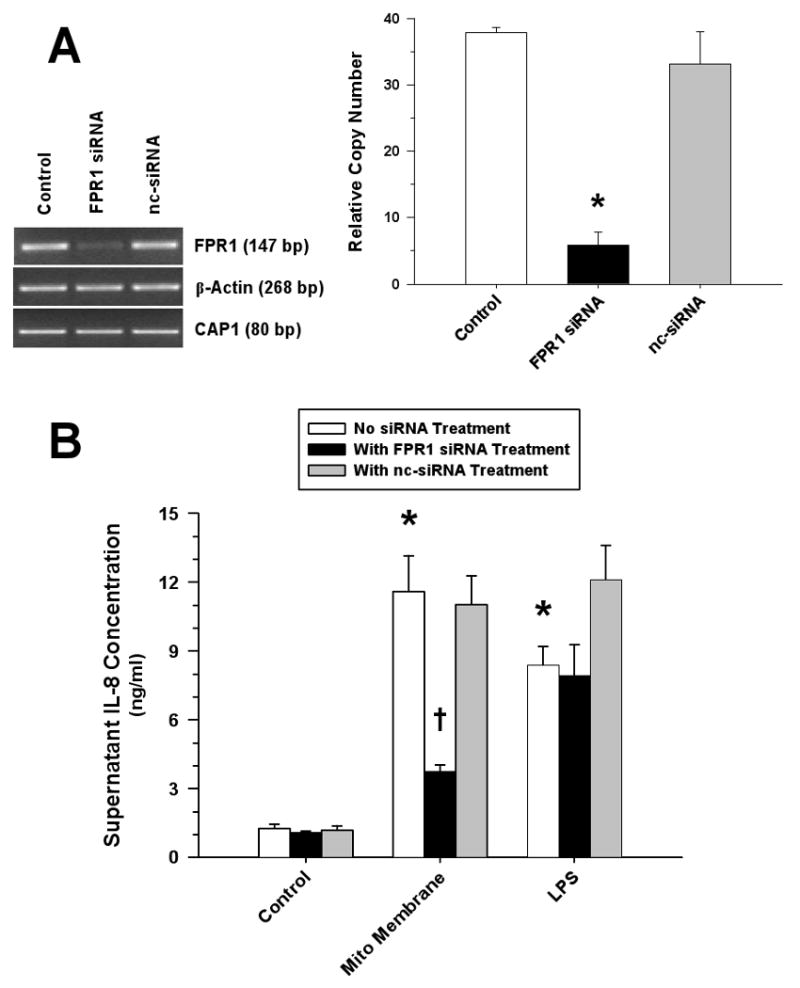
(A) RT-PCR (left) and real-time PCR (right) analyses demonstrated significant suppression of monocyte FPR mRNA after treatment with FPR siRNA; whereas, transfection with the non-silencing control siRNA (nc-siRNA) had no effect (*p<0.01, compared to matching Control). (B) Attenuation of monocyte IL-8 release following suppression of monocyte FPR mRNA in response to treatment with mitochondrial membrane proteins (500 ng/ml) but not to LPS (10 ng/ml) (*p<0.05, relative to matching Control; †p<0.05, compared to matching non-silencing treatment).
Bacterial and mitochondrial N-formyl peptides do not directly activate monocytes
To determine whether N-formyl peptides are sufficient to induce monocyte IL-8 release, synthetic bacterial N-formyl-Met-Leu-Phe (fMLP) or the immunogenic N-formyl peptide sequence (fMMYALF) of mitochondrial NADH dehydrogenase subunit 6 (14) were administered to human monocytes. Unexpectedly, these highly purified N-formyl peptides did not induce significant monocyte IL-8 release when administered alone (Figure 10B). Given that N-formyl peptides and FPR interactions are essential for monocyte IL-8 release in response to mitochondrial membrane proteins, as shown above, we reasoned that the FPR-induced response required a second pro-inflammatory stimulus, presumably in response to other immunogenic mitochondrial components. Thus, we determined whether mitochondrial DNA, which contains CpG DNA motifs, or mitochondrial transcription factor A (TFAM), which is homologous in structure and function to HMGB1 (18) and is localized to inner mitochondrial membranes in association with Complex I of the electron transport chain (24) (i.e., containing N-formyl peptides), act synergistically with N-formyl peptides to induce monocyte activation. After confirming that TFAM was concentrated in the mitochondrial membrane fraction of the cell (Figure 10A), we investigated whether TFAM or combinations of TFAM + N-formyl peptides would induce monocyte activation. Neither highly purified (endotoxin-free) recombinant human TFAM nor HMGB1 activated monocytes alone, except at concentrations exceeding 1.0 μg/ml. However, ng/ml concentrations of synthetic N-formyl peptides promoted monocyte activation in the presence of either HMGB1 or TFAM. The combination of N-formyl peptides and CpG DNA exhibited no significant synergy (data not shown) consistent with the earlier finding that depletion of DNA from mitochondrial protein samples had little effect upon their induction of monocyte IL-8 release (Figure 5). In addition, monocyte activation in response to the combination of TFAM + N-formyl peptides was inhibited by CsH (Figure 10B); whereas, IL-8 release was not influenced by the formyl peptide receptor like-1 inhibitor, WRW4 (data not shown).
Figure 10. N-formyl Peptides Alone Are Insufficient to Induce Monocyte IL-8 Release.

(A) Representative Western blot analysis showed TFAM to be concentrated in the mitochondrial membrane fraction of the cell (protein load: 30 μg). (B) Monocyte IL-8 responses to mitochondrial (fMMYALF) or bacterial (fMLP) N-formyl peptides (100 ng/ml) in the presence or absence of mitochondrial (TFAM) or nuclear (HMGB1) chromatin-binding proteins (5 μg/ml) and CsH (3 μM) treatment (*p<0.05, relative to Control; †p<0.05, compared to matching treatment with TFAM or HMGB1 alone; ‡p<0.05, relative to matching treatment in the absence of CsH). LPS (10 ng/ml) was used as a positive control.
Discussion
The innate immune response triggered by damaged tissues in the context of acute illness, such as pancreatitis, burns, or trauma, has important implications for the survival of the host. The systemic release of pro-inflammatory cytokines under these conditions promotes the recruitment and activation of various immune cells, which on the one hand facilitates the removal of necrotic debris and sanitizes the damaged tissue, while on the other hand contributing to further inflammation, resulting in progressive cell and tissue damage. The persistence of systemic inflammation and progression of organ dysfunction that commonly occurs in the setting of severe acute illnesses is highly predictive of an adverse outcome in critically ill patients (25, 26). However, other than supportive care, there are no effective treatments to interrupt this cycle of tissue damage and inflammation, and the mechanisms driving this sort of inflammation are not completely understood. This study provides evidence that the mitochondrial component of the cell, particularly mitochondrial membrane proteins (N-formyl peptides, TFAM), contributes significantly to the induction of the innate immune response to necrotic cells.
Recent studies have considered the mechanisms by which the innate immune response is regulated by damaged cells. As with autoimmune diseases wherein “sequestered antigens” released from dying cells at the site of inflammation trigger adaptive immune responses (27), self-antigens released from necrotic cells during acute illness are shown to promote innate immune responses (28). The same is not true for apoptotic cells, wherein potentially pro-inflammatory antigens, including proteins, DNA, and RNA, are effectively packaged and enzymatically degraded (29). Moreover, specific cell surface antigens expressed on apoptotic cell membranes actually suppress the responsiveness of phagocytes that are engaged in their clearance (30). Thus, apoptotic cell death is advantageous from the standpoint of minimizing inflammatory responses in the context of removing damaged or senescent cells.
Identification of the specific antigens released from necrotic cells and their corresponding immune cell receptors provides novel insight into the pathogenesis of local and systemic inflammation under conditions favoring cell necrosis. Investigations by Scaffidi et al suggested that the nuclear chromatin-binding protein, HMGB1, is primarily responsible for the activation of monocytes in the context of cell necrosis (1). However, more recent studies indicate that highly-purified (endotoxin-free) HMGB1 lacks pro-inflammatory actions, unless combined with other antigens, such as CpG dinucleotides (31) or bacterial lipopolysaccharides (32). Moreover, necrotic cells deficient in HMGB1 are shown to strongly induce inflammation in vivo (4). Thus, other intracellular antigens are incriminated.
The immune system of humans retains highly conserved “pattern-recognizing” receptors, including TLRs, NODs, and FPRs, which are extremely sensitive to the presence of potential environmental pathogens, including bacteria, fungi, and viruses. Like bacteria, eukaryotic mitochondria possess circular DNA, which contains CpG DNA motifs and encodes specific high-abundance N-formyl peptides. All 13 proteins encoded by mitochondrial DNA possess N-formyl peptide sequences, and these proteins are found exclusively in the mitochondrial inner membrane, where they participate in electron transport (33). As with their bacterial homologues, mitochondrial N-formyl peptides are potent chemotaxins for neutrophils (12) and are recognized by the high-affinity FPR (14). TFAM, a critical regulator of mitochondrial DNA transcription, is associated with the inner mitochondrial membrane and with mitochondrial DNA (24). TFAM is a structural and functional homologue of HMGB1; however, the amino acid sequence differs significantly (18). To the extent that TFAM and HMGB1 share a common pro-inflammatory active site, it follows that TFAM released from damaged cells may be recognized by the immune system through various TLRs (34) and/or RAGE (31), which would contribute to local (35) and systemic inflammation (36). Thus, the release of mitochondrial antigens in the context of cell and tissue damage (10, 37) could have important implications for the perpetuation of inflammation during acute illness.
Our data indicates that monocytes respond vigorously to mitochondrial proteins, and N-formyl peptides contribute significantly to this response. FPRs are critical for antibacterial responses, promoting neutrophil chemotaxis to either bacterial or mitochondrial N-formyl peptides (12). IL-8, a potent inducer of neutrophil chemotaxis, was the primary cytokine produced by monocytes upon exposure to low concentrations of mitochondrial proteins or necrotic cells. This finding is in keeping with the observed neutrophil recruitment in the acute phase of tissue damage (38), which serves the dual purpose of decontaminating the wound while removing necrotic debris. The immune response to necrotic cells resembles that mounted against potential invading organisms, wherein interactions between bacterial N-formyl peptides and host FPR promote the chemoattraction of phagocytes to the site of infection (39). Like monocytes, neutrophils exposed to bacterial N-formyl peptides exhibit no significant IL-8 production unless they are co-stimulated by another antigen, such as fibrinogen (40).
The role of TFAM and HMGB1 in the activation of monocytes and other cells participating in the innate immune response to necrotic cells is unclear. We show that TFAM is localized to the inner mitochondrial membrane and that monocytes respond most vigorously to the mitochondrial membrane fraction of the cell. Furthermore, TFAM, like HMGB1, is released from necrotic cells and synergizes with N-formyl peptides to promote monocyte activation. However, it is unclear whether TFAM is essential for the activation of monocytes in the context of cell necrosis, and the monocyte receptor(s) that respond to TFAM remain to be determined. Nonetheless, it is clear from these investigations that TFAM and/or HMGB1 are capable of interacting with bacterial or mitochondrial N-formyl peptides to promote monocyte IL-8 release, all of which are present in lysates of necrotic cells.
A specific role of FPR in monocyte cytokine responses to mitochondrial proteins is a new finding and is suggested by several lines of evidence. FPR is known to engage mitochondrial and bacterial N-formyl peptides, thereby promoting chemotaxis (14). However, to our knowledge, FPR activation by N-formyl peptides is not reported to regulate monocyte pro-inflammatory cytokine responses. We show that CsH, a highly selective inhibitor of FPR which stabilizes the receptor in its inactive state thereby interfering with G-protein binding, and Boc-FLFLF, a competitive and non-specific inhibitor of FPR which is less potent than CsH (41), significantly attenuate monocyte IL-8 release by mitochondrial proteins. Similar findings were observed in response to RNA silencing of FPR expression. FPR inhibition also reduced the activation of monocytes by necrotic cell lysates. Given the dependence of FPR on G-protein signaling (42), it is surprising that pertussis toxin did not effectively block monocyte IL-8 release. However, there are examples of induction of IL-8 release by pertussis toxin via Toll-like receptor (TLR)-4 and NF-κB-dependent signaling in HEK-293 and dendritic cells (42, 43). It is further possible that pertussis toxin blocks anti-inflammatory pathways that are G-protein dependent (44). Notwithstanding partial inhibition by pertussis toxin, the results obtained with specific FPR receptor inhibitors and FPR gene-silencing indicate that FPR plays an important role in the induction of monocyte activation by necrotic cells, particularly by mitochondrial N-formyl peptides.
We conclude that proteins localized to the inner mitochondrial membrane contribute significantly to the activation of monocytes in the context of acute cell necrosis. Interactions between mitochondrial N-formyl peptides and high affinity FPRs, together with other mitochondrial antigens, such as TFAM and perhaps others, strongly contribute to the activation of monocytes by mitochondrial antigens released from necrotic cells. Specific targeting of FPR may prove to be beneficial for interrupting the deadly cycle of tissue damage and inflammation that characterizes acute life-threatening illnesses.
Acknowledgments
Financial Support: NIH grant AI62740 and the OSU Medical Center Davis Developmental Grant. The authors have not disclosed any potential conflicts of interest.
References
- 1.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 2.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ. High mobility group 1 proteins (HMG-1) stimulate proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–1182. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 4.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 5.Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004;75:995–1000. doi: 10.1189/jlb.0703328. [DOI] [PubMed] [Google Scholar]
- 6.Cerri C, Chimenti D, Conti I, Neri T, Paggiaro P, Celi A. Monocyte/macrophage-derived microparticles up-regulate inflammatory mediator synthesis by human airway epithelial cells. J Immunol. 2006;177:1975–1980. doi: 10.4049/jimmunol.177.3.1975. [DOI] [PubMed] [Google Scholar]
- 7.Cowley RA, Mergner WJ, Fisher RS, Jones RT, Trump BF. The subcellular pathology of shock in trauma patients: studies using the immediate autopsy. Am Surg. 1979;45:255–269. [PubMed] [Google Scholar]
- 8.Crouser ED, Julian MW, Blaho DV, Pfeiffer DR. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med. 2002;30:276–284. doi: 10.1097/00003246-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Ahlemeyer B, Klumpp S, Krielstein J. Release of cytochrome c into the extracellular space contributes to neuronal apoptosis induced by staurosporine. Brain Res. 2002;934:107–116. doi: 10.1016/s0006-8993(02)02365-x. [DOI] [PubMed] [Google Scholar]
- 10.Crouser ED, Julian MW, Huff JE, Struck J, Cook CH. Carbamoyl phosphate synthase-1: a marker of mitochondrial damage and depletion in the liver during sepsis. Crit Care Med. 2006;34:2439–2446. doi: 10.1097/01.CCM.0000230240.02216.21. [DOI] [PubMed] [Google Scholar]
- 11.Alleyne T, Joseph J, Sampson V. Cytochrome c diction: a diagnostic marker for myocardial infarction. Appl Biochem Biotechnol. 2001;90:97–105. doi: 10.1385/abab:90:2:97. [DOI] [PubMed] [Google Scholar]
- 12.Carp H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med. 1982;155:264–275. doi: 10.1084/jem.155.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czapiga M, Gao JL, Kirk A, Lekstrom-Himes J. Human platelets exhibit chemotaxis using functional N-formyl peptide receptors. Exp Hematol. 2005;33:73–84. doi: 10.1016/j.exphem.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Rabiet MJ, Huet E, Boulay F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur J Immunol. 2005;35:2486–2495. doi: 10.1002/eji.200526338. [DOI] [PubMed] [Google Scholar]
- 15.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 16.Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 17.Thorson LM, Doxsee D, Scott MG, Wheeler P, Stokes RW. Effect of mycobacterial phospholipids on interaction of Mycobacterium tuberculosis with macrophages. Infect Immun. 2001;69:2172–2179. doi: 10.1128/IAI.69.4.2172-2179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parisi MA, Clayton DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. 1991;252:965–969. doi: 10.1126/science.2035027. [DOI] [PubMed] [Google Scholar]
- 19.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 20.Nair JR, McGuire JJ. Submitochondrial localization of the mitochondrial isoforms of folylpolyglutamate synthetase in CCRF-CEM human T-lymphoblastic leukemia cells. Biochim Biophys Acta. 2005;1746:38–44. doi: 10.1016/j.bbamcr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Bianchi ME, Beltrame M, Paonessa G. Specific recognition of cruciform DNA by nuclear protein HMG1. Science. 1989;243:1056–1059. doi: 10.1126/science.2922595. [DOI] [PubMed] [Google Scholar]
- 22.Stefanovsky VY, Langlois F, Bazett-Jones D, Pelletier G, Moss T. ERK modulates DNA bending and enhancesome structure by phosphorylating HMG1-boxes 1 and 2 of the RNA polymerase I transcription factor UBF. Biochemistry. 2006;45:3626–3634. doi: 10.1021/bi051782h. [DOI] [PubMed] [Google Scholar]
- 23.Folch J, Lees M, Sloane-Stanley GH. A simple method for isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 24.Wang Y, Bogenhagen DF. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J Biol Chem. 2006;281:25791–25802. doi: 10.1074/jbc.M604501200. [DOI] [PubMed] [Google Scholar]
- 25.Kajdacsy-Balla AC, Andrade FM, Moreno R, Artigas A, Cantraine F, Vincent JL. Use of sequential organ failure assessment score as a severity score. Intensive Care Med. 2005;31:243–249. doi: 10.1007/s00134-004-2528-6. [DOI] [PubMed] [Google Scholar]
- 26.Pettilä V, Hynninen M, Takkunen O, Kuusela P, Valtonen M. Predictive value of procalcitonin and interleukin 6 in critically ill patients with suspected sepsis. Intensive Care Med. 2002;28:1220–1225. doi: 10.1007/s00134-002-1416-1. [DOI] [PubMed] [Google Scholar]
- 27.Hyland KV, Leon JS, Daniels MD, Giafis N, Woods LM, Bahk TJ, Wang K, Engman DM. Modulation of autoimmunity by treatment of an infectious disease. Infect Immun. 2007;75:3641–3650. doi: 10.1128/IAI.00423-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, Lorenz HM. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ. 2008;15:183–191. doi: 10.1038/sj.cdd.4402239. [DOI] [PubMed] [Google Scholar]
- 29.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, Maderna P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178:4595–4605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- 31.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 32.El Mezayen R, El Gazzar M, Seeds MC, McCall CE, Dreskin SC, Nicolls MR. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett. 2007;111:36–44. doi: 10.1016/j.imlet.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taanman JM. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410:103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 34.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–C924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 35.Kokkola R, Li J, Sundberg E, Aveberger AC, Palmblad K, Yang H, Tracey KJ, Andersson U, Harris HE. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003;48:2052–2058. doi: 10.1002/art.11161. [DOI] [PubMed] [Google Scholar]
- 36.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radhakrishnan J, Wang S, Ayoub IM, Kolarova JD, Levine RF, Gazmuri RJ. Circulating levels of cytochrome c after resuscitation from cardiac arrest: a marker of mitochondrial injury and predictor of survival. Am J Physiol Heart Circ Physiol. 2007;292:H767–H775. doi: 10.1152/ajpheart.00468.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283–289. doi: 10.2741/1184. [DOI] [PubMed] [Google Scholar]
- 39.Fillion I, Quellet N, Simard M, Bergeron Y, Sato S, Bergeron MG. Role of chemokines and formyl peptides in pneumococcal pneumonia-induced monocyte/macrophage recruitment. J Immunol. 2001;166:7353–7361. doi: 10.4049/jimmunol.166.12.7353. [DOI] [PubMed] [Google Scholar]
- 40.Kuhns DB, Nelson EL, Alvord WG, Gallin JI. Fibrinogen induces IL-8 synthesis in human neutrophils stimulated with formyl-methionyl-leucyl-phenylalanine or leukotriene B(4) J Immunol. 2001;167:2869–2878. doi: 10.4049/jimmunol.167.5.2869. [DOI] [PubMed] [Google Scholar]
- 41.Wenzel-Seifert K, Hurt CM, Seifert R. High constitutive activity of human formyl peptide receptor. J Biol Chem. 1998;273:24181–24189. doi: 10.1074/jbc.273.37.24181. [DOI] [PubMed] [Google Scholar]
- 42.Wang ZY, Yang D, Chen Q, Leifer CA, Segal DM, Su SB, Caspi RR, Howard ZO, Oppenheim JJ. Induction of dendritic cell maturation by pertussis toxin and its B subunit differentially initiate Toll-like receptor 4-dependent signal transduction pathways. Exp Hematol. 2006;34:1115–1124. doi: 10.1016/j.exphem.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 43.Tonon S, Goriely S, Aksoy E, Pradier O, Del Giudice G, Trannoy E, Willems F, Goldman M, De Wit D. Bordetella pertussis toxin induces the release of inflammatory cytokines and dendritic cell activation in whole blood: impaired responses in newborns. Eur J Immunol. 2002;32:3118–3125. doi: 10.1002/1521-4141(200211)32:11<3118::AID-IMMU3118>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 44.Zhang N, Yang D, Dong H, Chen Q, Dimitrova DI, Rogers TJ, Sitkovsky M, Oppenheim JJ. Adenosine A2a receptors induce heterologous desensitization of chemokine receptors. Blood. 2006;108:38–44. doi: 10.1182/blood-2005-06-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]