

Abstract
Mesenchymal stem cells (MSCs) participate in the wound healing process in mammalians. Adhesion of MSCs to endothelium is a key step in the homing of MSCs circulating in the bloodstream to the sites of injury and inflammation. Because endothelial cells (ECs) may become apoptotic under certain pro-inflammatory conditions, we investigated the effects of pro-inflammatory, TNF-α and IL-1β, and pro-apoptotic agents, actinomycin D, cycloheximide, okadaic acid, wortmannin, and staurosporine, on human MSCs (hMSCs) adhesion to ECs. Treatment of ECs with pro-apoptotic agents markedly increased adhesion of hMSCs to ECs. This adhesion correlated with reduction of mitochondrial membrane potential, inhibition of NADH dehydrogenases, and release of von Willebrand factor (vWF) by ECs. Treatment of ECs with exogenous vWF also stimulated hMSC adhesion. These data provide evidence that apoptosis of ECs may regulate homing of hMSCs to the sites of tissue injury. These results are consistent with the hypothesis that activation of apoptotic signaling pathways in ECs releases vWF which regulates hMSC adhesion to ECs.
Mesenchymal stem cells (MSCs) are mobilized into the bloodstream in response to burns or skeletal muscle injury (Mansilla et al., 2006; Ramirez et al., 2006). When delivered intravenously isolated and cell culture expanded MSCs home to tissues injured by radiation, infarction, or trauma (Mosca et al., 2000; Devine et al., 2002; Wang et al., 2002; Barbash et al., 2003; Bittira et al., 2003; Chapel et al., 2003; Rombouts and Ploemacher, 2003; Ji et al., 2004; Kraitchman et al., 2005; Mouiseddine et al., 2007). Homing of MSCs circulating in the bloodstream to the sites of injury requires MSC adhesion to endothelial cells (ECs). During adhesion to ECs MSCs display coordinated rolling and adhesion behavior, which depends on the expression of P-selectin and VCAM-1/VLA-4 on the surface of ECs (Ruster et al., 2006). After firm adhesion MSCs can transmigrate through the endothelium barrier (Schmidt et al., 2006). Expression of CXCR4, the receptor for SDF-1, by MSCs appears to be important for selective recognition of hypoxic ECs in vitro (Potapova et al., 2008) and in vivo during homing to ischemic brain lesions (Wang et al., 2008) and bone marrow (Wynn et al., 2004). Only a small fraction (~11%) of human MSCs (hMSCs) natively circulating in the bloodstream are CXCR4 positive (Wang et al., 2006). Merely 1–3% of culture expanded MSCs express CXCR4 (Wynn et al., 2004; Wang et al., 2006; Seeger et al., 2007a; Potapova et al., 2008). MSCs freshly isolated from the bone marrow are 30–35% CXCR4 positive, but they lose expression of CXCR4 within 24 h after isolation (Seeger et al., 2007a). The loss of CXCR4 by hMSCs is not permanent and CXCR4 expression in hMSCs can be induced by culturing them as spherical aggregates—hMSC spheroids (Potapova et al., 2008); however, induced expression of CXCR4 by hMSCs is not sustainable as it is lost within 24 h dissociation of hMSCs from the spheroids (Potapova et al., 2008).
ECs are activated by inflammatory and stress/pro-apoptotic stimuli. Similar to inflammation the induction of apoptosis in ECs results in hyperstimulation of leukocyte and platelet adhesion (Cavender et al., 1987; Bombeli et al., 1999). Apoptosis-dependent adhesion of neutrophils to human umbilical vein endothelial cells (HUVECs) sensitized with phosphatidylinositol 3-kinase inhibitor LY294002 depends on activation of caspase-3 and caspase-8 (Chen and Easton, 2008). It was hypothesized that activation of apoptotic signaling pathways in ECs stimulates leukocyte adhesion; however, details of this process remain poorly understood (Chen and Easton, 2008). In contrast, the role of stress conditions plays in the platelet adhesion to ECs and injured vascular wall is well established and related to the secretion of von Willebrand factor (vWF) by ECs (Varga-Szabo et al., 2008). Apoptotic ECs are an integral part of the microenvironment existing at the sites of injury. Systemic inflammatory and ischemic conditions cause EC apoptosis (Nagata, 1997; Pohlman and Harlan, 2000; Gonzalez and Selwyn, 2003). Ischemia initiates EC apoptosis in isolated rat hearts, and reperfusion attenuates this process (Scarabelli et al., 2001). Exposure of ECs to tumor necrosis factor-α (TNF-α) or interleukin-1β (IL-1β) in vitro does not induce apoptosis of ECs, however, in combination with inhibitors of RNA and protein synthesis these pro-inflammatory cytokines provoke an apoptotic response in ECs (Pohlman and Harlan, 1989; Polunovsky et al., 1994; Pober, 1998). The role of EC apoptosis in MSC homing to injured tissues is unknown. For this reason, we investigated the effects of apoptosis of ECs on hMSC adhesion. HUVECs were treated with TNF-α and IL-1β, alone or in combination with actinomycin D (ActD), an inhibitor of RNA synthesis, or cycloheximide (CHX), an inhibitor of protein synthesis. Apoptosis in HUVECs was also induced by staurosporine, a broad range protein kinase inhibitor; wortmannin, a phosphatidylinositol 3-kinase inhibitor; or okadaic acid, a serine–threonine phosphatase inhibitor.
In the experiments presented below we used cell culture expanded, CXCR4-negative hMSCs. We observed a hyperadhesiveness of hMSCs toward HUVECs treated with pro-apoptotic agents. These data provide the first evidence that ECs undergoing apoptosis become pro-adhesive toward hMSCs.
Materials and Methods
Reagents
Recombinant human TNF-α, recombinant human IL-1β, ActD, and CHX were purchased from Sigma-Aldrich (St. Louis, MO). Staurosporine was from Cell Signaling Technology Inc. (Danvers, MA). Okadaic acid was obtained from EMD Calbiochem Brand (San Diego, CA). Wortmannin was purchased from Millipore (Danvers, MA).
Cell culture
hMSCs and HUVECs were purchased from Lonza Group Ltd. (Basel, Switzerland) and cultured in MSCGM Bullet Kit (Lonza) and in EGM-2 Bullet Kit (Lonza), accordingly. Passages 2–5 were used. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
hMSCs–HUVECs adhesion assay
hMSC adhesion to HUVECs was conducted as previously described (Potapova et al., 2008) with some modifications. Briefly, hMSCs grown as a monolayer were dissociated with trypsin–EDTA solution (Lonza), washed with Hank’s balanced solution (HBSS), and labeled with 4 μg/ml calcein AM (Molecular Probes Brand, Invitrogen Corporation, Carlsbad, CA) in HBSS for 45 min at 37°C and 5% CO2. After labeling, cells were washed with HBSS and resuspended in Dulbecco’s modified Eagle’s medium (DMEM; Sigma) supplied with 5% fetal bovine serum (Sigma). Confluent HUVECs grown on 96-well cell culture black plates (Corning Incorporated Life Sciences, Lowell, MA) in complete EGM-2 media at 37°C in a humidified atmosphere of 5% CO2 were treated with pro-inflammatory and pro-apoptotic agents for specified periods of time. Treatment of HUVECs with vWF was performed in HBSS. Before the assay, the medium was removed, cells were washed once with HBSS, and left in 50 μl of HBSS. hMSCs in suspension (50 μl, 10,000 cells per well) were added to HUVECs and incubated for 30 min at 37°C and 5% CO2. Fluorescence was determined using a POLARstar OPTIMA microplate reader (BMG Labtech Inc., Durham, NC) at excitation/emission wavelengths of 485/520 nm. Wells without hMSCs were used to assay background fluorescence. After incubation unbound hMSCs were aspirated and washed with 200 μl of HBSS two times. HBSS (100 μl per well) was added and plates were scanned using a POLARstar OPTIMA microplate reader at excitation/emission wavelengths of 485/520 nm. Percentage of bound cells was calculated as a ratio between the fluorescence of washed and unwashed wells after subtraction of background fluorescence from both values. At least six wells were used for each experimental condition. At least three independent experiments were conducted for each treatment.
Immunofluorescence staining and flow cytometric analysis of surface antigen expression in HUVECs
Analysis of cell surface molecules was performed as described (Potapova et al., 2008) with some modifications. After treatment with different substances for 4 h in complete EGM-2 media at 37°C in a humidified atmosphere of 5% CO2, cells were dissociated from a monolayer of HUVECs with Enzyme Free Cell Dissociation Solution (Millipore) and resuspended in the flow cytometry buffer consisting of 2% bovine serum albumin (Sigma) and 0.1% sodium azide (Sigma-Aldrich) in Dulbecco’s phosphate buffered saline (PBS; Sigma). Cells (2 × 105) were stained using the manufacturer’s suggested concentrations of fluorochrome-conjugated monoclonal antibodies for 30 min at room temperature in the dark. Antibodies to human CD31 (PECAM-1), CD34, CD44, CD51/CD61 (integrin αvβ3), CD54 (ICAM-1), CD62E (E-selectin), CD99, CD106 (VCAM-1), CD107a (LAMP-1), CD114, CD142 (tissue factor), CD146 (Mel-CAM), and CD166 (ALCAM) were from BD Biosciences (San Jose, CA). After staining, cells were washed with 5 ml of the flow cytometry buffer and resuspended in the flow cytometry buffer containing 1% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA). Background staining was assessed by incubation of cells with mouse fluorochrome- and isotype-matched immunoglobulins (isotype controls). Flow cytometric analysis of HUVECs was performed by analyzing 5,000 events on a FACScan flow cytometer (Becton–Dickinson). A CellQuest™ software package was used to process flow cytometry data. The cellular debris was assessed on the basis of forward and right angle scattering analysis and excluded from future analysis by the CellQuest™ software package.
Determination of the cytotoxic effects of apoptosis inducers on ECs
Cytotoxic effect of substances applied to induce apoptosis in HUVECs was measured by a WST-1 assay (Roche Diagnostics Corporation Roche Applied Science, Indianapolis, IN). Briefly, WST-1 is metabolized by mitochondrial NAD-dependent dehydrogenase into formazan, the colored product of WST-1. Generally, the amount of generated formazan dye correlates with the cell viability. Confluent HUVECs grown on 96-well cell culture black plates in complete EGM-2 media at 37°C in a humidified atmosphere of 5% CO2 were treated for 4 h with apoptosis inducers. The WST-1 reagent was added into the cell culture medium (10 μl per 100 μl medium), and cells were incubated for 4 h at 37°C and 5% CO2. The formazan absorbance was measured at 450 nm with the reference wavelength of 595 nm using a POLARstar OPTIMA microplate reader. At least six measurements were performed for each set of experimental conditions in three independent experiments.
Detection of changes in mitochondrial transmembrane potential
Confluent HUVECs grown on 96-well cell culture black plates in complete EGM-2 media were washed with PBS and incubated for 30 min at 37°C and 5% CO2 with JC-1 dye (MitoCapture reagent) according to manufacture’s recommendations (BioVision Inc., Mountain View, CA). Cells were washed with PBS and treated for 4 h at 37°C and 5% CO2 with apoptosis inducers in complete EGM-2 media. Green fluorescence was determined using a POLARstar OPTIMA microplate reader with eight measurements for each condition at excitation/emission wavelengths of 485/520 nm. Cell images were captured on an Olympus CK-40 inverted fluorescent microscope equipped with a digital camera (QImaging Inc., Surrey, BC, Canada).
vWF enzyme-linked immunosorbent assay (ELISA)
Confluent HUVECs were grown on 96-well cell culture black plates in complete EGM-2 media at 37°C in a humidified atmosphere of 5% CO2 and treated for 4 h with apoptosis inducers. Conditioned media were collected, and the release of vWF was measured by ELISA. Polyclonal rabbit anti-human vWF antibody (Dako Denmark A/S, Glostrup, Denmark) and peroxidase-conjugated polyclonal rabbit anti-human vWF antibody (Dako) were used according to manufacturer’s recommendations. The calibration curve was generated using human vWF—factor FVIII free (American Diagnostica, Inc., Mason, OH). The optical densities were measured at 450 nm with a 595 nm reference wavelength using a POLARstar OPTIMA microplate reader. Concentrations of vWF were calculated using data from eight measurements per set of experimental conditions.
Results
Apoptosis inducers enhance hMSC adhesion to HUVECs
HUVECs were treated for 4 h with 10 ng/ml TNF-α or 10 ng/ml IL-1β in the absence or presence of 5 μg/ml ActD or 20 μg/ml CHX, 0.05 μM okadaic acid, 5 μM wortmannin, or 0.25 μM staurosporine. HUVECs were washed before the addition of the hMSCs to the EC monolayers. Adhesion of hMSCs to HUVECs at these conditions is shown in Figure 1A.
Fig. 1.

Effects of pro-inflammatory and pro-apoptotic agents on hMSC adhesion to HUVECs. Panel A shows adhesion of hMSCs to untreated HUVECs (n =267) and HUVECs pretreated for 4 hours with 10 ng/ml TNF-α (n =48), 10 ng/ml IL-1β (n =54), 10 μg/ml actinomycin D (ActD, n =39), 20 μg/ml cycloheximide (CHX, n =76), 0.05 μM okadaic acid (n =30), 5 μM wortmannin (n =46), 0.25 μM staurosporine (n =31), or mixtures of TNF-α with ActD (n = 20), IL-1β with ActD (n =35), TNF-α with CHX (n = 51), IL-1β with CHX (n =43). Data are shown as mean ±SE of at least four independent experiments. Numbers in parenthesis show total number of measurements. Kruskal-Wallis analysis of variance on ranks was performed to establish statistical significance of observed differences. Asterisks mark statistically significant effects in comparison with untreated HUVECs (P-value ≤ 0.05). Panel B shows dose-response curves of hMSC adhesion to HUVECs pretreated with increasing concentrations of CHX, wortmannin, okadaic acid or staurosporine for 4 hours. Data are shown as mean ±SD of eight independent measurements. Panel C shows time courses of hMSC adhesion to HUVECs pretreated with 20 μg/ml CHX, 5 μM wortmannin, 0.05 μM okadaic acid or 0.25 μM staurosporine. Data are shown as mean ±SD of eight independent measurements.
Treatment of HUVECs with TNF-α resulted in a statistically significant 1.3-fold increase in hMSC adhesion. Exposure of HUVECs to IL-1β increased hMSC adhesion by 1.6-fold ( P-value <0.05). As detected by flow cytometric analysis, treatment with TNF-α or IL-1β increased expression of E-selectin (40- to 50-fold increase) and ICAM-1 (three to fourfold increase) on the surface of ECs (Table 1). Untreated HUVECs were CD106 (VCAM-1) and CD142 (tissue factor) negative, but they started to express VCAM-1 and tissue factor after treatment with TNF-α or IL-1β (Table 1). Flow cytometry demonstrated that the combined treatment of HUVECs with TNF-α or IL-1β and ActD or CHX abolished upregulation of CD54 (ICAM-1), CD62E (E-selectin), CD106 (VCAM-1), and CD142 (tissue factor) on the surface of ECs (Table 1). Adhesion of hMSCs to HUVECs treated with ActD was not different from hMSC adhesion to untreated HUVECs (Fig. 1A). Treatment of HUVECs with TNF-α or IL-1β in the presence of ActD resulted in statistically significant stimulation of hMSC adhesion 1.4- and 2.1-fold, respectively. CHX alone stimulated hMSC adhesion 1.7-fold ( P-value <0.05). In the presence of TNF-α or IL-1β, CHX significantly stimulated hMSC adhesion 1.9- and 2.2-fold, accordingly (Fig. 1A). These data suggest that stimulatory effects of TNF-α or IL-1β on hMSC adhesion may not relate to upregulation of adhesion molecules on the surface of ECs. We hypothesized that stimulation of hMSC adhesion to HUVECs depends on activation of apoptosis/stress signaling pathways in ECs treated with TNF-α or IL-1β in the presence of inhibitors of RNA or protein synthesis. It was thus possible that alternative methods to induce apoptosis in ECs would also stimulate hMSC adhesion. We tested the effects of okadaic acid, wortmannin, and staurosporine, the known inducers of stress and apoptosis in ECs, on hMSC adhesion. Okadaic acid, wortmannin, and staurosporine significantly stimulated hMSC adhesion 3-, 2.4- and 3.5-fold, respectively (Fig. 1A). Effects of CHX, okadaic acid, wortmannin, and staurosporine on hMSC adhesion were time- and dose-dependent. Dose–response curves for hMSC adhesion to HUVECs treated with CHX, wortmannin, okadaic acid, or staurosporine for 4 h are shown in Figure 1B. Time course curves for hMSC adhesion activation to HUVECs exposed to CHX, wortmannin, okadaic acid, or staurosporine are shown in Figure 1C. We tested activation of apoptosis in HUVECs treated with staurosporine and detected prominent activation of caspase-3 (Fig. 2A) and caspase-7 (Fig. 2B) as soon as 2 h after exposure of HUVECs to staurosporine.
TABLE 1.
Changes in antigen expression on the surface of HUVECs treated with pro-inflammatory and pro-apoptotic agentsa
| None | TNF-α | IL-1β | ActD | ActD +TNF-α | ActD +IL-1β | CHX | CHX +TNF-α | CHX +IL-1β | |
|---|---|---|---|---|---|---|---|---|---|
| CD31 (PECAM-1) | 1.0 | 0.9 ± 0.2 | 1.0 ± 1.0 | 0.95 ± 0.05* | 0.97 ± 0.2 | 0.8 ± 0.2* | 0.7 ± 0.1* | 0.7 ± 0.1* | 0.6 ± 0.1* |
| CD34 | 1.0 | 1.0 ± 0.2 | 1.0 ± 0.1 | 0.86 ± 0.2* | 1.0 ± 0.3 | 0.6 ± 0.2* | 0.74 ± 0.3* | 0.6 ± 0.2* | 0.4 ± 0.2* |
| CD44 | 1.0 | 0.8 ± 0.2* | 0.8 ± 0.2* | 1.0 ± 0.2 | 0.9 ± 0.1 | 0.8 ± 0.1* | 0.8 ± 0.2* | 0.7 ± 0.1* | 0.8 ± 0.1* |
| CD51/61 (αVβ3-complex) | 1.0 | 0.8 ± 0.1* | 1.0 ± 0.1 | 1.00 ± 0.03 | 0.95 ± 0.03* | 0.9 ± 0.2 | 0.81 ± 0.04* | 0.79 ± 0.02* | 0.7 ± 0.1* |
| CD54 (ICAM-1) | 1.0 | 3.0 ± 1* | 4.0 ± 1* | 0.9 ± 0.1* | 0.9 ± 0.2 | 0.6 ± 0.1* | 0.7 ± 0.2* | 0.6 ± 0.2* | 0.5 ± 0.2* |
| CD62E (E-selectin) | 1.0 | 40 ± 10* | 50 ± 13* | 0.6 ± 0.1* | 0.7 ± 0.2* | 0.5 ± 0.1* | 0.66 ± 0.09* | 2.0 ± 0.5* | 1.5 ± 0.2* |
| CD99 | 1.0 | 0.85 ± 0.08 | 0.92 ± 0.06 | 1.0 ± 0.08 | 1.02 ± 0.05 | 0.87 ± 0.02* | 0.8 ± 0.2 | 0.86 ± 0.08 | 0.70 ± 0.03* |
| CD106 (VCAM-1)b | 11 ± 2 | 66 ± 40* | 58 ± 31* | 10.4 ± 0.6 | 10.9 ± 0.5 | 8.8 ± 0.6 | 8.5 ± 1.5 | 8.3 ± 1.6 | 10.0 ± 5.3 |
| CD107a (LAMP-1)b | 18 ± 2 | 15 ± 3 | 17.3 ± 0.9 | 17 ± 3 | 18 ± 2 | 19 ± 4 | 15 ± 3 | 17 ± 5 | 18 ± 6 |
| CD114 | 1.0 | 0.8 ± 0.3 | 1.0 ± 0.2 | 0.8 ± 0.2 | 0.8 ± 0.1 | 0.63 ± 0.08* | 0.7 ± 0.1* | 0.61 ± 0.08* | 0.55 ± 0.06* |
| CD142b (tissue factor) | 19 ± 10 | 48 ± 9* | 74 ± 12* | 14 ± 6 | 16 ± 6 | 14 ± 7 | 14 ± 8 | 16 ± 6 | 18 ± 8 |
| CD146 (MCAM) | 1.0 | 0.8 ± 0.2 | 0.9 ± 0.1 | 0.8 ± 0.2* | 0.9 ± 0.1 | 0.75 ± 0.06* | 0.8 ± 0.1* | 0.8 ± 0.2 | 0.9 ± 0.3 |
| CD166 (ALCAM) | 1.0 | 0.83 ± 0.05* | 0.90 ± 0.06* | 1.0 ± 0.1 | 0.98 ± 0.04* | 0.9 ± 0.1 | 0.88 ± 0.05* | 0.9 ± 0.1 | 0.82 ± 0.07* |
Statistically significant changes in comparison with non-treated HUVECs (P-value <0.05).
Data shown in the table represent an average of at least four independent experiments.
Antigens CD106, 107a, and CD142 were not expressed on the surface of non-treated HUVECs. For these antigens, mean fluorescent values ±SD are shown. For other antigens changes relatively to non-treated HUVECs are presented.
Fig. 2.
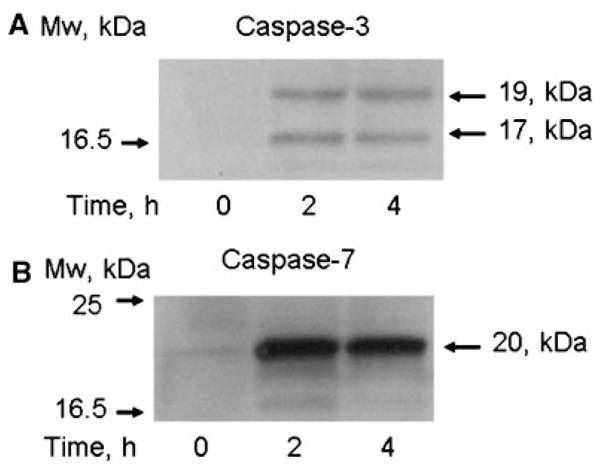
Activation of caspase-3 and caspase-7 in HUVECs treated with staurosporine. Cells were treated with 0.25 μM staurosporine for indicated periods of time. Activation of caspase-3 (Panel A) and caspase-7 (Panel B) was assayed by Western blot for specific proteolytic fragments of caspase-3 and caspase-7.
hMSC adhesion correlates with the inhibition of mitochondrial transmembrane potential and activity of NADH dehydrogenases in HUVECs
Since the reduction of mitochondrial transmembrane potential is one of the earliest indicators of apoptosis, we assayed mitochondria membrane dysfunction of HUVECs using MitoCapture cationic dye. MitoCapture accumulates in the mitochondria of healthy cells giving off a red fluorescence. In apoptotic cells, MitoCapture is released into the cytosole due to the reduction in the transmembrane potential of mitochondria and gives rise to green fluorescence (Reers et al., 1995). As shown in Figure 3A, the intensity of the green fluorescence was increased in cells treated with ActD in the presence of IL-1β and in cells treated with CHX in the presence or absence of TNF-α or IL-1β. Reduction in mitochondrial membrane potential was quantitatively confirmed by determining the time course of MitoCapture release into the cytosole of HUVECs with the microplate reader. HUVECs treated with CXH, CXH in the presence of TNF-α, CXH in the presence of IL-1β, okadaic acid, wortmannin, or staurosporine demonstrated the strongest reduction of the mitochondrial membrane potential (Fig. 3B). We used the data shown in Figures 1A and 3B to correlate hMSC adhesion with the inhibition of mitochondrial transmembrane potential in HUVECs. The correlation plot is shown in Figure 3C. HMSC adhesion strongly correlated with the inhibition of mitochondrial transmembrane potential of HUVECs (R = 0.85, P-value = 0.005).
Fig. 3.
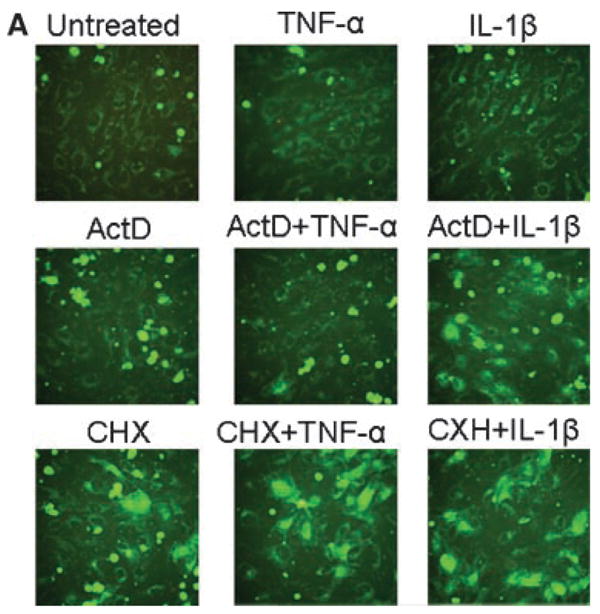

Effects of pro-inflammatory and pro-apoptotic agents on mitochondrial membrane potential in HUVECs. The correlation between adhesion of hMSCs and mitochondrial membrane potential in HUVECs. Changes in mitochondrial membrane potential were assayed in HUVEC cells loaded with MitoCapture dye. Panel A shows representative green fluorescence images of untreated HUVECs and HUVECs treated with 10 ng/ml TNF-α, 10 ng/ml IL-1β, 10 μg/ml actinomycin D (ActD), 20 μg/ml cycloheximide (CHX) or mixtures of TNF-α with ActD, IL-1β with ActD, TNF-α with CHX or IL-1β with CHX for 4 hours. Panel B shows the data from a quantitative analysis of MitoCapture release into the cytosole of untreated HUVECs and HUVECs treated with 10 ng/ml TNF-α, 10 ng/ml IL-1β, 10 μg/ml ActD, 20 μg/ml CHX, 0.05 μM okadaic acid, 5 μM wortmannin, 0.25 μM staurosporine, or mixtures of TNF-α with ActD, IL-1β with ActD, TNF-α with CHX, or IL-1β with CHX. Analysis was performed on a microplate reader. Data are shown as mean ±SD of eight independent measurements. Panel C shows Person’s correlation (R =0.85, P-value =0.005) between adhesion of hMSCs (data from Figure 1) and MitoCapture release into the cytosole of HUVECs.
The cytotoxicity of substances used in our experiments toward HUVECs was also measured using WST-1 reagent, the substrate for mitochondrial NADH dehydrogenases. Treatment of HUVECs with the pro-inflammatory and pro-apoptotic agents resulted in inhibition of NADH dehydrogenases for most of them, with the exception of ActD, where changes were not statistically significant (Fig. 4A). Okadaic acid, wortmannin, and staurosporine, the strongest stimulators of hMSC adhesion, were also the strongest inhibitors of NADH dehydrogenases. We analyzed the correlation between adhesion of hMSCs (data from Fig. 1A) and activity of NADH dehydrogenases in HUVECs. HMSC adhesion strongly correlated with the inhibition of HUVEC NADH dehydrogenases (R =−0.93, P-value = 0.00001). The correlation plot is shown in Figure 4B.
Fig. 4.

Effects of pro-inflammatory and pro-apoptotic agents on activity of NADH-dehydrogenases in HUVECs. The correlation between adhesion of hMSCs and activity of NADH-dehydrogenases in HUVECs. Activity of NADH-dehydrogenases in HUVECs was assayed using a WST-1 reagent. Panel A shows WST-1 assay data for untreated HUVECs and HUVECs treated with 10 ng/ml TNF-α, 10 ng/ml IL-1β, 10 μg/ml actinomycin D (ActD), 20 μg/ml cycloheximide (CHX) or mixtures of TNF-αwith ActD, IL-1βwith ActD, TNF-αwith CHX or IL-1β with CHX for 4 hours. Data are shown as mean ±SD of eight independent measurements. Asterisks mark statistically significant changes in comparison with untreated HUVECs (P-value ≤0.05). Panel B shows Person’s correlation (R =−0.93, P-value = 0.00001) between adhesion of hMSCs (data from Figure 1) and activity of NADH-dehydrogenases in HUVECs.
Thus, HMSC adhesion to HUVECs strongly correlated with the depolarization of the mitochondrial transmembrane potential and inhibition of NAD dehydrogenase activity in ECs.
vWF regulates hMSC adhesion to HUVECs
We measured accumulation of vWF, a marker of EC dysfunction, in media conditioned by HUVECs treated with the pro-inflammatory or pro-apoptotic agents (Fig. 5A). The secretion of vWF was stimulated by TNF-α and IL-1β. Concomitant treatment with ActD or CHX did not abolish TNF-α and IL-1β-dependent vWF secretion but rather increased the accumulation of vWF in media conditioned by HUVECs. Treatment of HUVECs with okadaic acid, wortmannin, or staurosporine also stimulated the secretion of vWF. We analyzed the correlation between hMSC adhesion (data from Fig. 1A) and vWF secretion by HUVECs. The correlation plot is shown in Figure 5B. There is a strong correlation between HMSC adhesion and the secretion of vWF by HUVECs (R = 0.86, P-value = 0.0003).
Fig. 5.

Effects of pro-inflammatory and pro-apoptotic agents on secretion of vWF by HUVECs. The correlation between adhesion of hMSCs and concentrations of vWF in media conditioned by HUVECs. Effects of HUVECs treatment with endogenous vWF on hMSC adhesion. Concentrations of vWF in media conditioned by HUVECs was measured by ELISA. Panel A shows concentrations of vWF in media conditioned by untreated HUVECs and HUVECs treated with 10 ng/ml TNF-α, 10 ng/ml IL-1β, 10 μg/ml actinomycin D (ActD), 20 μg/ml cycloheximide (CHX) or mixtures of TNF-α with ActD, IL-1β with ActD, TNF-α with CHX or IL-1β with CHX, for 4 hours. Data are shown as mean ±SD of eight independent measurements. Asterisks mark statistically significant changes in comparison with untreated HUVECs (P-value ≤0.05). Panel B shows Person’s correlation (R =0.86, P-value 0.0003) between adhesion of hMSCs (data from Figure 1) and secretion of vWF by HUVECs. Panel C show dose-response curves of hMSC adhesion to HUVECs pretreated with increasing concentrations of vWF for 3 hours. hMSC adhesion was assayed in the presence (○) or absence of vWF (●). Data are shown as mean ±SD of eight independent measurements.
The correlation between hMSC adhesion and vWF release by ECs suggests that vWF might be involved in the regulation of hMSC adhesion to ECs. To investigate the role of vWF on hMSC adhesion, HUVECs were pretreated with increasing concentrations of endogenous soluble vWF for 3 h, and hMSC adhesion was assayed in the presence or absence of vWF. Treatment of HUVECs with vWF resulted in stimulation of hMSC adhesion to ECs. Effects of vWF were dose-dependent. Dose–response curves for hMSC adhesion to HUVECs treated with vWF are shown in Figure 5C. The presence of vWF in the adhesion assay media was not required for the stimulation of hMSC adhesion (Fig. 5C) indicating that vWF acts primarily through modification of adhesion properties of EC surface.
Discussion
MSCs circulating in the bloodstream or expanded in vitro and delivered in the circulation accumulate at the sites of injury and inflammation (Devine et al., 2002; Barbash et al., 2003; Bittira et al., 2003; Chapel et al., 2003; Kraitchman et al., 2005; Mansilla et al., 2006; Ramirez et al., 2006). A growing body of evidence shows that injection of MSCs improves cardiac function after ischemia in animal models (Galli et al., 2005; Zhang et al., 2006; Mazhari and Hare, 2007) and clinical trials (Schachinger et al., 2006a,b; Assmus et al., 2007; Erbs et al., 2007; Dimmeler et al., 2008). As suggested, MSCs supply pro-angiogenic and anti-apoptotic factors promoting tissue repair in a paracrine manner. Previously, we have demonstrated that hMSCs secrete paracrine factors that stimulate EC migration, extracellular matrix invasion, proliferation, and survival (Potapova et al., 2007). For successful application of MSCs in the clinic, it is crucial to understand the mechanisms of MSC targeting and retention in injured tissues. Different stages of myocardial infarction are associated with the development of hypoxia, inflammatory response, and cell death. Apoptosis of ECs precedes myocyte cell apoptosis in ischemia/reperfusion injury (Scarabelli et al., 2001). Tissue repair may be optimized by transplantation of MSCs at the most “favorable” time. However, the first and the key step is MSC homing to injured tissues—hMSC–EC interaction remains poorly understood. A number of research groups have evaluated the role of chemokines and chemokine receptors in MSCs homing. Homing of MSCs to brain injuries depends on the expression of CXCR4 by MSCs and is regulated by local concentrations of SDF-1 in the ischemic lesion sites (Ji et al., 2004; Wang et al., 2008). CXCR4-positive hMSCs specifically adhere to hypoxic ECs in vitro (Potapova et al., 2008). Yet, with only a small fraction of isolated MSCs expressing CXCR4, these cells are considered homing impaired (Rombouts and Ploemacher, 2003; Seeger et al., 2007a). It has been suggested that cell enhancement strategies are required to improve hMSC homing (Seeger et al., 2007b). Several approaches were developed to obtain CXCR4-positive hMSCs that include an improved procedure for hMSCs isolation (Seeger et al., 2007a), upregulation of CXCR4 expression in hMSCs by culturing them as 3D aggregates (Potapova et al., 2008), and transfection of MSCs with viral vectors harboring CXCR4 (Cheng et al., 2008; Wang et al., 2008; Zhang et al., 2008).
Although CXCR4 plays a role in MSC homing, the CXCR4-dependent mechanisms may not encompass the entire spectrum of MSC homing. Dynamic MRI imaging has shown that systemically infused MSCs effectively accumulate in infarcted heart (Kraitchman et al., 2005) despite impairment in CXCR4 expression (Wynn et al., 2004; Seeger et al., 2007a,b; Potapova et al., 2008). It was reported that MSCs do not use CXCR4 for myocardial migration and engraftment (Ip et al., 2007). The disappearance of hMSCs natively present in the bloodstream of patients with myocardial infarction occurs 7 days after the infarction and does not correlate with the concentration of SDF-1 in the blood, which reaches its maximum 21 days post-infraction (Wang et al., 2006).
Our results show that pro-apoptotic agents markedly increased adhesion of hMSCs to ECs. HMSC adhesion correlated with the reduction of mitochondrial transmembrane potential of ECs and the inhibition of NADH dehydrogenases in ECs. The intrinsic apoptosis pathway is activated by the release of cytochrome c from the mitochondria due to a decrease in mitochondria transmembrane potential and an increase in the permeability of the mitochondrial membrane (Riedl and Salvesen, 2007). Our data suggest that hMSCs have a capacity to survey the energetic status of ECs and specifically respond to inhibition of mitochondrial functions in ECs.
The secretion of vWF by ECs is an indicator of a distressed endothelium. The plasma levels of vWF are elevated in response to endothelial damage (Blann, 1993), hypoxia, inflammatory cytokines, thrombin, and adrenalin (Spiel et al., 2008). VWF plays a pivotal role in platelet adhesion to ECs (Ruggeri and Ware, 1993). We demonstrated that the adhesion of hMSCs correlates with the release of vWF by ECs. Furthermore, treatment of ECs with endogenous vWF also stimulated hMSC adhesion. Since stimulatory effects of vWF may be attributed to activation of ECs, hMSCs, or both, we performed an hMSC adhesion assay to ECs treated with vWF in the presence or absence of vWF. Our results show that the presence of soluble vWF is not required for the stimulation of hMSC adhesion to ECs pretreated with vWF, suggesting that vWF modifies adhesion properties of the endothelium.
Overall, our data demonstrate that hMSCs have CXCR4-independent mechanisms of EC recognition related to activation of apoptosis/stress signaling pathways in ECs and the release of vWF. We hypothesize that apoptosis of ECs regulates homing of hMSCs to injured tissues.
Acknowledgments
This work was supported by an American Heart Association Scientist Development Grant to Sergey V. Doronin and NIH grants HL67101 and HL28958, and grant from NYSTEM.
Contract grant sponsor: American Heart Association Scientist Development.
Contract grant sponsor: NIH;
Contract grant numbers: HL67101, HL28958.
Contract grant sponsor: NYSTEM.
Literature Cited
- Assmus B, Fischer-Rasokat U, Honold J, Seeger FH, Fichtlscherer S, Tonn T, Seifried E, Schachinger V, Dimmeler S, Zeiher AM. Transcoronary transplantation of functionally competent BMCs is associated with a decrease in natriuretic peptide serum levels and improved survival of patients with chronic postinfarction heart failure: Results of the TOPCARE-CHD registry. Circ Res. 2007;100:1234–1241. doi: 10.1161/01.RES.0000264508.47717.6b. [DOI] [PubMed] [Google Scholar]
- Barbash IM, Chouraqui P, Baron J, Feinberg MS, Etzion S, Tessone A, Miller L, Guetta E, Zipori D, Kedes LH, Kloner RA, Leor J. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation. 2003;108:863–868. doi: 10.1161/01.CIR.0000084828.50310.6A. [DOI] [PubMed] [Google Scholar]
- Bittira B, Shum-Tim D, Al-Khaldi A, Chiu RCJ. Mobilization and homing of bone marrow stromal cells in myocardial infarction. Eur J Cardiothorac Surg. 2003;24:393–398. doi: 10.1016/s1010-7940(03)00325-7. [DOI] [PubMed] [Google Scholar]
- Blann A. von Willebrand factor and the endothelium in vascular disease. Br J Biomed Sci. 1993;50:125–134. [PubMed] [Google Scholar]
- Bombeli T, Schwartz BR, Harlan JM. Endothelial cells undergoing apoptosis become proadhesive for nonactivated platelets. Blood. 1999;93:3831–3838. [PubMed] [Google Scholar]
- Cavender D, Haskard D, Foster N, Ziff M. Superinduction of T lymphocyte-endothelial cell (EC) binding by treatment of EC with interleukin 1 and protein synthesis inhibitors. J Immunol. 1987;138:2149–2154. [PubMed] [Google Scholar]
- Chapel A, Bertho JM, Bensidhoum M, Fouillard L, Young RG, Frick J, Demarquay C, Cuvelier F, Mathieu E, Trompier F, Dudoignon N, Germain C, Mazurier C, Aigueperse J, Borneman J, Gorin NC, Gourmelon P, Thierry D. Mesenchymal stem cells home to injured tissues when co-infused with hematopoietic cells to treat a radiation-induced multi-organ failure syndrome. J Gene Med. 2003;5:1028–1038. doi: 10.1002/jgm.452. [DOI] [PubMed] [Google Scholar]
- Chen PL, Easton A. Apoptotic phenotype alters the capacity of tumor necrosis factor-related apoptosis-inducing ligand to induce human vascular endothelial activation. J Vasc Res. 2008;45:111–122. doi: 10.1159/000109880. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Ou L, Zhou X, Li F, Jia X, Zhang Y, Liu X, Li Y, Ward CA, Melo LG, Kong D. Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol Ther. 2008;16:571–579. doi: 10.1038/sj.mt.6300374. [DOI] [PubMed] [Google Scholar]
- Devine MJ, Mierisch CM, Jang E, Anderson PC, Balian G. Transplanted bone marrow cells localize to fracture callus in a mouse model. J Orthop Res. 2002;20:1232–1239. doi: 10.1016/S0736-0266(02)00051-7. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Burchfield J, Zeiher AM. Cell-based therapy of myocardial infarction. Arterioscler Thromb Vasc Biol. 2008;28:208–216. doi: 10.1161/ATVBAHA.107.155317. [DOI] [PubMed] [Google Scholar]
- Erbs S, Linke A, Schachinger V, Assmus B, Thiele H, Diederich K-W, Hoffmann C, Dimmeler S, Tonn T, Hambrecht R, Zeiher AM, Schuler G. Restoration of microvascular function in the infarct-related artery by intracoronary transplantation of bone marrow progenitor cells in patients with acute myocardial infarction: The Doppler substudy of the reinfusion of enriched progenitor cells and infarct remodeling in acute myocardial infarction (REPAIR-AMI) trial. Circulation. 2007;116:366–374. doi: 10.1161/CIRCULATIONAHA.106.671545. [DOI] [PubMed] [Google Scholar]
- Galli D, Innocenzi A, Staszewsky L, Zanetta L, Sampaolesi M, Bai A, Martinoli E, Carlo E, Balconi G, Fiordaliso F, Chimenti S, Cusella G, Dejana E, Cossu G, Latini R. Mesoangioblasts, vessel-associated multipotent stem cells, repair the infarcted heart by multiple cellular mechanisms: A comparison with bone marrow progenitors, fibroblasts, and endothelial cells. Arterioscler Thromb Vasc Biol. 2005;25:692–697. doi: 10.1161/01.ATV.0000156402.52029.ce. [DOI] [PubMed] [Google Scholar]
- Gonzalez MA, Selwyn AP. Endothelial function, inflammation, and prognosis in cardiovascular disease. Am J Med. 2003;115:99–106. doi: 10.1016/j.amjmed.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Ip JE, Wu Y, Huang J, Zhang L, Pratt RE, Dzau VJ. Mesenchymal stem cells use integrin beta1 not CXC chemokine receptor 4 for myocardial migration and engraftment. Mol Biol Cell. 2007;18:2873–2882. doi: 10.1091/mbc.E07-02-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji JF, He BP, Dheen ST, Tay SSW. Interactions of chemokines and chemokine receptors mediate the migration of mesenchymal stem cells to the impaired site in the brain after hypoglossal nerve injury. Stem Cells. 2004;22:415–427. doi: 10.1634/stemcells.22-3-415. [DOI] [PubMed] [Google Scholar]
- Kraitchman DL, Tatsumi M, Gilson WD, Ishimori T, Kedziorek D, Walczak P, Segars WPH, Chen H, Fritzges D, Izbudak I, Young RG, Marcelino M, Pittenger MF, Solaiyappan M, Boston RC, Tsui BMW, Wahl RL, Bulte JWM. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005;112:1451–1461. doi: 10.1161/CIRCULATIONAHA.105.537480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansilla E, Marin GH, Drago H, Sturla F, Salas E, Gardiner C, Bossi S, Lamonega R, Guzman A, Nucez A, Gil MA, Piccinelli G, Ibar R, Soratti C. Bloodstream cells phenotypically identical to human mesenchymal bone marrow stem cells circulate in large amounts under the influence of acute large skin damage: New evidence for their use in regenerative medicine. Transplant Proc. 2006;38:967–969. doi: 10.1016/j.transproceed.2006.02.053. [DOI] [PubMed] [Google Scholar]
- Mazhari R, Hare JM. Mechanisms of action of mesenchymal stem cells in cardiac repair: Potential influences on the cardiac stem cell niche. Nat Clin Pract Cardiovasc Med. 2007;4:S21–S26. doi: 10.1038/ncpcardio0770. [DOI] [PubMed] [Google Scholar]
- Mosca JD, Hendricks JK, Buyaner D, Davis-Sproul J, Chuang LC, Majumdar MK, Chopra R, Barry F, Murphy M, Thiede MA, Junker U, Rigg RJ, Forestell SP, Bohnlein E, Storb R, Sandmaier BM. Mesenchymal stem cells as vehicles for gene delivery. Clin Orthop Relat Res. 2000;379:S71–S90. doi: 10.1097/00003086-200010001-00011. [DOI] [PubMed] [Google Scholar]
- Mouiseddine M, Francois S, Semont A, Sache A, Allenet B, Mathieu N, Frick J, Thierry D, Chapel A. Human mesenchymal stem cells home specifically to radiation-injured tissues in a non-obese diabetes/severe combined immunodeficiency mouse model. Br J Radiol. 2007;80:S49–S55. doi: 10.1259/bjr/25927054. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Pober JS. Activation and injury of endothelial cells by cytokines. Pathol Biol (Paris) 1998;46:159–163. [PubMed] [Google Scholar]
- Pohlman TH, Harlan JM. Human endothelial cell response to lipopolysaccharide, interleukin-1, and tumor necrosis factor is regulated by protein synthesis. Cell Immunol. 1989;119:41–52. doi: 10.1016/0008-8749(89)90222-0. [DOI] [PubMed] [Google Scholar]
- Pohlman TH, Harlan JM. Adaptive responses of the endothelium to stress. J Surg Res. 2000;89:85–119. doi: 10.1006/jsre.1999.5801. [DOI] [PubMed] [Google Scholar]
- Polunovsky VA, Wendt CH, Ingbar DH, Peterson MS, Bitterman PB. Induction of endothelial cell apoptosis by TNF[alpha]: Modulation by inhibitors of protein synthesis. Exp Cell Res. 1994;214:584–594. doi: 10.1006/excr.1994.1296. [DOI] [PubMed] [Google Scholar]
- Potapova IA, Gaudette GR, Brink PR, Robinson RB, Rosen MR, Cohen IS, Doronin SV. Mesenchymal stem cells support migration, extracellular matrix invasion, proliferation, and survival of endothelial cells in vitro. Stem Cells. 2007;25:1761–1768. doi: 10.1634/stemcells.2007-0022. [DOI] [PubMed] [Google Scholar]
- Potapova IA, Brink PR, Cohen IS, Doronin SV. Culturing of human mesenchymal stem cells as 3D-aggregates induces functional expression of CXCR4 that regulates adhesion to endothelial cells. J Biol Chem. 2008;283:13100–13107. doi: 10.1074/jbc.M800184200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez M, Lucia A, Gomez-Gallego F, Esteve-Lanao J, Perez-Martinez A, Foster C, Andreu AL, Martin MA, Madero L, Arenas J, Garcia-Castro J. Mobilisation of mesenchymal cells into blood in response to skeletal muscle injury. Br J Sports Med. 2006;40:719–722. doi: 10.1136/bjsm.2006.028639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–417. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Salvesen GS. The apoptosome: Signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- Rombouts WJC, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia. 2003;17:160–170. doi: 10.1038/sj.leu.2402763. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM, Ware J. von Willebrand factor. FASEB J. 1993;7:308–316. doi: 10.1096/fasebj.7.2.8440408. [DOI] [PubMed] [Google Scholar]
- Ruster B, Gottig S, Ludwig RJ, Bistrian R, Muller S, Seifried E, Gille J, Henschler R. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood. 2006;108:3938–3944. doi: 10.1182/blood-2006-05-025098. [DOI] [PubMed] [Google Scholar]
- Scarabelli T, Stephanou A, Rayment N, Pasini E, Comini L, Curello S, Ferrari R, Knight R, Latchman D. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation. 2001;104:253–256. doi: 10.1161/01.cir.104.3.253. [DOI] [PubMed] [Google Scholar]
- Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, Holschermann H, Yu J, Corti R, Mathey DG, Hamm CW, Suselbeck T, Assmus B, Tonn T, Dimmeler S, Zeiher AM the R-AMII. Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N Engl J Med. 2006a;355:1210–1221. doi: 10.1056/NEJMoa060186. [DOI] [PubMed] [Google Scholar]
- Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, Holschermann H, Yu J, Corti R, Mathey DG, Hamm CW, Suselbeck T, Werner N, Haase J, Neuzner J, Germing A, Mark B, Assmus B, Tonn T, Dimmeler S, Zeiher AM for the R-AMII. Improved clinical outcome after intracoronary administration of bone-marrow-derived progenitor cells in acute myocardial infarction: Final 1-year results of the REPAIR-AMI trial. Eur Heart J. 2006b;27:2775–2783. doi: 10.1093/eurheartj/ehl388. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Ladage D, Steingen C, Brixius K, Schinköthe T, Klinz F-J, Schwinger RHG, Mehlhorn U, Bloch W. Mesenchymal stem cells transmigrate over the endothelial barrier. Eur J Cell Biol. 2006;85:1179–1188. doi: 10.1016/j.ejcb.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Seeger FH, Tonn T, Krzossok N, Zeiher AM, Dimmeler S. Cell isolation procedures matter: A comparison of different isolation protocols of bone marrow mononuclear cells used for cell therapy in patients with acute myocardial infarction. Eur Heart J. 2007a;28:766–772. doi: 10.1093/eurheartj/ehl509. [DOI] [PubMed] [Google Scholar]
- Seeger FH, Zeiher AM, Dimmeler S. Cell-enhancement strategies for the treatment of ischemic heart disease. Nat Clin Pract Cardiovasc Med. 2007b;4:S110–S113. doi: 10.1038/ncpcardio0734. [DOI] [PubMed] [Google Scholar]
- Spiel AO, Gilbert JC, Jilma B. von Willebrand factor in cardiovascular disease: Focus on acute coronary syndromes. Circulation. 2008;117:1449–1459. doi: 10.1161/CIRCULATIONAHA.107.722827. [DOI] [PubMed] [Google Scholar]
- Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28:403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- Wang L, Li Y, Chen J, Gautam SC, Zhang Z, Lu M, Chopp M. Ischemic cerebral tissue and MCP-1 enhance rat bone marrow stromal cell migration in interface culture. Exp Hematol. 2002;30:831–836. doi: 10.1016/s0301-472x(02)00829-9. [DOI] [PubMed] [Google Scholar]
- Wang Y, Johnsen HE, Mortensen S, Bindslev L, Ripa RS, Haack-Sorensen M, Jorgensen E, Fang W, Kastrup J. Changes in circulating mesenchymal stem cells, stem cell homing factor, and vascular growth factors in patients with acute ST elevation myocardial infarction treated with primary percutaneous coronary intervention. Heart. 2006;92:768–774. doi: 10.1136/hrt.2005.069799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Deng Y, Zhou GQ. SDF-1alpha/CXCR4-mediated migration of systemically transplanted bone marrow stromal cells towards ischemic brain lesion in a rat model. Brain Res. 2008;1195:104–112. doi: 10.1016/j.brainres.2007.11.068. [DOI] [PubMed] [Google Scholar]
- Wynn RF, Hart CA, Corradi-Perini C, O’Neill L, Evans CA, Wraith JE, Fairbairn LJ, Bellantuono I. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood. 2004;104:2643–2645. doi: 10.1182/blood-2004-02-0526. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ge J, Sun A, Xu D, Qian J, Lin J, Zhao Y, Hu H, Li Y, Wang K, Zou Y. Comparison of various kinds of bone marrow stem cells for the repair of infarcted myocardium: Single clonally purified non-hematopoietic mesenchymal stem cells serve as a superior source. J Cell Biochem. 2006;99:1132–1147. doi: 10.1002/jcb.20949. [DOI] [PubMed] [Google Scholar]
- Zhang D, Fan GC, Zhou X, Zhao T, Pasha Z, Xu M, Zhu Y, Ashraf M, Wang Y. Over-expression of CXCR4 on mesenchymal stem cells augments myoangiogenesis in the infarcted myocardium. J Mol Cell Cardiol. 2008;44:281–292. doi: 10.1016/j.yjmcc.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]