

Abstract
Chronic lung diseases such as asthma, chronic obstructive pulmonary disease and interstitial lung disease are characterized by inflammation and tissue remodeling processes that compromise pulmonary function. Adenosine is produced in the inflamed and damaged lung where it plays numerous roles in the regulation of inflammation and tissue remodeling. Extracellular adenosine serves as an autocrine and paracrine signaling molecule by engaging cell surface adenosine receptors. Preclinical and cellular studies suggest that adenosine plays an anti-inflammatory role in processes associated with acute lung disease, where activation of the A2AR and A2BR have promising implications for the treatment of these disorders. In contrast, there is growing evidence that adenosine signaling through the A1R, A2BR and A3R may serve pro-inflammatory and tissue remodeling functions in chronic lung diseases. This review discusses the current progress of research efforts and clinical trials aimed at understanding the complexities of this signaling pathway as they pertain to the development of treatment strategies for chronic lung diseases.
Keywords: asthma, fibrosis, emphysema, COPD, G-protein coupled receptors, adenosine receptors, inflammation
1. Introduction
1.1 Chronic lung disease
Chronic lung diseases are characterized by persistent inflammation and tissue remodeling that contribute to a progressive decline in lung function (Sime & O'Reilly, 2001; Thannickal, et al., 2004; Vestbo & Prescott, 1997). Examples of chronic lung disease include asthma, chronic obstructive pulmonary disease (COPD) and interstitial lung diseases exhibiting fibrosis. These disorders are prominent and deadly. Together, they are the third leading cause of death in the United States following cancer and cardiovascular disease. Although therapies are available to treat certain symptomatic features of these disorders, approaches to manage the extensive remodeling seen in the airways of these patients are not available. The causes of chronic lung disease are diverse, as are the inflammatory responses seen. However, a common feature of these disorders is the recruitment and activation of effector cells such as macrophages, neutrophils, eosinophils, fibroblasts, and epithelial cells, which release mediators that promote additional inflammation and/or tissue remodeling (Sime & O'Reilly, 2001; Thannickal et al., 2004; Tillie-Leblond et al., 1999; Vestbo & Prescott, 1997). Little is known about the mechanisms that promote the chronic nature of asthma, COPD and interstitial lung disease, and understanding the regulation of effector cell recruitment and activation will likely provide novel therapeutic approaches for these disorders.
1.2 Aberrant wound healing in chronic lung disease
Excessive extracellular matrix deposition or breakdown, epithelial cell remodeling, and angiogenesis are prominent pathological features seen in chronic lung diseases (Elias et al., 2003; Lynch & Toews, 1998; Thannickal et al., 2004). For example, basement membrane thickening, excessive collagen deposition and angiogenesis around the bronchial airways are prominent features of airway remodeling in asthma, whereas, excessive fibroblast proliferation and extensive extracellular matrix deposition in the alveolar airways are cardinal features of interstitial lung diseases such as idiopathic pulmonary fibrosis. In addition, the destruction of the alveolar airways that is often seen in patients with COPD is associated with imbalances in proteases and anti-proteases that regulate matrix metabolism. The regulation of inflammation, matrix deposition and angiogenesis are all features of a normal wound healing response (Thannickal et al., 2004). The presence of these features in the lungs of patients with chronic lung disease has led to the hypothesis that deregulated or overactive wound healing pathways contribute to the excessive remodeling responses that are seen in chronic lung diseases. Hence, understanding how pathways that serve roles in normal wound healing are over activated in chronic lung disease may provide novel approaches to treat these disorders.
Adenosine is a signaling molecule that is produced following injury and promotes processes important in wound healing and tissue protection such as angiogenesis, matrix production and the regulation of inflammation (Valls et al., 2008). Adenosine levels are elevated in the lungs of animal models (Blackburn et al., 2003; Ma et al., 2006; Volmer et al., 2006) and humans (Driver et al. 1993; Huszar et al., 2002) with chronic lung disease. Accumulating evidence suggest that the generation of adenosine in the injured lung plays important anti-inflammatory roles during acute lung injury, yet promotes tissue remodeling during chronic stages of lung disease. Understanding the specific mechanisms governing the tissue-protective and tissue-destructive properties of adenosine signaling will be critical for advancing the use of adenosine-based therapeutics to treat various pulmonary diseases. The focus of this review will be to discuss recent advances in the understanding of adenosine generation and signaling in cellular and animal models of lung disease as well as in patients with chronic lung disease.
2. Adenosine metabolism and signaling
Adenosine is a nucleoside signaling molecule produced as a result of cell stress or damage. Once present, adenosine signals through one of four G-protein coupled, seven membrane spanning, cell surface receptors (R) (A1R, A2AR, A2BR and A3R) (Fredholm et al., 2001). Engagement of these receptors largely leads to changes in intracellular cAMP and Ca++ levels (Bruns et al., 1986; Gessi et al., 2008; Jin et al., 1997; Ryzhov et al., 2006), though other effects on intracellular signaling pathways have been described. Under physiologic conditions, the downstream effects of engagement of these receptors include regulation of a variety of homeostatic and adaptive functions.
In the absence of pathology, concentrations of adenosine in the extracellular fluid vary from 40-600 nM (Arch & Newsholme, 1978). Under these conditions, adenosine is largely generated as a result of increased intracellular production leading to passive transfer through equilibrative nucleoside transporters (Baldwin et al., 2004). In contrast, in the setting of tissue injury, the predominant source of extracellular adenosine arises from the breakdown of liberated adenine nucleotides (Eltzschig et al., 2003; Volmer et al., 2006). ATP and ADP can be released from various channels present within the cell membrane [CFTR (Reisin et al., 1994), multiple drug resistance (Roman et al., 2001), connexin hemichannels (Cotrina et al., 1998), maxi-ion (Bell et al., 2003), stretch-activated (Braunstein et al., 2001), voltage-dependent anion (Okada et al., 2004)], through the process of vesicle fusion, or from infiltrating inflammatory cells [mast cells (Marquardt et al., 1984), eosinophils (Resnick et al., 1993), neutrophils (Madara et al., 1993)]. These nucleotides are then dephosphorylated to AMP by the ectonucleoside triphosphate diphosphohydrolase CD39 (Kaczmarek et al., 1996) and AMP is subsequently dephosphorylated to adenosine by the ecto-5′-nucleotidase CD73 (Resta et al., 1998). In addition to the release of adenine nucleotides from cells, there is also evidence that adenosine itself can be released from inflammatory mast cells during the degranulation process (Marquardt et al., 1984). This highlights the importance of inflammatory cells as a direct source of adenosine.
In addition to maintaining homeostasis, adenosine also has diverse effects on regulating the immune response in the setting of tissue injury. Acute human pathology such as sepsis or ischemia produces adenosine concentrations as high as 10 μM (Martin et al., 2000). Concentrations reaching 100 μM have also been described in chronic conditions such as arthritis (Sottofattori et al., 2001), asthma and COPD (Driver et al., 1993). Elevated adenosine levels during acute tissue injury generally accesses anti-inflammatory and tissue protective pathways. Conversely, sustained adenosine elevations in persistent inflammatory conditions, particularly chronic lung diseases such as asthma and COPD, may mediate tissue destructive and pro-inflammatory effects (Blackburn, 2003). The effects of adenosine on the processes of inflammation and wound healing have been well-studied and have important implications with regard to the pathogenesis of chronic lung diseases.
3. Adenosine and the regulation of inflammation and wound healing
During acute injury, extracellular adenosine is generated and serves to regulate the acute inflammatory response (Cronstein, 1994; Hasko et al., 2008). In the setting of ischemia (Sperlagh et al., 2000) and sepsis (Jabs et al., 1998), extracellular concentrations of adenosine rise and subsequently signal through specific receptors on inflammatory cells, which affords systemic protection from unnecessary damage to host tissue. Genetic and pharmacologic antagonism studies in ischemia reperfusion injury models pinpoint specific adenosine receptors involved in protection. The A2BR maintains tissue integrity in the heart (Eckle et al., 2007) and kidney (Grenz et al., 2008), while A2AR signaling offers widespread organ protection from ischemia including heart (Yang et al., 2006), lung (Reece et al., 2005), liver (Day et al., 2004), kidney (Day et al., 2003) and spinal cord (Li et al., 2006). Adenosine signaling also has anti-inflammatory effects in models of sepsis. Genetic knockout or antagonism of both the A1 and A3 receptors increase cecal ligation-induced systemic inflammation and mortality (Gallos et al., 2005; Lee et al., 2006). A mouse model of LPS-induced injury also demonstrates A2AR-mediated protective mechanisms (Moore et al., 2008). These findings suggest that adenosine is generated in situations of acute insults to prevent loss of tissue vitality.
Adenosine is also a potent regulator of processes involved in wound healing such as angiogenesis and matrix production. A2AR and A2BR stimulation yields matrix production from fibroblasts and differentiation consistent with wound healing (Chan et al., 2006; Chen et al., 2004; Zhong et al., 2005). Topical application of an A2AR agonist increases the rate of wound healing through production of matrix and angiogenesis (Montesinos et al., 1997). A2AR and A2BR stimulation also lead to increased angiogenesis by increasing endothelial cell proliferation (Feoktistov et al., 2002), production of VEGF (Leibovich et al., 2002), and down-regulation of thrombospondin-1 (Desai et al., 2005), an inhibitor of angiogenesis. In addition, adenosine elicits VEGF production from macrophages (Pinhal-Enfield et al., 2003) and the formation of granulation tissue, evidence of ongoing wound repair, is lacking in A2AR-deficient mice (Victor-Vega et al., 2002).
4. Adenosine signaling in humans with chronic lung disease
Several studies demonstrate elevated adenosine levels in patients with chronic lung disease. Driver and colleagues showed that adenosine levels were elevated in lavage fluid collected from asthmatics where concentrations were estimated to reach 100 μM (Driver et al., 1993). More recent studies have shown elevations in adenosine in the exhaled breath condensate of patients with allergic asthma (Huszar et al., 2002), where levels correlated well with the degree of inflammation as measured by nitric oxide production. Interestingly, plasma levels of adenosine increase by two fold in asthmatic subjects following bronchial provocation with allergen (Mann et al., 1986), and are elevated in the plasma of patients with exercise-induced asthma (Vizi et al., 2002). In addition to asthma, adenosine levels have been found to be elevated in sputum samples from cystic fibrosis patients (Li et al., 2006). To date, elevations in adenosine within the lungs of patients with interstitial lung disease and fibrosis have not been documented, likely due to difficulties in measuring this highly labile molecule. However, adenosine levels are elevated in various mouse models exhibiting features of interstitial lung disease and fibrosis (Blackburn et al., 2003; Ma et al., 2006; Volmer et al., 2006).
Adenosine elevations in the lungs of patients with chronic lung disease suggest that this signaling molecule may regulate aspects of lung disease. Indeed, there is substantial clinical evidence to support the hypothesis that adenosine can directly influence cellular and physiological processes in the lungs of asthmatics and patients with COPD (Fozard & Hannon, 1999). Prominent among these is the ability of inhaled adenosine to induce bronchoconstriction in asthmatics (Cushley et al., 1983) and patients with COPD (Oosterhoff et al., 1993), but not in normal individuals. In asthma, the bronchoconstrictive features of inhaled adenosine are thought to be mediated by the ability of adenosine to promote mast cell degranulation (Cushley et al., 1984). Engagement of the A2BR has been suggested to mediated this response (Holgate, 2005); however, in vivo data documenting this is absent from the literature. Both allergic and non-allergic asthmatics exhibit heightened responses to adenosine, calling into question the necessity of primed mast cells in this response. More research is needed to dissect the mechanisms of heightened adenosine sensitivity in asthmatics. However, the sensitivity of airways to adenosine more closely reflects an inflammatory process and the phenotype for allergic asthma than does sensitivity to other known bronchoprovocative agents such as methacholine and histamine (Holgate, 2002; Spicuzza et al., 2003), which suggests important diagnostic features for adenosine provocation. Although patients with COPD do show some degree of increase in sensitivity to inhaled adenosine, it is not clear whether this is more closely associated with the incidence of smoking in these patients as opposed to direct inflammatory structural changes in the airways of these patients.
The unique bronchial sensitivity of asthmatics and COPD patients to adenosine suggest a fundamental alteration in adenosine receptor signaling in the lungs of these patients. Studies examining adenosine receptor expression in the lungs of patients with chronic lung disease support this hypothesis. A study using tissue obtained at thoracotomy from nonsmoking subjects and subjects with inflammatory airway disorders associated with tobacco smoke or asthma, demonstrated increased expression of the A3R in mesenchymal cells and eosinophils within the lamina propria of the airways and the adventitia of blood vessels (Walker et al., 1997). A recent study conducting immunohistochemical analysis of A1R expression on biopsy sections from asthmatics revealed strong expression of the A1R in the bronchial epithelium and bronchial smooth muscle (Brown et al., 2008). In comparison, weak immunostaining was observed in biopsy specimens obtained from healthy subjects. Analysis of adenosine receptor expression as measured by receptor binding studies revealed that the affinity of the A1R, A2AR, and A3R was significantly decreased in patients with COPD compared with controls, whereas their density was increased. The affinity of A2BR was not altered, but the density was decreased in patients with COPD compared with the control group (Varani et al., 2006). Lastly, a recent study has shown that the A2BR is increased in remodeled airway epithelial cells in rapidly progressing idiopathic pulmonary fibrosis patients (Selman et al., 2007). Hence, adenosine receptor levels are altered in the lungs of patients with chronic lung disease.
5. Adenosine receptor activities in cells relevant to chronic lung disease
Perhaps the most compelling evidence that adenosine signaling may be playing important roles in the regulation of chronic lung disease comes from mechanistic investigations using primary and established cell lines. Adenosine signaling can influence the activity of many cell types that play central roles in chronic lung disease, including mast cells, eosinophils, neutrophils, macrophages, fibroblasts, neurons, lymphocytes, epithelial and smooth muscle cells.
5.1 Mast cells
Mast cells are bone marrow-derived inflammatory cells that can release mediators that have both immediate and chronic effects on airway constriction and inflammation (Prussin & Metcalfe, 2003). Upon stimulation, mast cells rapidly release preformed mediators such as histamine and tryptase, which are stored inside secretory granules. The first correlation between adenosine and mast cells was observed in 1978 when Marquardt et al. reported that adenosine, although ineffective alone, potentiated histamine release induced by anti-immunoglobulin E, concanavalin A, compound 40/80, and by the calcium ionophore A23187 in isolated rat mast cells (Marquardt et al., 1978). Adenosine was subsequently shown to modulate histamine release in human lung mast cells (Hughes et al., 1984). The effect of adenosine was also examined in lavage mast cells challenged with adenosine or adenosine receptor agonists both of which were shown to induce histamine release (Forsythe et al., 1999), supporting an indirect mechanism for adenosine-induced bronchoconstriction in asthma. In addition to promoting degranulation, adenosine can also stimulate the production of mediators that can influence chronic aspects of disease, including IL-8, IL-4, IL-13 and VEGF (Feoktistov et al., 2001; Ryzhov et al., 2006; Ryzhov et al., 2004; Ryzhov, et al., 2008a). Hence, adenosine can elicit responses from mast cells that impact acute and chronic features of lung disease.
Pharmacologic and genetic studies have been utilized to examine the specific adenosine receptor subtypes involved in adenosine-mediated mast cell degranulation and the expression of inflammatory mediators. In rodents, several studies have shown that the A3R is responsible for the adenosine-mediated degranulation of mast cells (Reeves et al., 1997; Salvatore et al., 2000; Zhong et al., 2003). In contrast, experiments using canine mast cells suggest that the A2BR mediates mast cell degranulation (Auchampach et al., 1997), and it has been suggested that the same may be true in human mast cells (Holgate, 2005); however, clear evidence linking the A2BR to the degranulation of human mast cells, particularly in lung mast cells, is absent in the literature. Recent studies in A2BR knockout mice demonstrate that the A2BR is not responsible for mast cell degranulation; in fact this receptor appears to limit the responsiveness of mouse mast cells to antigen (Hua et al., 2007). However, studies in mast cells isolated from A2BR knockout mice suggest that this receptor is responsible for the adenosine-elicited production of IL-13 and VEGF from mouse mast cells (Ryzhov et al., 2008). Thus, in mice, it appears that the A3R regulates adenosine-mediated mast cell degranulation while the A2BR is responsible for adenosine-mediated expression of factors that can influence features of chronic lung disease (Figure 1). It remains to be determined whether this holds true in human mast cells, particularly those found in the lungs of asthmatics and other patients with chronic lung disease.
Figure 1.
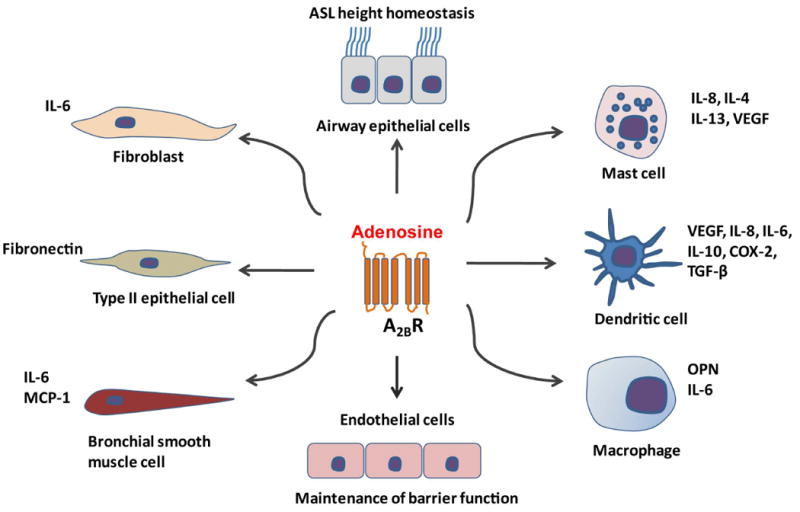
A2BR activities in cell types involved in chronic lung disease. Adenosine signaling through the A2BR can stimulate the production of IL-8, IL-4, IL-13 and VEGF from mast cells. A2BR signaling is an important factor of aberrant dendritic cell differentiation and generation of tolerogenic, angiogenic, and pro-inflammatory cells that produce VEGF, IL-8, IL-6, IL-10, COX-2 and TGF-β. In addition, A2BR engagement can promote the production of IL-6 and osteopontin (OPN) from macrophages; IL-6 and MCP-1 from bronchial smooth muscles; increases in IL-6 release from fibroblasts; induces myofibroblasts differentiation; and induces the expression of fibronectin in type II lung epithelial cells. A2BR signaling also contributes to the maintenance of ASL height in airway epithelial cells and vascular barrier function in endothelial cells.
5.2 Eosinophils
Eosinophils are closely associated with many allergic diseases including asthma, where they infiltrate tissues and release mediators that cause tissue damage and inflammation (Gleich, 1990). Using purified eosinophils, RT-PCR showed that the A3R is abundantly expressed on both human (Kohno et al., 1996) and mouse eosinophils (Young et al., 2004). This is supported by findings demonstrating A3R expression on eosinophils within the airways of patients with chronic lung disease (Walker et al., 1997) and the lungs of mice with allergic lung inflammation (Young et al., 2004). Studies on isolated human (Knight et al., 1997; Walker et al., 1997) and mouse (Young et al., 2004) eosinophils suggest that A3R activation inhibits chemokine-induced migration. Furthermore, A3R activation can inhibit eosinophil degranulation and superoxide anion release (Ezeamuzie & Philips, 1999). These findings suggest the A3R may serve an anti-inflammatory role; however, genetic removal or pharmacological blockade of the A3R in the ADA-deficient mouse model of adenosine-mediated lung disease resulted in decreased airway eosinophilia while not affecting the degree of circulating eosinophilia (Young et al., 2004), suggesting A3R signaling is important in eosinophil trafficking in this model. Moreover, a recent study demonstrated the absence of eosinophil peroxidase levels in the airways of A3R-deficient mice exposed to bleomycin (Morschl et al., 2008), suggesting that this receptor is needed for eosinophil degranulation in vivo. The discrepancies between findings in human cellular systems and those in animal models might be due to species differences or the inability to adequately assess the function of the A3R on eosinophils in vivo in humans.
5.3 Other immune cells
Other key immune cells associated with chronic lung diseases include lymphocytes, neutrophils, dentritic cells and macrophages. These cells all express adenosine receptors and have been implicated in regulating various aspects of both innate and adaptive immunity (Hasko & Cronstein, 2004; Hasko et al., 2008). Direct analysis of adenosine receptor signaling on these cells in the context of chronic lung disease are few; however, investigations into the functions of these receptors in the context of other diseases have provided evidence that adenosine signaling on these cells likely plays an important role in these disorders (reviewed in (Hasko et al., 2008)). For example, signaling through the A2AR can promote anti-inflammatory activities on dendritic cells (Panther et al., 2003; Schnurr et al., 2004), various lymphocyte populations including T regulatory cells (Deaglio et al., 2007; Lappas et al., 2005; Naganuma et al., 2006), macrophages (Hasko et al., 2000) and neutrophils (Cronstein et al., 1983). These observations suggest that activation of the A2AR on these cells could serve anti-inflammatory roles in chronic lung disease. Preclinical studies have validated this notion (see below) (Fozard et al., 2002). In contrast, there is evidence that signaling through the A3R (Morschl et al., 2008) and A2BR (Ryzhov et al., 2008b; Sun et al., 2006) can promote the production of mediators from macrophages that play a role in the progression of chronic lung disease (Figure 1).
5.4 Pulmonary cells
Bronchial smooth muscle cells contribute to the pathophysiologic changes that occur in asthma (Hirst, 2000). The A2BR is expressed in cultured human airway smooth muscle cells (Mundell et al., 2001). Adenosine, via engagement of the A2BR, increases the release of IL-6 and monocyte chemotactic protein-1 from bronchial smooth muscle cells (Zhong et al., 2004) providing a mechanism whereby adenosine acts as a pro-inflammatory mediator in the airway (Figure 1). More recently, Ethier et al. found that the A1R mediates mobilization of calcium in human bronchial smooth muscle cells, suggesting that adenosine has direct effects on contractile signaling pathways (Ethier & Madison, 2006) and suggesting a mechanism for bronchoconstriction.
Chronic lung diseases are associated with activation of subepithelial fibroblasts and differentiation of fibroblasts to myofibroblasts (Roche, 1991). Activated fibroblasts and myofibroblasts play a key role in airway remodeling by producing extracellular matrix components and releasing pro-inflammatory cytokines and chemokines. Activation of the A2BR on human lung fibroblasts increases the release of IL-6 and induces differentiation into myofibroblasts suggesting that adenosine, via A2BR participates in the remodeling process occurring in chronic lung diseases (Figure 1) (Zhong et al., 2005).
In human airway epithelial cells, adenosine signaling regulates a variety of physiologic activities including ion channels, mucin secretion, mucociliary mobility and clearance, airway surface liquid (ASL) height and matrix production. A2BR activation stimulates cystic fibrosis transmembrane conductance regulator (CFTR) activity and Cl- secretion in Calu-3 human airway epithelial cells (Cobb et al., 2003; Huang et al., 2001). In addition, the ATP-induced stimulation of Na+ absorption in a human bronchiolar epithelial cell line is mediated by A2AR and A2BR activation (Chambers et al., 2006). A2BR is the major adenosine receptor subtype detected in cultured primary human bronchial epithelial cells, and inhibition of this receptor with specific A2BR antagonists results in ASL height collapse and a failure to effect ASL height homeostasis (Rollins et al., 2008). In the alveolar airway epithelium, the A1R plays an important role in fluid clearance (Factor et al., 2007). Together these findings suggest that adenosine engagement of various receptors plays important roles in key physiological processes in the airway epithelium.
Cellular systems have also been used to define potential roles for adenosine receptor activation in injury repair and pathological processes that involve the airway epithelium. Adenosine signaling through the A2AR can promote epithelial migration in an in vitro wound healing assay (Allen-Gipson et al., 2007; Allen-Gipson et al. 2006). By engaging the A1R, adenosine can activate a Ca2+-activated Cl- channel to induce MUC2 mucin gene expression in human bronchial epithelial cells (McNamara et al., 2004), a process that may have important implications in asthma. In addition, adenosine can induce the expression of fibronectin in A549 lung epithelial cells through the A2BR (Roman et al., 2006), which has important implications in lung injury and remodeling situations that involve fibrosis (Figure 1). Finally, Sun et al. found that in Calu-3 cells, apical adenosine elicited robust release of IL-6 and IL-8, suggesting a pro-inflammatory role for adenosine (Sun et al., 2008)
6. Adenosine signaling in animal models of chronic lung disease
6.1 Adenosine deaminase deficient mice as a model of adenosine mediated lung disease
The adenosine deaminase deficient (ADA-deficient) mouse is an established model designed to study the consequences of adenosine elevations in vivo (Blackburn, 2003). ADA is an enzyme responsible for the degradation of adenosine and deoxyadenosine. Consequently, genetic removal of this enzyme results in profound elevations in systemic concentrations of these substrates, which results in multi-organ pathology (Blackburn et al. 1998; Blackburn et al., 2000a). Accumulations of deoxyadenosine in the thymus and spleen of ADA-deficient mice result in a severe combined immunodeficiency (Blackburn et al., 2000b; Blackburn et al. 1998), while adenosine accumulations are associated with the development of pulmonary inflammation and changes in lung architecture that resemble various aspects of human chronic lung disease (Blackburn et al., 2000a). Findings consistent with asthma include mast cell degranulation, eosinophilia, mucous cell metaplasia and airway hyperreactivity (Blackburn et al., 2000a; Chunn, et al., 2001; Zhong et al., 2001). Features consistent with COPD include the influx and activation of neutrophils and alveolar macrophages, and alveolar airway enlargement (Blackburn et al., 2000a; Sun et al., 2006). In addition, there is an up-regulation of cytokines and mediators commonly elevated in association with asthma and COPD including serum IgE and TH2 cytokines (Blackburn et al., 2000a). Similarly, ADA-deficient mice and patients with COPD exhibit increases in cytokines and chemokines that recruit neutrophils and macrophages to the airways (Banerjee et al., 2002; Blackburn et al., 2000a). Both also show up-regulations in mediators of airway enlargement such as matrix metalloproteases and cathepsins (Banerjee et al., 2002; Blackburn et al., 2000a). The observation that adenosine levels outside the normal physiologic range accompany pathologic changes suggests that adenosine is a driving force of these disease processes.
Studies utilizing ADA enzyme replacement therapy to lower systemic adenosine levels support the claim that features of the pulmonary phenotype are directly mediated by adenosine. Administration of ADA covalently linked to polyethylene glycol (PEG-ADA) to ADA-deficient mice will prevent and reverse many aspects of the pulmonary phenotype and indefinitely maintain the life of the animal (Blackburn et al., 2000a; Blackburn et al., 2000b). These findings display a clear correlation between adenosine levels, the presence of lung pathology, and its contribution to the overall morbidity in this model.
The use of PEG-ADA also enables the study of pulmonary fibrosis in the ADA-deficient model (Chunn et al., 2006; Chunn et al., 2005). ADA-deficient mice without PEG-ADA will die from presumed respiratory complications at three weeks of age before fibrosis develops. By utilizing a tapered regimen of PEG-ADA, adenosine levels gradually increase and the lifespan of the mouse is prolonged allowing for the development of pulmonary fibrosis. Features include increased alveolar and peribronchial myofibroblasts accumulation and collagen deposition (Chunn et al., 2005). Concurrently, measurable increases in soluble collagen are also present in the airways. Finally, lungs of these mice exhibit increases in known pro-fibrotic mediators including TGF-β1, IL-1β, MMP-2, IL-13, Pai-1, and osteopontin (OPN). Along with histological findings, these results show that adenosine signaling can access pro-fibrotic pathways contributing to the overall chronic pulmonary disease phenotype these mice display.
As with the asthma and COPD-like features, characteristics of pulmonary fibrosis can be prevented and reversed by maintaining low adenosine levels with PEG-ADA (Chunn et al., 2005). Overall, adenosine levels clearly correlate with many chronic lung disease features in ADA-deficient mice and these studies highlight the importance of understanding the role of adenosine in driving the chronic nature of these disease processes.
Genetic and pharmacologic studies in the ADA-deficient model suggest that adenosine signaling through specific receptors may directly influence features of chronic lung disease. A3R levels are elevated in the lungs of ADA-deficient mice where expression is localized to mast cells (Zhong et al., 2003), eosinophils and mucin producing bronchial airway epithelial cells (Young et al., 2004). Genetic removal or pharmacological blockade of the A3R results in the attenuation of mast cell degranulation (Zhong et al., 2003), lavage eosinophilia and mucous cell metaplasia (Young et al., 2004), suggesting A3R signaling plays an important role in the ability of adenosine elevations to influence these important features of chronic lung disease.
The A2BR is also elevated in the lungs of ADA-deficient mice (Chunn et al. 2001; Sun et al., 2005) and treatment of these mice with a selective antagonist of the A2BR, CVT-6883, during active stages of disease resulted in the attenuation of key features of chronic lung disease (Sun et al., 2006). CVT-6883 treatment resulted in reduced neutrophil and macrophage influx into the lungs of ADA-deficient mice in association with diminished cytokine and chemokine levels. A2BR antagonism also prevented alveolar airway destruction in conjunction with the regulation of matrix metalloprotease levels. In addition, CVT-6883 treatment resulted in an attenuation of pulmonary fibrosis in ADA-deficient mice, which was associated with diminished production of fibrotic mediators such as IL-6, TGF-β1 and osteopontin (Sun et al., 2006). These findings demonstrate that many of the pathological features seen in the lungs of ADA-deficient mice are mediated through activation of the A2BR.
In contrast to the pro-inflammatory and pathological features attributed to the A3R and A2BR, genetic studies revealed a tissue protective role for the A1R and A2AR in the pulmonary disease seen in ADA-deficient mice. Specifically, genetic knockout of the A1R (Sun et al., 2005) and A2AR (Mohsenin et al., 2007) in ADA-deficient mice leads to enhanced pulmonary inflammation, mucous cell metaplasia and airway damage in association with enhanced production of pro-inflammatory cytokines.
Taken together, these results suggest that the A3R and A2BR are playing a pathologic role, while the A1R and A2AR play a tissue protective role (Figure 2). It is interesting to note that the A1R and A2AR have a relatively high affinity for adenosine, while the A2BR and A3R require higher levels of adenosine for activation (Fredholm, 2007). This suggests that the A2BR and A3R may have increased activity under pathological conditions where adenosine levels are elevated. The findings in the ADA-deficient model contribute to the school of thought that selective adenosine receptor agonists and antagonists will be beneficial in the treatment of chronic lung diseases. This underscores the importance of understanding the effects of individual adenosine receptor signaling pathways in other models of lung disease and in humans in the context of elevated adenosine levels.
Figure 2.
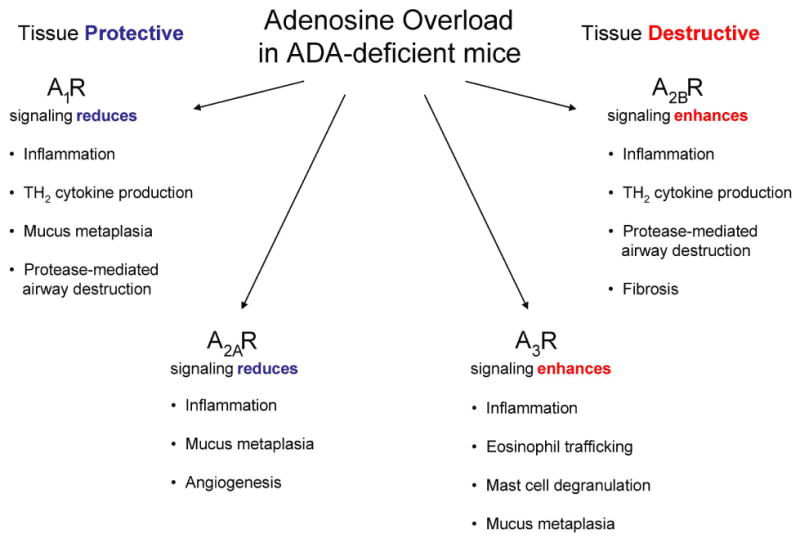
Adenosine receptor activities in the lungs of ADA-deficient mice. Genetic and pharmacologic approaches in the ADA-deficient model suggest that adenosine receptor signaling influences both tissue protective and tissue destructive pathways in the lungs.
6.2 Models of Th2-driven and allergic lung disease
Elaboration of Th2 cytokines is seen in lung disease where there is excessive tissue remodeling, particularly asthma and interstitial lung disease where fibrosis is present (Elias et al., 2003; Vestbo & Prescott, 1997). In particular, IL-13 and IL-4 have been shown to be critical inducers of pathways that lead to remodeling in the lung (Elias et al., 2003). Studies in transgenic mice suggest that adenosine signaling may contribute to the Th2-induced remodeling in the lung. Adenosine levels are substantially elevated in the lungs of mice exhibiting lung inflammation and damage driven by the over expression of either IL-13 (Blackburn et al., 2003) or IL-4 (Ma et al., 2006). Interestingly, mRNA and enzymatic activity of ADA was down regulated in the lungs of these mice, suggesting Th2 cytokines can promote an environment that is conducive to adenosine elevations. Remarkably, treatment of mice over expressing IL-13 or IL-4 with PEG-ADA to lower adenosine levels resulted in the attenuation of pulmonary inflammation and key features of airway remodeling including mucous cell metaplasia and the deposition of collagen in the airways (Blackburn et al., 2003; Ma et al., 2006). Interestingly, levels of the A3R and A2BR were elevated in the lungs of these mice similar to what was seen in ADA-deficient mice; however, the function of these receptors has not been tested in this system. These findings suggest that adenosine signaling plays a role in mediating aspects of Th2-induced pulmonary inflammation and damage.
Animal models of allergic lung inflammation that are associated with Th2 inflammation have been used to assess the role of specific adenosine receptors in processes associated with asthma. Aerosolized exposure of adenosine in mice sensitized and challenged with ragweed resulted in increased airway resistance that could be reversed by treatment with theophylline, a broad spectrum adenosine receptor antagonist (Fan & Mustafa, 2002). Similar findings were found in Brown Norway rats sensitized and challenged with ovalbumin (Fozard & Hannon, 2000), and in isolated parenchymal strips from the lungs of sensitized rats (Wolber & Fozard, 2005). These findings suggest that adenosine-induced bronchoconstriction is mediated by adenosine receptors. Mast cell degranulation appears to play an important role in adenosine-induced bronchoconstriction in both models (Hannon et al., 2001; Oldenburg & Mustafa, 2005).
A rabbit model has been utilized to assess the role of the A1R in allergic lung disease. Treatment with A1R antagonists (Ali et al., 1994) and antisense oligodeoxynucleotides (Nyce, et al., 1997) against the A1R attenuated bronchoconstrictor responses in this model, and a more recent study demonstrates that treatment with the A1R antagonist L-97-1 can attenuate pulmonary inflammation as well as bronchoconstriction (Nadeem et al., 2006). The site and mechanism of action of A1R blockade has not been identified, but these studies suggest there may be clinical benefit in the use of A1R antagonists in asthma.
Substantial evidence suggests that the A2AR mediates anti-inflammatory activities on numerous cells, implying A2AR agonists may have benefit in attenuating inflammation in many disorders (for review see reference (Hasko et al., 2008; Hasko & Pacher, 2008)). Given the central role of inflammation in asthma and COPD, preclinical studies have pursued the use of A2AR agonists in models of allergic airway inflammation. Fozard and colleagues demonstrated that treatment with the A2AR agonist CGS21680 inhibits airway inflammation in allergen sensitized and challenged Brown Norway rats (Fozard et al., 2002). Similar findings were reported in a mouse model of ovalbumin sensitization and challenge where CGS21680 treatment inhibited inflammatory cell recruitment in bronchoalveolar lavage fluid (Bonneau et al., 2006). This study also demonstrated mixed benefits of A2AR agonist treatment on airway inflammation seen following LPS or cigarette smoke exposure. Consistent with these findings, A2AR knockout mice sensitized and challenged with ragweed exhibit enhanced airway inflammation and airway hyperresponsiveness (Nadeem et al., 2007). These findings suggest that treatment with A2AR agonists may have benefit in the treatment of asthma and/or COPD.
As discussed earlier, the activation of the A2BR can promote the production of inflammatory mediators associated with chronic lung disease suggesting that A2BR antagonists might be useful in the treatment chronic lung diseases including allergic lung diseases such as asthma. Recent preclinical data in a mouse model of allergic lung inflammation support this hypothesis. Treatment with the A2BR antagonist CVT-6883 attenuated airway inflammation and airway hyperreactivity in a mouse model of ragweed sensitization and challenge (Mustafa et al., 2007). This supports earlier studies using sensitized Brown Norway rats that implicated the A2BR in adenosine-induced airway hyperreactivity (Fozard & Hannon, 2000; Hannon et al., 2002). Contrary to these findings, a study using tracheas isolated from guinea pigs suggest that the A2BR may mediate smooth muscle relaxation (Breschi et al., 2007). The analysis of A2BR knockout mice in models of allergic lung disease have not emerged in the literature. Such studies will provide important preclinical information into the usefulness of A2BR as a target in asthma.
Pharmacological studies also support a role for the A3R in animal models of allergic lung inflammation. A study in sensitized guinea pigs demonstrated that MRS-1220, an A3R antagonist could attenuate the influx of eosinophils and macrophages into the lungs following AMP challenge (Spruntulis & Broadley, 2001). Interestingly, treatment with MRS-1220 did not attenuate airway hyperreactivity in this model. In mice sensitized and challenged with ragweed, the A3R agonist, Cl-IB-MECA induced airway hyperreactivity that was blocked by the A3R antagonist MRS-1523 (Fan et al., 2003). In this study, MRS-1523 did not block the airway hyperreactivity to NECA that was induced by allergen, suggesting the involvement of additional adenosine receptors. Treatment with the A2BR antagonist enpropfylline together with MRS-1523 completely blocked hyperreactivity to NECA induced by allergen in this model suggesting a role for both A3R and A2BR (Fan et al., 2003). Similar findings have been characterized in the Brown Norway rat model (Hannon et al., 2002; Wolber & Fozard, 2005). Evidence also exists suggesting the A3R mediates mucin secretion in mice with allergic lung disease. The A3R is markedly elevated in mucin producing bronchial airway epithelial cells in several models of Th2-mediated airway inflammation (Blackburn et al., 2003; Ma et al., 2006; Young et al., 2004). Ovalbumin sensitized and challenged mice over expressing a transgene for the A3R in the bronchial epithelium exhibit enhanced mucin secretion following A3R agonist stimulation, whereas A3R-deficient mice fail to demonstrate mucous secretion follow A3R agonist exposure (Young et al., 2006). These studies suggest that A3R antagonists can attenuate features of asthma in animal models. Whether this will translate into similar effects in human disease is questionable given species differences in expression of this receptor.
Collectively, preclinical assessment of adenosine receptors in models of Th2-driven and allergen-driven lung disease suggest that adenosine is serving pro-inflammatory roles. There is evidence to support the use of antagonists to the A1R, A2BR and A3R, while some studies support the use of A2AR agonists in these models. Though logical and compelling in nature, continued efforts are needed to understand the mechanisms underlying the effects of specific adenosine receptor engagement in these models, and more importantly, efforts to examine the function of these receptors in humans with chronic lung disease.
6.3 Acute lung injury models
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are life-threatening disorders that can develop in the course of different clinical conditions such as pneumonia, acid aspiration, major trauma, or prolonged mechanical ventilation, and contribute significantly to critical illness (Ware et al., 2001). Despite the large impact of ALI on morbidity and mortality in critically ill patients, many episodes of ALI are self-limiting, and resolve spontaneously through unknown mechanisms. Whereas adenosine appears to play a largely detrimental role in the regulation of chronic lung disease, there is substantial evidence to suggest that extracellular adenosine signaling trough its receptors plays important anti-inflammatory roles in ALI.
Experimental and clinical studies have demonstrated that the pathogenesis of Th1 inflammation and ALI are characterized by pulmonary trafficking of neutrophils into the lungs (Abraham et al., 2000). Pulmonary exposure to lipopolysaccharide (LPS) is a useful model for the assessment of neutrophil trafficking in ALI (Reutershan et al., 2006). The main source of extracellular adenosine stems from a coordinated two-step enzymatic conversion of precursor nucleotides via CD39 and CD73. A recent study by Reutershan and colleagues demonstrated that CD39 and CD73 levels are elevated in the lungs following LPS exposure (Reutershan et al., 2008). Furthermore, LPS-induced accumulation of neutrophils in the lungs was enhanced in mice treated with a nonspecific ecto-nucleosidetriphosphate diphosphohydrolases inhibitor and in CD39 and CD73-deficient mice (Reutershan et al., 2008). These increases in neutrophil trafficking were accompanied by enhanced alveolar-capillary leakage and lung damage. These findings demonstrate that extracellular adenosine plays important anti-inflammatory functions in LPS-induced lung injury. Pharmacologic and genetic studies have identified the A2AR as a major signaling pathway in mediating the anti-inflammatory properties of adenosine in LPS-induced lung injury (Reutershan et al., 2007). A recent study demonstrated that inhalation of the selective A2AR agonist ATL202 reduced LPS-induced neutrophil migration, microvascular permeability, and chemokine release, suggesting a possible therapeutic approach for the treatment of ALI (Reutershan et al., 2007).
Activation of the A2AR has also been shown to limit the inflammatory response to ischemia-reperfusion injury in multiple different organ systems, including spinal cord, renal, and cardiac models (Cargnoni et al., 1999; Okusa et al., 1999; Reece et al., 2005; Reece et al., 2004). Consistent with these findings, A2AR agonists have demonstrated significant attenuation of ischemia-reperfusion injury in lung transplantation models. In an isolated, whole blood-perfused, ventilated rabbit lung model, A2AR activation blocked the neutrophil-mediated inflammatory response and reduced lung reperfusion injury following transplantation (Ross et al., 1999). Using mature pig lungs, A2AR activation during early reperfusion attenuated lung inflammation and preserved pulmonary function (Reece et al., 2005). In non-transplantation lung reperfusion injury models, A2AR agonists attenuated injury (Ellman et al., 2008), and a similar protective role was observed in a cardiopulmonary bypass-induced lung injury model (Lisle et al., 2008). These studies demonstrate that engagement of the A2AR can impart potent anti-inflammatory pathways in ischemia-reperfusion injury in the lung.
Anti-inflammatory pathways in ischemia-reperfusion injury in the lung have also been described for the A3R (Rivo et al., 2004), where A3R agonist treatment conferred protection against reperfusion lung injury in association with enhanced ERK activation and decreased apoptosis (Matot et al., 2006; Rivo et al., 2004). The role of A1R signaling in lung ischemia-reperfusion injury remains controversial. Inhibition of the A1R has been reported to prevent ischemia-reperfusion injury of the lung (Neely & Keith, 1995), while activation of A1Rs have been suggested to mediate the protective properties of ischemic preconditioning and adenosine preconditioning on ischemia-reperfusion injury in the lung (Yildiz et al., 2007).
Ventilator-induced lung injury is another model of ALI. CD39 and CD73 are induced during ventilator-induced lung injury and mediate the production of adenosine (Eckle et al., 2007). Furthermore, pharmacological and genetic approaches demonstrated that the A2BR mediates protective effects of adenosine in this model. In wild type mice, treatment with an A2BR-selective antagonist resulted in enhanced pulmonary inflammation, edema, and attenuated gas exchange, while an A2BR agonist attenuated injury. Deletion of the A2BR gene was associated with reduced survival time and increased pulmonary albumin leakage after injury. Moreover, measurement of alveolar fluid clearance demonstrated that A2BR signaling enhanced amiloride-sensitive fluid transport and elevation of pulmonary cAMP levels following injury, suggesting that A2BR agonist treatment protects by promoting fluid regulation in the lung (Eckle et al., 2008). Similar findings have been demonstrated in mice exposed to hypoxic conditions (Eckle et al., 2008). Collectively, these studies suggest that adenosine signaling through the A2BR plays important anti-inflammatory and tissue protective roles in ALI.
7. Differences in chronic and acute lung injuries
Observations in cellular and animal models suggest that adenosine generation and its subsequent signaling through adenosine receptors serves anti-inflammatory and tissue protective roles in ALI, whereas adenosine can serve pro-inflammatory and tissue destructive roles during chronic stages of disease. The specific mechanisms behind these differences are still not clear, but likely relate to the difference in cytokine and mediator environments inherent to these different diseases. Acute lung injuries are associated with a largely Th1 dominated environment, where elevations in IL-12, IFNγ and TNFα play prominent roles in the progression of inflammation and tissue damage. In contrast, chronic lung diseases such as asthma, certain classifications of COPD, and interstitial lung diseases exhibiting pulmonary fibrosis are associated with elevations in Th2 cytokines such as IL-4, IL-5 and IL-13 (Elias et al., 2003). Adenosine signaling serves to down regulate the production of Th1 cytokines (Hasko et al., 2008). This likely represents a major mechanism whereby adenosine exerts its anti-inflammatory effects in ALI. On the contrary, adenosine acting largely through the low affinity A2BR can increase the production of Th2 cytokines (Feoktistov and Biaggioni, 1995; Ryzhov et al., 2004; Ryzhov et al., 2008a; Ryzhov, et al., 2008b), providing a mechanism whereby adenosine contributes to the amplification or progression of chronic lung disease. Interestingly, the A2AR can down regulate the production of IL-12 (Hasko et al., 2000), which in turn promotes the development of a Th2 cytokine environment, suggesting a mechanism for adenosine-mediated transition from an acute to chronic disease phenotype. These studies suggest that the cytokine environment may play an important role in orchestrating the actions of adenosine in the lung (Figure 3).
Figure 3.

Relationship between adenosine and cytokines. Pulmonary adenosine levels increase in the lung following injury where subsequent engagement of adenosine receptors can influence aspects of inflammation, tissue protection and pathological remodeling. In acute lung injury, the A2AR and A2BR serve important anti-inflammatory and tissues protective roles, central to which are the inhibition of Th1 cytokine production and the promotion of endothelial barrier function. In chronic lung disease, the A2BR, A1R and A3R contribute to the progression of disease by increasing Th2 cytokine production and activating effector cells such as mast cells, macrophages and fibroblasts that can drive disease progression. Alterations in the expression levels of enzymes of adenosine metabolism (CD73, ADA), transport proteins (ENT) or adenosine receptors (ADA), a process known as purinergic remodeling, may influence the progression of acute and chronic lung diseases.
The notion that the local cytokine milieu plays an important role in regulating the anti- or pro-inflammatory actions of adenosine is supported further by studies demonstrating that cytokines can regulate the levels of adenosine metabolic enzymes and adenosine receptors in inflammatory environments. Cytokines such as TNFα (Nguyen et al., 2003) and INFγ (Xaus et al., 1999) can promote the expression of adenosine receptors on cells. Moreover, over expression of the Th2 cytokines in the lungs is associated with increased levels of the A1R, A2BR and the A3R (Blackburn et al., 2003; Ma et al., 2006). These studies also demonstrated that increases in Th2 cytokines in the lung were associated with a tissue-specific down regulation of ADA. These findings suggest that Th2 cytokines can promote both an environment for the accumulation of adenosine and enhance adenosine signaling. This purinergic remodeling could provide an important avenue for the promotion of chronic lung diseases (Figure 3).
Disruption of endothelial and epithelial barrier function is a component of ALI (Eckle et al., 2008). As mentioned above, the generation of extracellular adenosine (Eltzschig et al., 2003) and subsequent signaling through the A2BR (Eckle et al., 2008; Eckle et al., 2008) plays an important role in the maintenance of pulmonary barrier function (Figure 1). In this sense, the A2BR plays an anti-inflammatory role. Consistent with this, a recent study has demonstrated that genetic removal of the A2BR from ADA-deficient mice leads to enhanced inflammation and damage in association with enhanced loss of pulmonary barrier function (Zhou et al., 2009). Thus, although antagonism of the A2BR plays a role in regulating chronic aspects of pulmonary disease (Sun et al., 2006), it also plays important protective roles in acute stages of injury. Therefore, it will be important to monitor the specific stage of disease in order to determine whether to treat with A2BR agonist or antagonist. In addition, care should be taken not to provide exposures to A2BR antagonist that will completely block activity. Collectively, these findings emphasize the important distinction that adenosine signaling has both anti- and proinflammatory features that are disease and disease stage specific.
8. Adenosine-based therapies for the treatment of chronic lung disease
One hypothesis that can be put forth from the models presented above is that chronic lung diseases such as asthma, COPD and pulmonary fibrosis may benefit from treatment with agonist that stimulate the A2AR, or therapeutic approaches to antagonize the A1R, A3R or A2BR. Indeed, there is substantial preclinical data to support this hypothesis. In addition, pharmaceutical and biotechnology companies are developing compounds with high selectivity towards the adenosine receptors and clinical trials assessing the safety and efficacy of adenosine-based therapeutics have begun to emerge. A recent study examined the efficacy of inhalation of the A2AR agonist, GW328267X, in atopic asthmatics (Luijk et al., 2008). In this study, the A2AR agonist failed to protect against allergen-induced early or late asthmatic reactions, or the associated inflammatory response in the induced sputum. In another study, the dual A2BR/A3R antagonist, QAF 805, failed to attenuate bronchial hyperresponisveness to inhaled AMP in AMP-sensitive asthmatics (Pascoe et al., 2007). Although neither GW328267X of QAF 805 showed impressive efficacy, the studies were limited by concerns over off target hemodynamic affects that limited the use of higher doses that were likely necessary for optimal effectiveness. The selective A2BR antagonist, CVT 6883 has completed Phase 1 clinical trials with no adverse events reported (Kalla & Zablocki, 2009); however, efficacy in phase II trials has not yet been shown. In addition, a 21-mer antisense oligodeoxynucleotide targeting the human A1R (Ball et al., 2003), EPI-2020 has shown some efficacy in a small asthma trial, but failed to show benefit in a phase II trial (Langley et al., 2005). Thus, the current status of the use of adenosine-based therapeutics for the treatment of at least asthma has not fared well to date. However, many of the trials were focused on a small number of asthma patients and other chronic lung diseases have not been assessed. Additional clinical trials using larger asthma patient cohorts and extending trials to other chronic lung disorders are clearly needed to move the field of adenosine-based therapeutics forward.
9. Conclusion
Lung injury and remodeling is a complex process that is altered in pulmonary diseases. Adenosine is produced in response to injury to help orchestrate tissue protection and the initiation of wound healing. Consistent with this, preclinical and cellular studies suggest that adenosine plays an anti-inflammatory role in processes associated with ALI, where activation of the A2AR and A2BR have promising implications for the treatment of acute lung injuries. In contrast, there is growing evidence that adenosine signaling through the A1R, A2BR and A3R serves pro-inflammatory and tissue remodeling functions in chronic lung diseases such as asthma, COPD and pulmonary fibrosis. Additional studies in models are needed to understand the specific mechanisms involved in adenosine signaling in various pulmonary disorders. Moreover, larger and better controlled clinical trials are needed to assess the efficacy of new generations of adenosine receptor agonists and antagonists in the treatment of acute and chronic lung diseases.
Acknowledgments
Work in the laboratory of M. R. Blackburn is supported by NIH grants AI43572, HL70952 and HL095403 and funding from Battelle Memorial Institute. D. J. Schneider was funded by Award Number TL1RR024147 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.”
Abbreviations
- COPD
chronic obstructive pulmonary disease
- R
adenosine receptor
- ADA
adenosine deaminase
- LPS
lipopolysaccharide
- VEGF
vascular endothelial growth factor
- IL
interleukin
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- ASL
airway surface liquid
- CFTR
cystic fibrosis transmembrane conductance regulator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1137–1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- Ali S, Mustafa SJ, Metzger WJ. Adenosine-induced bronchoconstriction and contraction of airway smooth muscle from allergic rabbits with late-phase airway obstruction: evidence for an inducible adenosine A1 receptor. J Pharmacol Exp Ther. 1994;268:1328–1334. [PubMed] [Google Scholar]
- Allen-Gipson DS, Spurzem K, Kolm N, Spurzem JR, Wyatt TA. Adenosine promotion of cellular migration in bronchial epithelial cells is mediated by the activation of cyclic adenosine monophosphate-dependent protein kinase A. J Investig Med. 2007;55:378–385. doi: 10.2310/6650.2007.00019. [DOI] [PubMed] [Google Scholar]
- Allen-Gipson DS, Wong J, Spurzem JR, Sisson JH, Wyatt TA. Adenosine A2A receptors promote adenosine-stimulated wound healing in bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L849–855. doi: 10.1152/ajplung.00373.2005. [DOI] [PubMed] [Google Scholar]
- Arch JRS, Newsholme EA. The control of the metabolism and the hormonal role of adenosine. Essays in Biochem. 1978;14:82–103. [PubMed] [Google Scholar]
- Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J. Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol Pharmacol. 1997;52:846–860. doi: 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004;447:735–743. doi: 10.1007/s00424-003-1103-2. [DOI] [PubMed] [Google Scholar]
- Ball HA, Sandrasagra A, Tang L, Van Scott M, Wild J, Nyce JW. Clinical potential of respirable antisense oligonucleotides (RASONs) in asthma. Am J Pharmacogenomics. 2003;3:97–106. doi: 10.2165/00129785-200303020-00003. [DOI] [PubMed] [Google Scholar]
- Banerjee SK, Young HW, Volmer JB, Blackburn MR. Gene expression profiling in inflammatory airway disease associated with elevated adenosine. Am J Physiol Lung Cell Mol Physiol. 2002;282:L169–182. doi: 10.1152/ajplung.00243.2001. [DOI] [PubMed] [Google Scholar]
- Bell PD, Lapointe JY, Sabirov R, Hayashi S, Peti-Peterdi J, Manabe K, et al. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc Natl Acad Sci U S A. 2003;100:4322–4327. doi: 10.1073/pnas.0736323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR. Too much of a good thing: adenosine overload in adenosine-deaminase-deficient mice. Trends Pharmacol Sci. 2003;24:66–70. doi: 10.1016/S0165-6147(02)00045-7. [DOI] [PubMed] [Google Scholar]
- Blackburn MR, Volmer JB, Thrasher JL, Zhong H, Crosby JR, Lee JJ, et al. Metabolic consequences of adenosine deaminase deficiency in mice are associated with defects in alveogenesis, pulmonary inflammation, and airway obstruction. J Exp Med. 2000a;192:159–170. doi: 10.1084/jem.192.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR, Aldrich M, Volmer JB, Chen W, Zhong H, Kelly S, et al. The use of enzyme therapy to regulate the metabolic and phenotypic consequences of adenosine deaminase deficiency in mice. Differential impact on pulmonary and immunologic abnormalities. J Biol Chem. 2000b;275:32114–32121. doi: 10.1074/jbc.M005153200. [DOI] [PubMed] [Google Scholar]
- Blackburn MR, Chun GL, Young HWJ, Chunn JL, Banerjee SK, Elias JA. Adenosine mediates IL-13-induced inflammation and remodeling in the lung: evidence for an IL-13-adenosine amplification pathway. J Clin Invest. 2003;112:332–344. doi: 10.1172/JCI16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR, Datta SK, Kellems RE. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem. 1998;273:5093–5100. doi: 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- Bonneau O, Wyss D, Ferretti S, Blaydon C, Stevenson CS, Trifilieff A. Effect of adenosine A2A receptor activation in murine models of respiratory disorders. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1036–1043. doi: 10.1152/ajplung.00422.2005. [DOI] [PubMed] [Google Scholar]
- Braunstein GM, Roman RM, Clancy JP, Kudlow BA, Taylor AL, Shylonsky VG, et al. Cystic fibrosis transmembrane conductance regulator facilitates ATP release by stimulating a separate ATP release channel for autocrine control of cell volume regulation. J Biol Chem. 2001;276:6621–6630. doi: 10.1074/jbc.M005893200. [DOI] [PubMed] [Google Scholar]
- Breschi MC, Blandizzi C, Fogli S, Martinelli C, Adinolfi B, Calderone V, et al. In vivo adenosine A(2B) receptor desensitization in guinea-pig airway smooth muscle: implications for asthma. Eur J Pharmacol. 2007;575:149–157. doi: 10.1016/j.ejphar.2007.07.051. [DOI] [PubMed] [Google Scholar]
- Brown RA, Clarke GW, Ledbetter CL, Hurle MJ, Denyer JC, Simcock DE, et al. Elevated expression of adenosine A1 receptor in bronchial biopsy specimens from asthmatic subjects. Eur Respir J. 2008;31:311–319. doi: 10.1183/09031936.00003707. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Lu GH, Pugsley TA. Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol Pharmacol. 1986;29:331–346. [PubMed] [Google Scholar]
- Cargnoni A, Ceconi C, Boraso A, Bernocchi P, Monopoli A, Curello S, et al. Role of A2A receptor in the modulation of myocardial reperfusion damage. J Cardiovasc Pharmacol. 1999;33:883–893. doi: 10.1097/00005344-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Chambers LA, Constable M, Clunes MT, Olver RE, Ko WH, Inglis SK, et al. Adenosine-evoked Na+ transport in human airway epithelial cells. Br J Pharmacol. 2006;149:43–55. doi: 10.1038/sj.bjp.0706822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan ES, Fernandez P, Merchant AA, Montesinos MC, Trzaska S, Desai A, et al. Adenosine A2A receptors in diffuse dermal fibrosis: pathogenic role in human dermal fibroblasts and in a murine model of scleroderma. Arthritis Rheum. 2006;54:2632–2642. doi: 10.1002/art.21974. [DOI] [PubMed] [Google Scholar]
- Chen Y, Epperson S, Makhsudova L, Ito B, Suarez J, Dillmann W, et al. Functional effects of enhancing or silencing adenosine A2b receptors in cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2004;287:H2478–2486. doi: 10.1152/ajpheart.00217.2004. [DOI] [PubMed] [Google Scholar]
- Chunn JL, Mohsenin A, Young HW, Lee CG, Elias JA, Kellems RE, et al. Partially adenosine deaminase-deficient mice develop pulmonary fibrosis in association with adenosine elevations. Am J Physiol Lung Cell Mol Physiol. 2006;290:L579–587. doi: 10.1152/ajplung.00258.2005. [DOI] [PubMed] [Google Scholar]
- Chunn JL, Molina JG, Mi T, Xia Y, Kellems RE, Blackburn MR. Adenosine-dependent pulmonary fibrosis in adenosine deaminase-deficient mice. J Immunol. 2005;175:1937–1946. doi: 10.4049/jimmunol.175.3.1937. [DOI] [PubMed] [Google Scholar]
- Chunn JL, Young HW, Banerjee SK, Colasurdo GN, Blackburn MR. Adenosine-dependent airway inflammation and hyperresponsiveness in partially adenosine deaminase-deficient mice. J Immunol. 2001;167:4676–4685. doi: 10.4049/jimmunol.167.8.4676. [DOI] [PubMed] [Google Scholar]
- Cobb BR, Fan L, Kovacs TE, Sorscher EJ, Clancy JP. Adenosine receptors and phosphodiesterase inhibitors stimulate Cl- secretion in Calu-3 cells. Am J Respir Cell Mol Biol. 2003;29:410–418. doi: 10.1165/rcmb.2002-0247OC. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Lin JH, Nedergaard M. Cytoskeletal assembly and ATP release regulate astrocytic calcium signaling. J Neurosci. 1998;18:8794–8804. doi: 10.1523/JNEUROSCI.18-21-08794.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol. 1994;76:5–13. doi: 10.1152/jappl.1994.76.1.5. [DOI] [PubMed] [Google Scholar]
- Cronstein BN, Kramer SB, Weissmann G, Hirschhorn R. Adenosine: a physiological modulator of superoxide anion generation by human neutrophils. J Exp Med. 1983;158:1160–1177. doi: 10.1084/jem.158.4.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushley MJ, Tattersfield AE, Holgate ST. Inhaled adenosine and guanosine on airway resistance in normal and asthmatic subjects. Br J Clin Pharmacol. 1983;15:161–165. doi: 10.1111/j.1365-2125.1983.tb01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushley MJ, Tattersfield AE, Holgate ST. Adenosine-induced bronchoconstriction in asthma. Antagonism by inhaled theophylline. Am Rev Respir Dis. 1984;129:380–384. doi: 10.1164/arrd.1984.129.3.380. [DOI] [PubMed] [Google Scholar]
- Day YJ, Huang L, McDuffie MJ, Rosin DL, Ye H, Chen JF, et al. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J Clin Invest. 2003;112:883–891. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day YJ, Marshall MA, Huang L, McDuffie MJ, Okusa MD, Linden J. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: inhibition of chemokine induction. Am J Physiol Gastrointest Liver Physiol. 2004;286:G285–G293. doi: 10.1152/ajpgi.00348.2003. [DOI] [PubMed] [Google Scholar]
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Victor-Vega C, Gadangi S, Montesinos MC, Chu CC, Cronstein BN. Adenosine A2A receptor stimulation increases angiogenesis by down-regulating production of the antiangiogenic matrix protein thrombospondin 1. Mol Pharmacol. 2005;67:1406–1413. doi: 10.1124/mol.104.007807. [DOI] [PubMed] [Google Scholar]
- Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis. 1993;148:91–97. doi: 10.1164/ajrccm/148.1.91. [DOI] [PubMed] [Google Scholar]
- Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–3315. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, et al. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- Elias JA, Lee CG, Zheng T, Ma B, Homer RJ, Zhu Z. New insights into the pathogenesis of asthma. J Clin Invest. 2003;111:291–297. doi: 10.1172/JCI17748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman PI, Reece TB, Law MG, Gazoni LM, Singh R, Laubach VE, et al. Adenosine A2A activation attenuates nontransplantation lung reperfusion injury. J Surg Res. 2008;149:3–8. doi: 10.1016/j.jss.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethier MF, Madison JM. Adenosine A1 receptors mediate mobilization of calcium in human bronchial smooth muscle cells. Am J Respir Cell Mol Biol. 2006;35:496–502. doi: 10.1165/rcmb.2005-0290OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeamuzie CI, Philips E. Adenosine A3 receptors on human eosinophils mediate inhibition of degranulation and superoxide anion release [In Process Citation] Br J Pharmacol. 1999;127:188–194. doi: 10.1038/sj.bjp.0702476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Factor P, Mutlu GM, Chen L, Mohameed J, Akhmedov AT, Meng FJ, et al. Adenosine regulation of alveolar fluid clearance. Proc Natl Acad Sci U S A. 2007;104:4083–4088. doi: 10.1073/pnas.0601117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan M, Mustafa SJ. Adenosine-mediated bronchoconstriction and lung inflammation in an allergic mouse model. Pulm Pharmacol Ther. 2002;15:147–155. doi: 10.1006/pupt.2001.0329. [DOI] [PubMed] [Google Scholar]
- Fan M, Qin W, Mustafa SJ. Characterization of adenosine receptor(s) involved in adenosine-induced bronchoconstriction in an allergic mouse model. Am J Physiol Lung Cell Mol Physiol. 2003;284:L1012–1019. doi: 10.1152/ajplung.00353.2002. [DOI] [PubMed] [Google Scholar]
- Feoktistov I, Biaggioni I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J Clin Invest. 1995;96:1979–1986. doi: 10.1172/JCI118245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistov I, Garland EM, Goldstein AE, Zeng D, Belardinelli L, Wells JN, et al. Inhibition of human mast cell activation with the novel selective adenosine A(2B) receptor antagonist 3-isobutyl-8-pyrrolidinoxanthine (IPDX)(2) Biochem Pharmacol. 2001;62:1163–1173. doi: 10.1016/s0006-2952(01)00765-1. [DOI] [PubMed] [Google Scholar]
- Feoktistov I, Goldstein AE, Ryzhov S, Zeng D, Belardinelli L, Voyno-Yasenetskaya T, et al. Differential expression of adenosine receptors in human endothelial cells: role of A2B receptors in angiogenic factor regulation. Circ Res. 2002;90:531–538. doi: 10.1161/01.res.0000012203.21416.14. [DOI] [PubMed] [Google Scholar]
- Forsythe P, McGarvey LP, Heaney LG, MacMahon J, Ennis M. Adenosine induces histamine release from human bronchoalveolar lavage mast cells. Clin Sci (Colch) 1999;96:349–355. [PubMed] [Google Scholar]
- Fozard JR, Ellis KM, Villela Dantas MF, Tigani B, Mazzoni L. Effects of CGS 21680, a selective adenosine A2A receptor agonist, on allergic airways inflammation in the rat. Eur J Pharmacol. 2002;438:183–188. doi: 10.1016/s0014-2999(02)01305-5. [DOI] [PubMed] [Google Scholar]
- Fozard JR, Hannon JP. Adenosine receptor ligands: potential as therapeutic agents in asthma and COPD. Pulm Pharmacol Ther. 1999;12:111–114. doi: 10.1006/pupt.1999.0191. [DOI] [PubMed] [Google Scholar]
- Fozard JR, Hannon JP. Species differences in adenosine receptor-mediated bronchoconstrictor responses. Clin Exp Allergy. 2000;30:1213–1220. doi: 10.1046/j.1365-2222.2000.00894.x. [DOI] [PubMed] [Google Scholar]
- Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14:1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am J Physiol Renal Physiol. 2005;289:F369–376. doi: 10.1152/ajprenal.00470.2004. [DOI] [PubMed] [Google Scholar]
- Gessi S, Merighi S, Varani K, Leung E, Mac Lennan S, Borea PA. The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol Ther. 2008;117:123–140. doi: 10.1016/j.pharmthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Gleich GJ. The eosinophil and bronchial asthma: current understanding. J Allergy Clin Immunol. 1990;85:422–436. doi: 10.1016/0091-6749(90)90151-s. [DOI] [PubMed] [Google Scholar]
- Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, et al. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 2008;5:e137. doi: 10.1371/journal.pmed.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon JP, Tigani B, Williams I, Mazzoni L, Fozard JR. Mechanism of airway hyperresponsiveness to adenosine induced by allergen challenge in actively sensitized Brown Norway rats. Br J Pharmacol. 2001;132:1509–1523. doi: 10.1038/sj.bjp.0703961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon JP, Tigani B, Wolber C, Williams I, Mazzoni L, Howes C, et al. Evidence for an atypical receptor mediating the augmented bronchoconstrictor response to adenosine induced by allergen challenge in actively sensitized Brown Norway rats. Br J Pharmacol. 2002;135:685–696. doi: 10.1038/sj.bjp.0704516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–39. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, et al. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. Faseb J. 2000;14:2065–2074. doi: 10.1096/fj.99-0508com. [DOI] [PubMed] [Google Scholar]
- Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G, Pacher P. A2A receptors in inflammation and injury: lessons learned from transgenic animals. J Leukoc Biol. 2008;83:447–455. doi: 10.1189/jlb.0607359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst SJ. Airway smooth muscle as a target in asthma. Clin Exp Allergy. 2000;30:54–59. [PubMed] [Google Scholar]
- Holgate ST. Adenosine provocation: a new test for allergic type airway inflammation. Am J Respir Crit Care Med. 2002;165:317–318. doi: 10.1164/ajrccm.165.3.2112045a. [DOI] [PubMed] [Google Scholar]
- Holgate ST. The Quintiles Prize Lecture 2004. The identification of the adenosine A2B receptor as a novel therapeutic target in asthma. Br J Pharmacol. 2005;145:1009–1015. doi: 10.1038/sj.bjp.0706272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Kovarova M, Chason KD, Nguyen M, Koller BH, Tilley SL. Enhanced mast cell activation in mice deficient in the A2b adenosine receptor. J Exp Med. 2007;204:117–128. doi: 10.1084/jem.20061372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Lazarowski ER, Tarran R, Milgram SL, Boucher RC, Stutts MJ. Compartmentalized autocrine signaling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proc Natl Acad Sci U S A. 2001;98:14120–14125. doi: 10.1073/pnas.241318498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PJ, Holgate ST, Church MK. Adenosine inhibits and potentiates IgE-dependent histamine release from human lung mast cells by an A2-purinoceptor mediated mechanism. Biochem Pharmacol. 1984;33:3847–3852. doi: 10.1016/0006-2952(84)90050-9. [DOI] [PubMed] [Google Scholar]
- Huszar E, Vass G, Vizi E, Csoma Z, Barat E, Molnar VG, et al. Adenosine in exhaled breath condensate in healthy volunteers and in patients with asthma. Eur Respir J. 2002;20:1393–1398. doi: 10.1183/09031936.02.00005002. [DOI] [PubMed] [Google Scholar]
- Jabs CM, Sigurdsson GH, Neglen P. Plasma levels of high-energy compounds compared with severity of illness in critically ill patients in the intensive care unit. Surgery. 1998;124:65–72. [PubMed] [Google Scholar]
- Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest. 1997;100:2849–2857. doi: 10.1172/JCI119833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek E, Koziak K, Sevigny J, Siegel JB, Anrather J, Beaudoin AR, et al. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem. 1996;271:33116–33122. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- Kalla RV, Zablocki J. Progress in the discovery of selective, high affinity A(2B) adenosine receptor antagonists as clinical candidates. Purinergic Signal. 2009;5:21–29. doi: 10.1007/s11302-008-9119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight D, Zheng X, Rocchini C, Jacobson M, Bai T, Walker B. Adenosine A3 receptor stimulation inhibits migration of human eosinophils. J Leukoc Biol. 1997;62:465–468. doi: 10.1002/jlb.62.4.465. [DOI] [PubMed] [Google Scholar]
- Kohno Y, Ji X, Mawhorter SD, Koshiba M, Jacobson KA. Activation of A3 adenosine receptors on human eosinophils elevates intracellular calcium. Blood. 1996;88:3569–3574. [PMC free article] [PubMed] [Google Scholar]
- Langley SJ, Allen DJ, Houghton C, Woodcock A. An inhaled respirable antisense oligonucleotide: RASON. Am J Resp Crit Care Med. 2005;171:A360. [Google Scholar]
- Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol. 2005;174:1073–1080. doi: 10.4049/jimmunol.174.2.1073. [DOI] [PubMed] [Google Scholar]
- Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol. 2006;291:R959–969. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- Leibovich SJ, Chen JF, Pinhal-Enfield G, Belem PC, Elson G, Rosania A, et al. Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am J Pathol. 2002;160:2231–2244. doi: 10.1016/S0002-9440(10)61170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Oskouian RJ, Day YJ, Rieger JM, Liu L, Kern JA, et al. Mouse spinal cord compression injury is reduced by either activation of the adenosine A2A receptor on bone marrow-derived cells or deletion of the A2A receptor on non-bone marrow-derived cells. Neuroscience. 2006;141:2029–2039. doi: 10.1016/j.neuroscience.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang W, Parker W, Clancy JP. Adenosine regulation of cystic fibrosis transmembrane conductance regulator through prostenoids in airway epithelia. Am J Respir Cell Mol Biol. 2006;34:600–608. doi: 10.1165/rcmb.2005-0421OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisle TC, Gazoni LM, Fernandez LG, Sharma AK, Bellizzi AM, Schifflett GD, et al. Inflammatory lung injury after cardiopulmonary bypass is attenuated by adenosine A(2A) receptor activation. J Thorac Cardiovasc Surg. 2008;136:1280–1287. doi: 10.1016/j.jtcvs.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijk B, van den Berge M, Kerstjens HA, Postma DS, Cass L, Sabin A, et al. Effect of an inhaled adenosine A2A agonist on the allergen-induced late asthmatic response. Allergy. 2008;63:75–80. doi: 10.1111/j.1398-9995.2007.01557.x. [DOI] [PubMed] [Google Scholar]
- Lynch J, Toews G. Idiopathic pulmonary fibrosis. In: Fishman AP, Elias JA, Fishman JA, Grippi MA, Kaiser LR, Senior RM, editors. Pulmonary Diseases and Disorders. 3. Vol. 1. New york: McGraw-Hill; 1998. pp. 1069–1084. [Google Scholar]
- Ma B, Blackburn MR, Lee CG, Homer RJ, Liu W, Flavell RA, et al. Adenosine metabolism and murine strain-specific IL-4-induced inflammation, emphysema, and fibrosis. J Clin Invest. 2006;116:1274–1283. doi: 10.1172/JCI26372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara JL, Patapoff TW, Gillece-Castro B, Colgan SP, Parkos CA, Delp C, et al. 5′-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J Clin Invest. 1993;91:2320–2325. doi: 10.1172/JCI116462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann JS, Holgate ST, Renwick AG, Cushley MJ. Airway effects of purine nucleosides and nucleotides and release with bronchial provocation in asthma. J Appl Physiol. 1986;61:1667–1676. doi: 10.1152/jappl.1986.61.5.1667. [DOI] [PubMed] [Google Scholar]
- Marquardt DL, Gruber HE, Wasserman SI. Adenosine release from stimulated mast cells. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:6192–6196. doi: 10.1073/pnas.81.19.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt DL, Parker CW, Sullivan TJ. Potentiation of mast cell mediator release by adenosine. J Immunol. 1978;120:871–878. [PubMed] [Google Scholar]
- Martin C, Leone M, Viviand X, Ayem ML, Guieu R. High adenosine plasma concentration as a prognostic index for outcome in patients with septic shock. Crit Care Med. 2000;28:3198–3202. doi: 10.1097/00003246-200009000-00014. [DOI] [PubMed] [Google Scholar]
- Matot I, Weiniger CF, Zeira E, Galun E, Joshi BV, Jacobson KA. A3 adenosine receptors and mitogen-activated protein kinases in lung injury following in vivo reperfusion. Crit Care. 2006;10:R65. doi: 10.1186/cc4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara N, Gallup M, Khong A, Sucher A, Maltseva I, Fahy J, et al. Adenosine up-regulation of the mucin gene, MUC2, in asthma. Faseb J. 2004;18:1770–1772. doi: 10.1096/fj.04-1964fje. [DOI] [PubMed] [Google Scholar]
- Mohsenin A, Mi T, Xia Y, Kellems RE, Chen JF, Blackburn MR. Genetic removal of the A2A adenosine receptor enhances pulmonary inflammation, mucin production, and angiogenesis in adenosine deaminase-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2007;293:L753–761. doi: 10.1152/ajplung.00187.2007. [DOI] [PubMed] [Google Scholar]
- Montesinos MC, Gadangi P, Longaker M, Sung J, Levine J, Nilsen D, et al. Wound healing is accelerated by agonists of adenosine A2 (G alpha s-linked) receptors. J Exp Med. 1997;186:1615–1620. doi: 10.1084/jem.186.9.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CC, Martin EN, Lee GH, Obrig T, Linden J, Scheld WM. An A2A adenosine receptor agonist, ATL313, reduces inflammation and improves survival in murine sepsis models. BMC Infect Dis. 2008;8:141. doi: 10.1186/1471-2334-8-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morschl E, Molina JG, Volmer JB, Mohsenin A, Pero RS, Hong JS, et al. A3 adenosine receptor signaling influences pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol. 2008;39:697–705. doi: 10.1165/rcmb.2007-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundell SJ, Olah ME, Panettieri RA, Benovic JL, Penn RB. Regulation of G protein-coupled receptor-adenylyl cyclase responsiveness in human airway smooth muscle by exogenous and autocrine adenosine. Am J Respir Cell Mol Biol. 2001;24:155–163. doi: 10.1165/ajrcmb.24.2.4243. [DOI] [PubMed] [Google Scholar]
- Mustafa SJ, Nadeem A, Fan M, Zhong H, Belardinelli L, Zeng D. Effect of a specific and selective A(2B) adenosine receptor antagonist on adenosine agonist AMP and allergen-induced airway responsiveness and cellular influx in a mouse model of asthma. J Pharmacol Exp Ther. 2007;320:1246–1251. doi: 10.1124/jpet.106.112250. [DOI] [PubMed] [Google Scholar]
- Nadeem A, Fan M, Ansari HR, Ledent C, Jamal Mustafa S. Enhanced airway reactivity and inflammation in A2A adenosine receptor-deficient allergic mice. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1335–1344. doi: 10.1152/ajplung.00416.2006. [DOI] [PubMed] [Google Scholar]
- Nadeem A, Obiefuna PC, Wilson CN, Mustafa SJ. Adenosine A1 receptor antagonist versus montelukast on airway reactivity and inflammation. Eur J Pharmacol. 2006;551:116–124. doi: 10.1016/j.ejphar.2006.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB. Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. 2006;177:2765–2769. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- Neely CF, Keith IM. A1 adenosine receptor antagonists block ischemia-reperfusion injury of the lung. Am J Physiol. 1995;268:L1036–1046. doi: 10.1152/ajplung.1995.268.6.L1036. [DOI] [PubMed] [Google Scholar]
- Nguyen DK, Montesinos MC, Williams AJ, Kelly M, Cronstein BN. Th1 cytokines regulate adenosine receptors and their downstream signaling elements in human microvascular endothelial cells. J Immunol. 2003;171:3991–3998. doi: 10.4049/jimmunol.171.8.3991. [DOI] [PubMed] [Google Scholar]
- Nyce JW, Metzger WJ. DNA antisense therapy for asthma in an animal model. Nature. 1997;385:721–725. doi: 10.1038/385721a0. [DOI] [PubMed] [Google Scholar]
- Okada SF, O'Neal WK, Huang P, Nicholas RA, Ostrowski LE, Craigen WJ, et al. Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J Gen Physiol. 2004;124:513–526. doi: 10.1085/jgp.200409154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okusa MD, Linden J, Macdonald T, Huang L. Selective A2A adenosine receptor activation reduces ischemia-reperfusion injury in rat kidney. Am J Physiol. 1999;277:F404–412. doi: 10.1152/ajprenal.1999.277.3.F404. [DOI] [PubMed] [Google Scholar]
- Oldenburg PJ, Mustafa SJ. Involvement of mast cells in adenosine-mediated bronchoconstriction and inflammation in an allergic mouse model. J Pharmacol Exp Ther. 2005;313:319–324. doi: 10.1124/jpet.104.071720. [DOI] [PubMed] [Google Scholar]
- Oosterhoff Y, de Jong JW, Jansen MA, Koeter GH, Postma DS. Airway responsiveness to adenosine 5′-monophosphate in chronic obstructive pulmonary disease is determined by smoking. Am Rev Respir Dis. 1993;147:553–558. doi: 10.1164/ajrccm/147.3.553. [DOI] [PubMed] [Google Scholar]
- Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, et al. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood. 2003;101:3985–3990. doi: 10.1182/blood-2002-07-2113. [DOI] [PubMed] [Google Scholar]
- Pascoe SJ, Knight RH, Woessner R. An a2b/A3 adenosine receptor antagonist does not attenuate AMP challenge in subjects with athma. Am J Res Crit Care Med. 2007;175:A682. [Google Scholar]
- Pinhal-Enfield G, Ramanathan M, Hasko G, Vogel SN, Salzman AL, Boons GJ, et al. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am J Pathol. 2003;163:711–721. doi: 10.1016/S0002-9440(10)63698-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prussin C, Metcalfe DD. 4. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2003;111:S486–494. doi: 10.1067/mai.2003.120. [DOI] [PubMed] [Google Scholar]
- Reece TB, Ellman PI, Maxey TS, Crosby IK, Warren PS, Chong TW, et al. Adenosine A2A receptor activation reduces inflammation and preserves pulmonary function in an in vivo model of lung transplantation. J Thorac Cardiovasc Surg. 2005;129:1137–1143. doi: 10.1016/j.jtcvs.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Reece TB, Okonkwo DO, Ellman PI, Warren PS, Smith RL, Hawkins AS, et al. The evolution of ischemic spinal cord injury in function, cytoarchitecture, and inflammation and the effects of adenosine A2A receptor activation. J Thorac Cardiovasc Surg. 2004;128:925–932. doi: 10.1016/j.jtcvs.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Reeves JJ, Jones CA, Sheehan MJ, Vardey CJ, Whelan CJ. Adenosine A3 receptors promote degranulation of rat mast cells both in vitro and in vivo. Inflamm Res. 1997;46:180–184. doi: 10.1007/s000110050169. [DOI] [PubMed] [Google Scholar]
- Reisin IL, Prat AG, Abraham EH, Amara JF, Gregory RJ, Ausiello DA, et al. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J Biol Chem. 1994;269:20584–20591. [PubMed] [Google Scholar]
- Resnick MB, Colgan SP, Patapoff TW, Mrsny RJ, Awtrey CS, Delp-Archer C, et al. Activated eosinophils evoke chloride secretion in model intestinal epithelia primarily via regulated release of 5′-AMP. J Immunol. 1993;151:5716–5723. [PubMed] [Google Scholar]
- Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev. 1998;161:95–109. doi: 10.1111/j.1600-065x.1998.tb01574.x. [DOI] [PubMed] [Google Scholar]
- Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol. 2007;179:1254–1263. doi: 10.4049/jimmunol.179.2.1254. [DOI] [PubMed] [Google Scholar]
- Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, et al. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest. 2006;116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reutershan J, Vollmer I, Stark S, Wagner R, Ngamsri KC, Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. Faseb J. 2009;23:473–482. doi: 10.1096/fj.08-119701. [DOI] [PubMed] [Google Scholar]
- Rivo J, Zeira E, Galun E, Matot I. Activation of A3 adenosine receptors attenuates lung injury after in vivo reperfusion. Anesthesiology. 2004;101:1153–1159. doi: 10.1097/00000542-200411000-00015. [DOI] [PubMed] [Google Scholar]
- Roche WR. Fibroblasts and asthma. Clin Exp Allergy. 1991;21:545–548. doi: 10.1111/j.1365-2222.1991.tb00845.x. [DOI] [PubMed] [Google Scholar]
- Rollins BM, Burn M, Coakley RD, Chambers LA, Hirsh AJ, Clunes MT, et al. A2B adenosine receptors regulate the mucus clearance component of the lung's innate defense system. Am J Respir Cell Mol Biol. 2008;39:190–197. doi: 10.1165/rcmb.2007-0450OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman J, Rivera HN, Roser-Page S, Sitaraman SV, Ritzenthaler JD. Adenosine induces fibronectin expression in lung epithelial cells: implications for airway remodeling. Am J Physiol Lung Cell Mol Physiol. 2006;290:L317–325. doi: 10.1152/ajplung.00118.2005. [DOI] [PubMed] [Google Scholar]
- Roman RM, Lomri N, Braunstein G, Feranchak AP, Simeoni LA, Davison AK, et al. Evidence for multidrug resistance-1 P-glycoprotein-dependent regulation of cellular ATP permeability. J Membr Biol. 2001;183:165–173. doi: 10.1007/s00232-001-0064-7. [DOI] [PubMed] [Google Scholar]
- Ross SD, Tribble CG, Linden J, Gangemi JJ, Lanpher BC, Wang AY, et al. Selective adenosine-A2A activation reduces lung reperfusion injury following transplantation. J Heart Lung Transplant. 1999;18:994–1002. doi: 10.1016/s1053-2498(99)00066-2. [DOI] [PubMed] [Google Scholar]
- Ryzhov S, Goldstein AE, Biaggioni I, Feoktistov I. Cross-talk between G(s)- and G(q)-coupled pathways in regulation of interleukin-4 by A(2B) adenosine receptors in human mast cells. Mol Pharmacol. 2006;70:727–735. doi: 10.1124/mol.106.022780. [DOI] [PubMed] [Google Scholar]
- Ryzhov S, Goldstein AE, Matafonov A, Zeng D, Biaggioni I, Feoktistov I. Adenosine-activated mast cells induce IgE synthesis by B lymphocytes: an A2B-mediated process involving Th2 cytokines IL-4 and IL-13 with implications for asthma. J Immunol. 2004;172:7726–7733. doi: 10.4049/jimmunol.172.12.7726. [DOI] [PubMed] [Google Scholar]
- Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy SV, Blackburn MR, Biaggioni I, et al. Effect of A2B adenosine receptor gene ablation on adenosine-dependent regulation of proinflammatory cytokines. J Pharmacol Exp Ther. 2008a;324:694–700. doi: 10.1124/jpet.107.131540. [DOI] [PubMed] [Google Scholar]
- Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy SV, Dikov MM, Blackburn MR, et al. Effect of A2B Adenosine Receptor Gene Ablation on Proinflammatory Adenosine Signaling in Mast Cells. J Immunol. 2008b;180:7212–7220. doi: 10.4049/jimmunol.180.11.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA. Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem. 2000;275:4429–4434. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- Schnurr M, Toy T, Shin A, Hartmann G, Rothenfusser S, Soellner J, et al. Role of adenosine receptors in regulating chemotaxis and cytokine production of plasmacytoid dendritic cells. Blood. 2004;103:1391–1397. doi: 10.1182/blood-2003-06-1959. [DOI] [PubMed] [Google Scholar]
- Selman M, Carrillo G, Estrada A, Mejia M, Becerril C, Cisneros J, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS ONE. 2007;2:e482. doi: 10.1371/journal.pone.0000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sime PJ, O'Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- Sottofattori E, Anzaldi M, Ottonello L. HPLC determination of adenosine in human synovial fluid. J Pharm Biomed Anal. 2001;24:1143–1146. doi: 10.1016/s0731-7085(00)00574-4. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Doda M, Baranyi M, Hasko G. Ischemic-like condition releases norepinephrine and purines from different sources in superfused rat spleen strips. J Neuroimmunol. 2000;111:45–54. doi: 10.1016/s0165-5728(00)00365-9. [DOI] [PubMed] [Google Scholar]
- Spicuzza L, Bonfiglio C, Polosa R. Research applications and implications of adenosine in diseased airways. Trends Pharmacol Sci. 2003;24:409–413. doi: 10.1016/S0165-6147(03)00193-7. [DOI] [PubMed] [Google Scholar]
- Spruntulis LM, Broadley KJ. A3 receptors mediate rapid inflammatory cell influx into the lungs of sensitized guinea-pigs. Clin Exp Allergy. 2001;31:943–951. doi: 10.1046/j.1365-2222.2001.01087.x. [DOI] [PubMed] [Google Scholar]
- Sun CX, Young HW, Molina JG, Volmer JB, Schnermann J, Blackburn MR. A protective role for the A(1) adenosine receptor in adenosine-dependent pulmonary injury. J Clin Invest. 2005;115:35–43. doi: 10.1172/JCI22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, et al. Role of A2B receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest. 2006;116:1–10. doi: 10.1172/JCI27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wu F, Sun F, Huang P. Adenosine promotes IL-6 release in airway epithelia. J Immunol. 2008;180:4173–4181. doi: 10.4049/jimmunol.180.6.4173. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- Tillie-Leblond I, Pugin J, Marquette CH, Lamblin C, Saulnier F, Brichet A, et al. Balance between proinflammatory cytokines and their inhibitors in bronchial lavage from patients with status asthmaticus. Am J Respir Crit Care Med. 1999;159:487–494. doi: 10.1164/ajrccm.159.2.9805115. [DOI] [PubMed] [Google Scholar]
- Valls MD, Cronstein BN, Montesinos MC. Adenosine receptor agonists for promotion of dermal wound healing. Biochem Pharmacol. 2009;77:1117–1124. doi: 10.1016/j.bcp.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani K, Caramori G, Vincenzi F, Adcock I, Casolari P, Leung E, et al. Alteration of adenosine receptors in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:398–406. doi: 10.1164/rccm.200506-869OC. [DOI] [PubMed] [Google Scholar]
- Vestbo J, Prescott E. Update on the “Dutch hypothesis” for chronic respiratory disease. Lancet. 1997;350:1431–1434. doi: 10.1136/thx.53.2008.s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor-Vega C, Desai A, Montesinos MC, Cronstein BN. Adenosine A2A receptor agonists promote more rapid wound healing than recombinant human platelet-derived growth factor (Becaplermin gel) Inflammation. 2002;26:19–24. doi: 10.1023/a:1014417728325. [DOI] [PubMed] [Google Scholar]
- Vizi E, Huszar E, Csoma Z, Boszormenyi-Nagy G, Barat E, Horvath I, et al. Plasma adenosine concentration increases during exercise: a possible contributing factor in exercise-induced bronchoconstriction in asthma. J Allergy Clin Immunol. 2002;109:446–448. doi: 10.1067/mai.2002.121955. [DOI] [PubMed] [Google Scholar]
- Volmer JB, Thompson LF, Blackburn MR. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol. 2006;176:4449–4458. doi: 10.4049/jimmunol.176.7.4449. [DOI] [PubMed] [Google Scholar]
- Walker BA, Jacobson MA, Knight DA, Salvatore CA, Weir T, Zhou D, et al. Adenosine A3 receptor expression and function in eosinophils. Am J Respir Cell Mol Biol. 1997;16:531–537. doi: 10.1165/ajrcmb.16.5.9160835. [DOI] [PubMed] [Google Scholar]
- Ware LB, Conner ER, Matthay MA. von Willebrand factor antigen is an independent marker of poor outcome in patients with early acute lung injury. Crit Care Med. 2001;29:2325–2331. doi: 10.1097/00003246-200112000-00016. [DOI] [PubMed] [Google Scholar]
- Wolber C, Fozard JR. The receptor mechanism mediating the contractile response to adenosine on lung parenchymal strips from actively sensitised, allergen-challenged Brown Norway rats. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:158–168. doi: 10.1007/s00210-004-1012-8. [DOI] [PubMed] [Google Scholar]
- Xaus J, Mirabet M, Lloberas J, Soler C, Lluis C, Franco R, et al. IFN-gamma up-regulates the A2B adenosine receptor expression in macrophages: a mechanism of macrophage deactivation. J Immunol. 1999;162:3607–3614. [PubMed] [Google Scholar]
- Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–2064. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- Yildiz G, Demiryurek AT, Gumusel B, Lippton H. Ischemic preconditioning modulates ischemia-reperfusion injury in the rat lung: role of adenosine receptors. Eur J Pharmacol. 2007;556:144–150. doi: 10.1016/j.ejphar.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Young HW, Molina JG, Dimina D, Zhong H, Jacobson M, Chan LNL, et al. A3 adenosine receptor signaling contributes to airway inflammation and mucus production in adenosine deaminase-deficient mice. J Immunol. 2004;173(3):1380–1389. doi: 10.4049/jimmunol.173.2.1380. [DOI] [PubMed] [Google Scholar]
- Young HW, Sun CX, Evans CM, Dickey BF, Blackburn MR. A3 adenosine receptor signaling contributes to airway mucin secretion after allergen challenge. Am J Respir Cell Mol Biol. 2006;35:549–558. doi: 10.1165/rcmb.2006-0060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Belardinelli L, Maa T, Feoktistov I, Biaggioni I, Zeng D. A(2B) adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am J Respir Cell Mol Biol. 2004;30:118–125. doi: 10.1165/rcmb.2003-0118OC. [DOI] [PubMed] [Google Scholar]
- Zhong H, Belardinelli L, Maa T, Zeng D. Synergy between A2B Adenosine Receptors and Hypoxia in Activating Human Lung Fibroblasts. Am J Respir Cell Mol Biol. 2005;32:2–8. doi: 10.1165/rcmb.2004-0103OC. [DOI] [PubMed] [Google Scholar]
- Zhong H, Chunn JL, Volmer JB, Fozard JR, Blackburn MR. Adenosine-mediated mast cell degranulation in adenosine deaminase- deficient mice. J Pharmacol Exp Ther. 2001;298:433–440. [PubMed] [Google Scholar]
- Zhong H, Sergiy S, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, et al. Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol. 2003;170:338–345. doi: 10.4049/jimmunol.171.1.338. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Morschl E, Mohsenin A, Young HWJ, Molina JG, Ma W, et al. Enhanced airway inflammation and remodeling in adenosine deaminase deficient mice lacking the A2B adenosine receptor. J Immunol. 2009 doi: 10.4049/jimmunol.0900515. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]