

Abstract
We developed a cryo-imaging system to provide single-cell detection of fluorescently labeled cells in mouse, with particular applicability to stem cells and metastatic cancer. The Case cryo-imaging system consists of a fluorescence microscope, robotic imaging positioner, customized cryostat, PC-based control system, and visualization/analysis software. The system alternates between sectioning (10−40 μm) and imaging, collecting color brightfield and fluorescent block-face image volumes >60GB. In mouse experiments, we imaged quantum-dot labeled stem cells, GFP-labeled cancer and stem cells, and cell-size fluorescent microspheres. To remove subsurface fluorescence, we used a simplified model of light-tissue interaction whereby the next image was scaled, blurred, and subtracted from the current image. We estimated scaling and blurring parameters by minimizing entropy of subtracted images. Tissue specific attenuation parameters were found [uT : heart (267 ± 47.6 μm), liver (218 ± 27.1 μm), brain (161 ± 27.4 μm)] to be within the range of estimates in the literature. “Next image” processing removed subsurface fluorescence equally well across multiple tissues (brain, kidney, liver, adipose tissue, etc.), and analysis of 200 microsphere images in the brain gave 97±2% reduction of subsurface fluorescence. Fluorescent signals were determined to arise from single cells based upon geometric and integrated intensity measurements. Next image processing greatly improved axial resolution, enabled high quality 3D volume renderings, and improved enumeration of single cells with connected component analysis by up to 24%. Analysis of image volumes identified metastatic cancer sites, found homing of stem cells to injury sites, and showed microsphere distribution correlated with blood flow patterns.
We developed and evaluated cryo-imaging to provide single-cell detection of fluorescently labeled cells in mouse. Our cryo-imaging system provides extreme (>60GB), micron-scale, fluorescence, and bright field image data. Here we describe our image pre-processing, analysis, and visualization techniques. Processing improves axial resolution, reduces subsurface fluorescence by 97%, and enables single cell detection and counting. High quality 3D volume renderings enable us to evaluate cell distribution patterns. Applications include the myriad of biomedical experiments using fluorescent reporter gene and exogenous fluorophore labeling of cells in applications such as stem cell regenerative medicine, cancer, tissue engineering, etc.
Keywords: image processing, block face imaging, fluorescence imaging, in vivo cellular imaging, stem cell imaging, cryo-imaging
Introduction
Stem cell therapies are promising treatments for many diseases, and preclinical trials in ischemic heart disease show enhanced recovery of function. While mechanisms for homing are being defined, exogenous stem cell therapies are being implemented in clinical trials, despite a limited understanding of mechanisms. Due to limitations of current methods such as histology and QPCR, one cannot reliably determine efficiency and dose response of exogenous stem cell therapy. In order to determine dose response of stem cell treatments, imaging must be performed with single cell sensitivity and resolution over an entire specimen.
Cryo-imaging consists of a modified, bright-field/fluorescence microscope; a robotic imaging system positioner; a customized, whole mouse motorized cryomicrotome; control system; and analysis/visualization software [1]. By alternately sectioning and imaging the specimen, the system acquires brightfield color and fluorescent image volumes, providing micron-scale resolution, detailed views of anatomy and cells labeled with fluorescent reporter genes or exogenous fluorophores[2-5]. Cryo-imaging provides single cell resolution and sensitivity over an entire specimen, which is not possible with in vivo, small animal imaging systems such as CT, MRI, PET, SPECT, intra-vital imaging or bioluminescence. Optical imaging modalities such as intra-vital imaging [6] do not offer the field of view (FOV) or depth of field of cryo-imaging. By imaging with high resolution and sensitivity, it is possible to identify fluorescently-labeled single cells or cell clusters within a mouse. Once cells are identified, cell locations can be mapped relative to the tissue anatomy in the high contrast, 3D cryo-image color volumes.
Once images have been collected of an entire mouse at sufficient resolution to detect individual fluorescently labeled cells, image processing methods must be developed to increase axial resolution sufficiently through the removal of subsurface fluorescence to enable the accurate measurement of labeled volumes. Further image processing algorithms are also required to detect labeled cells and cell aggregates and quantify the number of cells present in an aggregate.
In this paper, we develop and evaluate processing methods for enumerating fluorescently labeled stem cells in a mouse. We first apply “next-image” processing for reducing out-of-plane fluorescence in cryo-imaging. We then apply image processing algorithms for segmenting cell clusters and estimating the number of cells per cluster. In experiments, we image homogeneous phantoms and whole mice injected with fluorescent microspheres or cells. In sections that follow, we develop the algorithms, describe the software and parameter estimation, process cryo-image volumes, and compare 3D reconstructions of original and processed data.
Next-Image Algorithm for Reduction of Out-of-plane Fluorescence
Subsurface fluorescence presents a challenge to single cell detection in cryo-imaging. Typically, in epifluorescence imaging, an objective lens with a high numerical aperture lens is used, which in turn has a small focal depth, limiting the out-of-plane fluorescence [7]. To satisfy field of view and depth of focus constraints, we often use a 0.11 NA objective lens giving a 477 μm depth of focus and leading to inclusion of out-of-plane fluorescence. Subsurface fluorescence gives a haze in 2D images and an elongation artifact along the z-axis that appears as a “comet tail” in 3D. The “comet tail” artifact can connect separate structures, leading to quantification errors, particularly when counting fluorescently-labeled cells, such as stem cells.
Optical blurring is negligible in cryo-imaging, because we use a microscope with a long focal distance (527 μm at 27X and .036 NA). While optical blurring is negligible, there is a cryo-imaging, scattering PSF which is very asymmetrical. The scattering PSF is one sided because once a fluorophore is sectioned off, its associated signal is removed. To correct optical blurring, a common ad hoc approach is to use nearest neighbor or multi-neighbor processing [8-10]. Because the plane of interest contains fluorescence from nearby planes, filtered by the PSF of the microscope, nearby planes are blurred and subtracted from the plane of interest. This is fast but can raise noise levels and reduce SNR because the entire signal is not used. However, by removing blurred light, CNR, (IA-IB)/σBKG, is increased. Our “next image” algorithm is an adaptation of this concept.
To remove subsurface fluorescence in “next-image” processing we model the formation of a cryo-image. Consider a series of block face images i = 1, 2, 3, ..., where increasing i indicates subsequent tissue slicing down through the block. Equations below describe the block face images, where Io is the incident light intensity, Tat is transmission across the air to tissue interface, Fi is a factor which converts excitation light intensity to fluorescence from slice i and which depends upon fluorophore concentration, μ is the attenuation coefficient which depends upon absorption and scattering in the tissue, s is the slice thickness, and Tta is transmission from tissue to air.
| [Eq. 3] |
From a series of block face images, we take the next block face image, attenuate it, and subtract it from the current image, giving an image which reflects the fluorescence in the tissue section; i.e.,
| [Eq. 4] |
We further modified Eq 4 to include blurring to account for light scatter using a Gaussian blurring kernel, h(x,y),
| [Eq. 5] |
In initial experiments, we obtained 20 μm thick cryo-images of fluorescent beads (15 μm) in phantom material (OCT) and in tissue following tail vein injections. We found that μ depends upon tissue type and that blurring is minimal with a Gaussian of σ = 1.2 μm, < 1 pixel. Processing of microspheres in a tissue phantom removes “comet tails” otherwise present when viewing the image volume from the side (Fig 1). This allows one to image single fluorescently labeled blood borne cells in a parabiosis model (Figs 2), and 3D vessels (Fig 3).
Fig 1.

Side projection of cryo-image stack before (a) and after (b) processing to remove subsurface fluorescence. Before processing, spheres are visible for ≈100 μm in this semitranslucent phantom.
Fig 2.
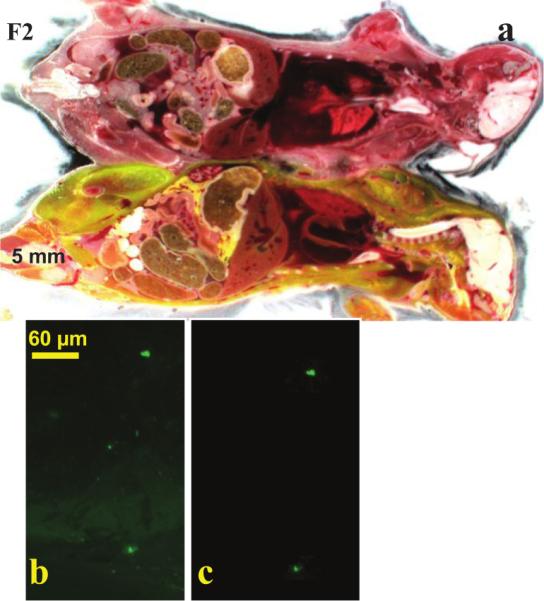
Parabiosis mouse model experiments with Dr. Muschler (Orthopedic Surgery, CCF). A GFP-positive mouse was surgically conjoined with a recipient (a) and the tibia was fractured. GFP-labeled blood born cells were recruited from the donor to the injury site, as shown in a high resolution fluorescent image of the fracture site (b). With next-image processing, sub-surface fluorescence was removed, and cells were clearly identified (c).
Fig 3.

Cryo-imaging of micro-vessels in a mouse brain perfused with India ink. Low (left) and high (middle) magnifications show dark blood vessels. Vessels were segmented using an interactive 3D region growing algorithm. A 3D surface rendering of blood vessels (right) is shown. Vessels were segmented down to a size of about 10 μm in diameter.
Algorithm for Quantification of Cells/Microspheres
Typical cellular quantification algorithms range in sophistication from simple manual dot counting, to deformable model [11] and neural network based segmentation [12]. All of these methods require images of sufficient resolution, such that the cell boundary is distinguishable. In order to limit the number of images required to image an entire mouse, cryo-imaging is performed with a 15.6 μm pixel size, as compared to the smallest pixel size of 1.2 μm. At this resolution cells are approximately the size of a single pixel, making it impossible to resolve cell boundaries in large clusters of cells. For this reason it is necessary to utilize quantification algorithms that do not rely on the imaging resolution to perform quantification.
Model based quantification algorithms are commonly used when the size and shape of cells varies greatly and when the boundary between cells is indistinguishable due to low resolution or contrast [13]. The cell model is used to incorporate prior information about cell size, shape and intensity, as well as the transfer function of the imaging system[14]. A common model of a single cell and image formation is a Gaussian [13;15;16]. Cells are approximately the size of a pixel, which appears as a single bright point. This point is then blurred during imaging due to pixilation, the transfer function of the imaging system, and light scattering by the tissue. These blurring properties may be approximated as convolution with a Gaussian. By modeling a cell as a Gaussian we are accounting for the spherical shape of the cells, as well as blurring due to the imaging process.
Model based cell quantification is typically performed in two segments; detection/segmentation and cell estimation [16]. To perform cell quantification, it is first necessary to detect and segment cell clusters. To segment cell clusters we use hysteresis thresholding. Hysteresis thresholding first segments the image by a ‘hard’ threshold, leaving only the brightest pixels in each cluster of cells. A ‘soft’ threshold is then applied, which includes all pixels in the cell clusters, as well as background and autofluorescence. Pixels selected by the ‘soft’ threshold are retained if they are connected to a pixel selected by the ‘hard’ threshold. This segmentation successfully removes autofluorescence from the images, while leaving cluster intensity unaffected (Fig. 4). Connected component analysis is then performed to uniquely label subsets of connected components remaining in the hysteresis thresholded image [17]. From these uniquely labeled connected components, statistical properties such as volume, integrated intensity, center of mass, and bounding box are determined. After feature properties have been determine for segmented clusters, these features are used in conjunction with a model based approach to quantify the number of cells in a cluster.
Fig 4.

Cryo-imaging of mouse skeletal muscle after cardiac injection with fluorescent microspheres. The original image shows background autofluorescence (a). After hysteresis thresholding (b) autofluorescent signal has been removed, leaving only the fluorescent microspheres.
Histogram analysis of cluster properties, e.g. volume and integrated intensity, is performed to estimate the number of cells per cluster. Cells are separated prior to injection into the specimen, and therefore single cells will form the largest peak on a histogram of cluster integrated intensity and volume. Cell aggregates contained within the largest peak are assumed to be single cells, and are used to determine the model properties for a single cell. Each single cell cluster is fit to a Gaussian with free parameters; σ, integrated intensity (It), and the center of the Gaussian (xc and yc). The optimal parameters are determined for each single cell cluster using a least-squares error objective function and a nonlinear optimizer [18]. The model parameters for a single cell are set to be the average and average It of all Gaussian fits.
Model based analysis is performed to estimate the number of cells per cluster for all detected clusters in the volume. For any given cluster, we first estimate the number of cells in a cluster by determining the integrated intensity of the cluster and dividing this by It, determined above. A range of integer numbers of cells around this estimate is used to find which number of cells best fits the cluster images. For each integer number of cells, the optimal placement of the cells is determined in 3D that most closely resembles the 3D image of the cluster. Optimal placement is determined through minimization of an objective function using a nonlinear optimizer [18]. The number of cells that best matches the 3D cluster image, as determined through least square error is designated as the number of cells in the cluster.
Results
Prior to next-image processing, fluorescent microspheres appeared as “comet tails” when viewed from the “side” in a 3D view (Fig. 1a). Subsurface fluorescence is more apparent in the imaging phantom than in tissue due to its relatively low absorption and scattering and its lack of autofluorescence background. As a result, the phantom is more difficult to successfully process, and it presents good data for evaluation and testing. In the next-image processed data (Fig. 1b) 97 ± 2% of subsurface fluorescence was removed.
Next-image processing removed large autofluorescent background haze caused by the bone, further improving single cell contrast in cryo-images of fluorescent cells in a parabiosis experiment (Fig. 2). Images were taken of a parabiosis mouse model in which a GFP positive transgenic mouse and a GFP negative littermate had surgically joined circulatory system. Cryo-images show fluorescently labeled cells at a fracture site in a mouse (Fig. 2b). Subsurface fluorescence was successfully removed leaving in-plane fluorescence from two surface GFP positive cells (Fig. 2c).
Next-image processing removed subsurface haze, improving 3D surface renderings of brain micro-vasculature (Fig. 3). High resolution images were taken of the brain of a mouse perfused with India ink (Fig. 3a). Subsurface haze caused by the India ink obscured cryo-images prior to next-image processing (Fig. 3b). After next-image processing vasculature in the brain was segmented utilizing 3D seeded region growing and surfaced rendered, showing local blood vessel branching (Fig. 3c).
Background autofluorescence in fluorescent cryo-images can lead to errors in quantification, due to the inclusion of false positives. Autofluorescence in the skeletal muscle of a mouse perfused with 15 μm microspheres is visible prior to hysteresis thresholding (Fig. 4a). Hysteresis thresholding successfully removed autofluorescence, leaving the fluorescent microspheres intensity unchanged (Fig. 4b).
Conclusion
Cryo-imaging provides both color brightfield and fluorescence images with single cell resolution and sensitivity over an entire mouse. The sensitivity of cryo-imaging allows for the identification of fluorescently-labeled single cells or cell clusters within a mouse and to determine their precise location relative to the tissue anatomy with micron-scale resolution. Utilizing image processing techniques described in this paper we are able to remove subsurface fluorescence, segment cells/clusters, and estimate the number of cells per cluster.
Acknowledgements
This work was supported by NIH 1T32EB007509-01 (Interdisciplinary Biomedical Imaging Training Program).
Bibliography
- 1.Roy D, Steyer G, Gargesha M, Stone M, Wilson D. 3D Cryo-Imaging: A Very High-resolution View of the Whole Mouse. 2009. [DOI] [PMC free article] [PubMed]
- 2.Kenzie-Graham A, Lee EF, Dinov ID, Bota M, Shattuck DW, Ruffins S, Yuan H, Konstantinidis F, Pitiot A, Ding Y, Hu GG, Jacobs RE, Toga AW. A multimodal, multidimensional atlas of the C57BL/6J mouse brain. Journal of Anatomy. 2004 Feb.204(2):93–102. doi: 10.1111/j.1469-7580.2004.00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nissanov J, Bertrand L, Tretiak O. Cryosectioning distortion reduction using tape support. Microscopy Research and Technique. 2001 May;53(3):239–240. doi: 10.1002/jemt.1089. [DOI] [PubMed] [Google Scholar]
- 4.Spaan JAE, ter Wee R, van Teeffelen JWGE, Streekstra G, Siebes M, Kolyva C, Vink H, Fokkema DS, VanBavel E. Visualisation of intramural coronary vasculature by an imaging cryomicrotome suggests compartmentalisation of myocardial perfusion areas. Medical & Biological Engineering & Computing. 2005 Jul;43(4):431–435. doi: 10.1007/BF02344722. [DOI] [PubMed] [Google Scholar]
- 5.Weninger WJ, Mohun T. Phenotyping transgenic embryos: a rapid 3-D screening method based on episcopic fluorescence image capturing. Nature Genetics. 2002 Jan.30(1):59–65. doi: 10.1038/ng785. [DOI] [PubMed] [Google Scholar]
- 6.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nature Reviews Cancer. 2003 Dec.3(12):921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 7.Wallace W, Schaefer LH, Swedlow JR. A workingperson's guide to deconvolution in light microscopy. Biotechniques. 2001 Nov.31(5):1076–+. doi: 10.2144/01315bi01. [DOI] [PubMed] [Google Scholar]
- 8.McNally JG, Karpova T, Cooper J, Conchello JA. Three-dimensional imaging by deconvolution microscopy. Methods-A Companion to Methods in Enzymology. 1999 Nov.19(3):373–385. doi: 10.1006/meth.1999.0873. [DOI] [PubMed] [Google Scholar]
- 9.Park SC, Park MK, Kang MG. Super-resolution image reconstruction: A technical overview. Ieee Signal Processing Magazine. 2003 May;20(3):21–36. [Google Scholar]
- 10.Magain P, Courbin F, Sohy S. Deconvolution with correct sampling. Astrophysical Journal. 1998 Feb.494(1):472–+. [Google Scholar]
- 11.Garrido A, de la Blanca NP. Applying deformable templates for cell image segmentation. Pattern Recognition. 2000 May;33(5):821–832. [Google Scholar]
- 12.Sjostrom PJ, Frydel BP, Wahlberg LU. Artificial neural network-aided image analysis system for cell counting. Cytometry. 1999 May;36(1):18–26. doi: 10.1002/(sici)1097-0320(19990501)36:1<18::aid-cyto3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 13.Wörz S, Heinzer S, Weiss M, Rohr K. Model-based segmentation and quantification of subcellular structures in 2D and 3D fluorescent microscopy images. 6916 ed 2008. [Google Scholar]
- 14.Bernard R, Kanduser M, Pernus F. Model-based automated detection of mammalian cell colonies. Physics in Medicine and Biology. 2001 Nov.46(11):3061–3072. doi: 10.1088/0031-9155/46/11/320. [DOI] [PubMed] [Google Scholar]
- 15.Bernard SL, Ewen JR, Barlow CH, Kelly JJ, McKinney S, Frazer DA, Glenny RW. High spatial resolution measurements of organ blood flow in small laboratory animals. American Journal of Physiology-Heart and Circulatory Physiology. 2000 Nov.279(5):H2043–H2052. doi: 10.1152/ajpheart.2000.279.5.H2043. [DOI] [PubMed] [Google Scholar]
- 16.Noordmans HJ, Smeulders AWM. Detection and characterization of isolated and overlapping spots. Computer Vision and Image Understanding. 1998 Apr.70(1):23–35. [Google Scholar]
- 17.Olabarriaga SD, Smeulders AW. Interaction in the segmentation of medical images: a survey. Med. Image Anal. 2001 Jun;5(2):127–142. doi: 10.1016/s1361-8415(00)00041-4. [DOI] [PubMed] [Google Scholar]
- 18.Nelder J. A simplex method for function minimization. 7 ed 1965. [Google Scholar]