

Cocaine use affects more than 30 million people in this country, and is responsible for the majority of deaths ascribed to drug overdose.1 While the pharmacodynamics of cocaine toxicity are increasingly understood, the molecular determinants that define fatal outcome remain largely unknown. Indeed, in cocaine poisoning the mainstay of management is supportive, limited predominantly to sedation and monitoring of vital signs.2 Here, we report that the susceptibility to cocaine-induced mortality is genetically controlled. By identifying the KCNJ11 gene as a safeguard element in the response to cocaine overdose, the present study unmasks a previously unrecognized framework for targeted therapy of cocaine intoxication.
An overdriven activation of the sympathetic nervous system, which results in a mismatch of demand and supply producing seizures, heart rhythm disorders and cardiopulmonary collapse, is an established substrate of cocaine toxicity.3 By inhibiting reuptake of catecholamines in presynaptic neurons, cocaine induces an excessive accumulation of sympathetic neurotransmitters, resulting in a general state of hyperadrenergic stimulation.4 Distributed throughout the body, ATP-sensitive K+ (KATP) channels have been recently implicated in securing tolerance to systemic stress imposed by a sympathetic surge.5 KATP channels operate as unique molecular rheostats, capable of adjusting membrane potential-dependent functions to match cellular metabolic demands.6 Underscoring the critical role for KATP channels in vital homeostatic functions, targeted disruption of KCNJ11, the gene encoding the pore-forming Kir6.2 subunit of KATP channels, precipitates generalized seizure,7 heart failure8 and arrhythmia under stress.9 Therefore, we tested here whether KATP channel function provides a protective mechanism in the setting of cocaine toxicity.
In response to systemic administration of a toxic dose of cocaine, Kir6.2-knockout (Kir6.2-KO) mice7–9 demonstrated a marked survival disadvantage compared to sex- and age-matched wild-type (WT) controls (Figure 1a). Specifically, at 24-h follow-up, a single injection of cocaine (100mgkg−1 i.p.) produced a 67% mortality in the Kir6.2-KO (n = 15) compared to 15% mortality in the WT (n = 14, P < 0.01; Figure 1a). In fact, the absence of functional KATP channels in the Kir6.2-deficient cohort predisposed to a prompt and massive lethality, with this propensity to aggravated mortality over WT also observed in the Kir6.2-KO following repeated cocaine abuse (P = 0.008, WT vs Kir6.2-KO; Figure 1b). Thus, the presence of intact KATP channels critically defines survivorship in cocaine overdose.
Figure 1.
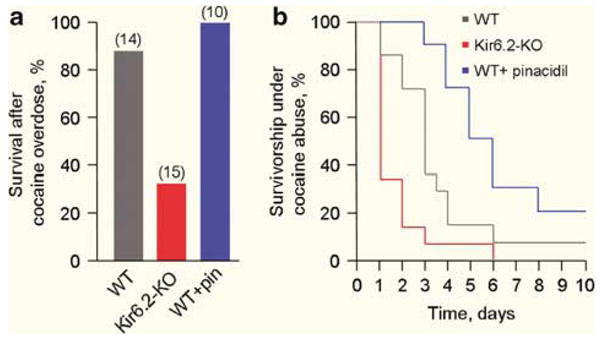
KATP channels protect against cocaine-induced death. (a) Compared to wild type (WT), knockout (KO) of KATP channels (Kir6.2-KO) produced survival disadvantage after single injection of cocaine (100mgkg−1 i.p.; P < 0.01). Conversely, pretreatment with the KATP channel opener pinacidil (pin, 15mgkg−1, i.p.) abrogated mortality in WT (P < 0.05). Cohort size provided in parentheses. (b) Aggravated mortality trends following repeated cocaine abuse (100mgkg−1, i.p. once daily) in Kir6.2-KO vs WT (P = 0.008). Pretreatment with pinacidil (15mgkg−1, i.p., 30 min before each cocaine injection) abrogated cumulative mortality with improved long-term survival (P < 0.01) over WT (without pinacidil). Statistical analysis was performed using Pearson’s χ2 or log-rank tests as appropriate.
Despite protection afforded by KATP channels, WT mice demonstrated a significant vulnerability to cocaine (Figures 1a and b). Promotion of KATP channel activity in the WT through pretreatment with the prototypic and clinically available potassium channel opening drug, pinacidil (15mgkg−1, i.p.),10 abrogated acute and cumulative mortality with improvement in long-term survival (n = 10, P < 0.05; Figures 1a and b). Thus, use of a potassium channel opener imparts enhanced protection, identifying pharmacological targeting of KATP channel function as a novel strategy for prevention of death in cocaine overdose.
Cocaine-induced mortality is a leading cause of deaths ascribed to drug abuse; yet, limited information is presently available regarding the identity of the genetic determinants defining prognosis. The present study, using the Kir6.2-KO model, provides first evidence linking the status of KATP channels with outcome in cocaine abuse, establishing in vivo a novel susceptibility mechanism in cocaine toxicity. KATP channels are uniquely positioned to serve as stress-responsive elements as they harness the capacity to translate signals of cellular distress into protective stress adaptation.11,12 While the intimate events underlying the beneficial role of KATP channels in cocaine intoxication remain to be established, KATP channel activation provides a feedback control necessary for preservation of cellular homeostasis under stress by regulating action potential duration and thus Ca2+ influx, preventing the deleterious consequences of Ca2+ overload associated with hyperadrenergic states.5,12 While the hyperadrenergic effect of cocaine sets the conditions for KATP channel opening, a direct inhibitory effect of cocaine could result in suboptimal activation of the channel.13 In fact, use of KATP channel opening drugs, known to reduce intracellular Ca2+ loading,10 were discovered here as effective in abrogating the lethal effect of cocaine overdose, opening a new therapeutic avenue in managing substance abuse.
The present demonstration that KATP channels protect against cocaine-induced death provides a foundation for translational investigation of the role of these cytoprotective channels in the population exposed to cocaine use. Recognition of individuals with a genetic background predisposing to increased risk, and further evaluation of the effectiveness of KATP channel-based therapeutic strategies will provide a critical step towards a personalized approach to prevention and management of life-threatening consequences of cocaine overdose.
Acknowledgments
This work was supported by grants from the National Institutes of Health, Marriott Heart Disease Research Program, Marriott Foundation, Ted Nash Long Life Foundation and Japanese Ministry of Education, Science, Sports, Culture and Technology.
Footnotes
Conflict of interest
The authors declared that they have no competing interests.
References
- 1.Hahn IH, Hoffman RS. EmergMed Clin North Am. 2001;19:493–510. doi: 10.1016/s0733-8627(05)70197-8. [DOI] [PubMed] [Google Scholar]
- 2.Tintinalli JE, Kellen GD, Stapczynski JS, Ma OJ, Cline DM. Emergency Medicine. 6. McGraw-Hill; New York: 2004. [Google Scholar]
- 3.Benowitz N. Pharmacol Toxicol. 1993;72:3–12. doi: 10.1111/j.1600-0773.1993.tb01331.x. [DOI] [PubMed] [Google Scholar]
- 4.Mendelson JH, Mello NK. N Engl J Med. 1996;334:965–972. doi: 10.1056/NEJM199604113341507. [DOI] [PubMed] [Google Scholar]
- 5.Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, et al. Proc Natl Acad Sci USA. 2002;99:13278–13283. doi: 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zingman LV, Hodgson D, Alekseev AE, Terzic A. Mol Psychiatry. 2003;8:253–254. doi: 10.1038/sj.mp.4001323. [DOI] [PubMed] [Google Scholar]
- 7.Yamada K, Ji JJ, Yuan H, Miki T, Sato S, Horimoto N, et al. Science. 2001;292:1543–1546. doi: 10.1126/science.1059829. [DOI] [PubMed] [Google Scholar]
- 8.Kane GC, Behfar A, Dyer RB, O’Cochlain DF, Liu XK, Hodgson DM, et al. Hum Mol Genet. 2006;15:2285–2297. doi: 10.1093/hmg/ddl154. [DOI] [PubMed] [Google Scholar]
- 9.Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, et al. Nat Clin Pract Cardiovasc Med. 2007;4:110–116. doi: 10.1038/ncpcardio0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jahangir A, Terzic A. J Mol Cell Cardiol. 2005;39:99–112. doi: 10.1016/j.yjmcc.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alekseev AE, Hodgson DM, Karger AB, Park S, Zingman LV, Terzic A. J Mol Cell Cardiol. 2005;38:895–905. doi: 10.1016/j.yjmcc.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kane GC, Liu XK, Yamada S, Olson TM, Terzic A. J Mol Cell Cardiol. 2005;38:937–943. doi: 10.1016/j.yjmcc.2005.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu SN, Chang HD, Sung RJ. Basic Clin Pharmacol Toxicol. 2006;98:510–517. doi: 10.1111/j.1742-7843.2006.pto_354.x. [DOI] [PubMed] [Google Scholar]