

Summary
The inhibins are secreted α:β heterodimers of the TGF-β superfamily that are mainly synthesized in Sertoli cells and granulosa cells, and are critical regulators of testicular and ovarian development and function. Mice homozygous for a targeted deletion of the inhibin α subunit gene (Inha-/-) develop sex cord-stromal tumors in a gonadotropin-dependent manner. Here, we determine the contribution of LH to gonadal tumorigenesis by generating mice deficient in both inhibins and LH. Inha-/-Lhb-/- mice have increased survival and delayed tumor progression, and these observations correlate with lower serum FSH and estradiol levels compared to Inha-/- controls. Double mutant testicular tumors demonstrate decreased expression of cyclin D2, while double mutant ovarian tumors have elevated expression of p15INK4b and trend toward higher levels of p27Kip1. We conclude that LH is not required for tumor formation in the absence of inhibins but promotes tumor progression, likely through alterations in serum hormone levels and cell cycle regulators.
Keywords: inhibin, luteinizing hormone, ovarian cancer, testicular cancer
Introduction
The hypothalamic-pituitary-gonadal axis is central to mammalian reproductive physiology. Hypothalamic GnRH stimulates the synthesis and secretion of the pituitary gonadotropins FSH and LH, heterodimeric glycoproteins each composed of a common α subunit noncovalently linked to a hormone-specific β subunit. FSH signals through receptors on Sertoli cells in the testis and granulosa cells in the ovary to regulate spermatogenesis and early follicle development, respectively. FSH-deficient (Fshb-/-) male mice are fertile despite having decreased testis size, sperm count, and sperm motility, while FSH-deficient females are infertile with small ovaries and a block in follicle maturation prior to antrum formation (Kumar et al., 1997). In contrast to FSH, LH binds to receptors on testicular Leydig cells and ovarian theca cells to promote gonadal steroidogenesis. LH receptors are also expressed on granulosa cells during late follicle development, when LH signaling is critical for ovulation and formation of the corpus luteum. LH-deficient (Lhb-/-) mice are hypogonadal and infertile, with males displaying reduced testosterone levels and a block in spermatogenesis at the round spermatid stage, and females demonstrating reduced serum estradiol and progesterone, and no preovulatory follicles or corpora lutea (Ma et al., 2004).
To maintain the balance of growth and differentiation during spermatogenesis and folliculogenesis, the pituitary gonadotropins cooperate with inhibins and activins, peptide hormones of the transforming growth factor-β (TGF-β) superfamily that also includes bone morphogenetic proteins, growth and differentiation factors, and Müllerian inhibiting substance. Inhibins (α:βA, α:βB) and activins (βA:βA, βB:βB, βA:βB) are dimers secreted from Sertoli cells and granulosa cells that suppress (i.e., inhibins) or stimulate (i.e., activins) pituitary production and secretion of FSH. To define the specific functions of inhibins, we previously generated mice homozygous for a targeted deletion of the inhibin α subunit gene (Inha-/-) and therefore completely inhibin-deficient (Matzuk et al., 1992). These mice develop sex cord-stromal tumors (e.g., granulosa/Sertoli cell tumors) with 100% penetrance in both males and females as early as 4 weeks of age (Matzuk et al., 1992). The tumors secrete estradiol (Matzuk et al., 1996) and activins (Matzuk et al., 1994) into the circulation, with the elevated serum activins causing a cancer cachexia-like syndrome marked by severe weight loss and multiple extragonadal defects from which the mice eventually die (Coerver et al., 1996; Matzuk et al., 1994). These studies identified inhibins as secreted tumor suppressors with gonadal specificity, and provided a valuable model to investigate in vivo the mechanisms of gonadal tumorigenesis and identify various modifiers. Intercrossing inhibin α mutants with mice deficient in GnRH [i.e., the hypogonadal (Gnrh1hpg) model], in which FSH and LH levels are suppressed, led to double homozygous mutants that were strikingly free of tumors and the cancer cachexia-like syndrome (Kumar et al., 1996). Thus, these genetic studies confirmed that gonadotropins are critical modifiers of sex cord-stromal tumor development in inhibin-deficient mice.
Consistent with the known function of inhibins to suppress FSH, Inha-/- mice have increased serum FSH levels compared to wild type mice (Matzuk et al., 1992). To delineate the individual contribution of FSH to gonadal tumorigenesis, we previously generated mice deficient in both inhibins and FSH (Kumar et al., 1999). These double mutant mice demonstrated increased survival because of slower growing and less invasive tumors (Kumar et al., 1999). Although Inha-/-Fshb-/- males also have decreased incidence of tumors, all double homozygous mutant females eventually develop tumors and the accompanying wasting syndrome (Kumar et al., 1999). This genetic model lacking inhibin and only FSH, but not LH, suggests that FSH promotes sex cord-stromal tumor progression but is not strictly required for tumor formation.
The distinct phenotypes observed in Inha-/-Gnrh1hpg/hpg mice and Inha-/-Fshb-/- mice support the hypothesis that LH also contributes to the pathogenesis of ovarian and testicular cancers. Several gain-of-function experiments further support this notion and provide additional insight into LH regulation of gonadal tumors. Chronic hypersecretion of LH in transgenic female mice (bLHβ-CTP) causes strain-dependent formation of granulosa cell tumors with 100% penetrance by 5 months of age (Keri et al., 2000; Risma et al., 1995). Transgenic mice expressing SV40 T-antigen driven by the Inha promoter (Inhα/Tag) invariably develop granulosa cell tumors by 5-6 months of age (Kananen et al., 1995), and notably, double transgenic bLHβ-CTP/Inhα/Tag mice display earlier tumor formation and more rapid progression (Mikola et al., 2003). In another transgenic mouse model, simultaneous overexpression of both the α and β subunits of hCG, an LH analog that functions through binding to the LH receptor, results in over 1000-fold excess of bioactive hCG/LH and the development of ovarian germ cell tumors (Huhtaniemi et al., 2005). Chronic hCG treatment also causes the formation of Leydig cell tumors in male rats (Neumann, 1991).
In the present study, we generated mice deficient in both inhibins and LH to further define the unique and independent roles of LH in gonadal tumorigenesis. We demonstrate that LH is not required for tumor formation in the absence of inhibins but promotes tumor progression, most likely through alterations in serum hormone levels and cell cycle regulators.
Materials and Methods
Experimental animals
Generation and genotyping of Inhα (Inha) and LHβ (Lhb) mutant mice by PCR or Southern blot have been described (Ma et al., 2004; Matzuk et al., 1992; Pierson et al., 2000). Inha+/- mice were initially crossed to Lhb+/- mice to obtain Inha+/-Lhb+/- mice. The double heterozygotes were intercrossed to generate Inha-/-, Inha-/-Lhb+/-, Inha-/-Lhb-/-, and Lhb-/-mutants, all of which were recovered at the expected Mendelian ratios. Mice were maintained as described in the NIH Guide for the Care and Use of Laboratory Animals and according to approved institutional animal protocols.
Survival curves
Mice were weighed weekly to monitor for weight loss due to the cancer cachexia-like syndrome. Inha-/- and Inha-/-Lhb-/- mice were sacrificed upon reaching 14.0-16.0 grams, a range at which they were unlikely to survive for an additional week based on previous studies (Kumar et al., 1996; Matzuk et al., 1994). These mice are referred to as ‘end-stage’ mutants and span the following ages: Inha-/- males (8-31 weeks old); Inha-/-Lhb-/- males (6-60 weeks old); Inha-/- females (10-33 weeks old); Inha-/-Lhb-/- females (11-53 weeks old).
Morphological and histological analysis
Testes, ovaries, stomachs, and livers were collected from mutants of each sex and genotype. Specimens were fixed in 10% neutral buffered formalin (Richard-Allan Scientific, Kalamazoo, MI) and subsequent processing, embedding, sectioning, and staining with periodic acid-Schiff/hematoxylin or hematoxylin/eosin were performed by standard procedures in the Baylor College of Medicine Pathology Core Services laboratory. For each tissue, at least 5 independent specimens were examined from each sex and genotype. Liver weights were also measured.
Serum analysis
Mice were anesthetized by isoflurane inhalation (Abbott Laboratories, North Chicago, IL), and blood was collected in Microtainer tubes (Becton Dickinson, Franklin Lakes, NJ) by closed cardiac puncture. Serum was separated by centrifugation and stored at -20°C until further use. FSH, estradiol, and testosterone measurements were made by the University of Virginia Ligand Assay and Analysis Core (Specialized Cooperative Centers Program in Reproduction Research NICHD/NIH U54 HD28934). Assay information is available at http://www.healthsystem.virginia.edu/internet/crr/ligand.cfm. Serum profiles are represented as box plots because of the intrinsic variability in hormone levels between mice within the same group.
RNA isolation and quantitative real-time PCR (QPCR)
Testicular and ovarian tumors from end-stage Inha-/- and Inha-/-Lhb-/- mutants were collected and preserved in RNAlater (Ambion, Austin, TX), and total RNA was isolated from 20-80 mg of preserved tissue using TRIzol Reagent (Invitrogen, Carlsbad, CA) followed by cleanup using the RNeasy Mini Kit (Qiagen, Valencia, CA) with on-column DNase digestion using the RNase-Free DNase Set (Qiagen), all according to the manufacturers' protocols. One microgram of total RNA was reverse-transcribed in a 50 μl reaction using 250 U Superscript II reverse transcriptase (Invitrogen) and random hexamers (Invitrogen). Samples were diluted 10-fold and 10 μl were used for each QPCR reaction. QPCR was performed on the ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA) using predesigned TaqMan Gene Expression Assays (ABI) and eukaryotic 18S rRNA as an endogenous control. The following TaqMan assays were used: Star, Mm00441558; Cyp11a1, Mm00490735; Cyp17a1, Mm00484040; Ccnd2, Mm00438071; Cdkn1b, Mm00438167; Cdkn2b, Mm00483241; Inhba, Mm00434338; Inhbb, Mm01286587; and eukaryotic 18S rRNA, 4319413E. TaqMan PCR was performed using the TaqMan Universal PCR Master Mix (ABI) in 21 μl. The reaction conditions were as follows: 10 min hold at 95°C followed by 40 cycles of: 15 sec at 95°C (denaturation); 1 min at 60°C (annealing/extension). Each sample was analyzed in duplicate or triplicate (n = 4-5 independent mice per genotype). Two nontemplate control (ribonuclease-free water) samples were included on each plate for each primer-probe set. The relative quantity (RQ) of transcript was calculated using the 2-ΔΔCT method (Livak and Schmittgen, 2001).
Statistical analysis
Statistical analysis was performed using the JMP version 7.0.1 statistical package (SAS Institute, Cary, NC). Survival curves were compared using the log-rank test. Liver weights and 6-week-old serum hormone data were evaluated using one-way ANOVA followed by Tukey's Honestly Significant Differences (HSD) test. End-stage serum hormone measurements and QPCR data were evaluated using the Student's t-test. p<0.05 was considered statistically significant.
Results
Inhibin-deficient mice live longer in the absence of LH
To investigate the potential contribution of LH to the pathogenesis of gonadal cancers, we generated mice deficient in both inhibins and LH. Inha-/-Lhb-/- male and female mice demonstrated increased survival in comparison to Inha-/- and Inha-/-Lhb+/- controls (Fig. 1). The median survival for Inha-/- males was 11 weeks, an age at which 78% of Inha-/-Lhb-/- males were alive (Fig. 1A). When Inha-/-Lhb-/- males reached their median survival at 16 weeks of age, only 27% of Inha-/- males were alive. For Inha-/- females, the median survival was 17 weeks, an age at which 63% of Inha-/-Lhb-/- females were alive (Fig. 1B). From this age onward, double knockout females died at a slower rate in comparison to controls. When double knockout females reached their median survival at 22 weeks, only 13% of Inha-/- females were alive. In addition, both male and female Inha-/-Lhb-/- mutants had maximum lifespans over one year of age, while the longest living Inha-/- male and female mice were 31 and 33 weeks old, respectively. In contrast to the other mutants, Lhb-/- male and female control mice demonstrated 100% survival.
Fig. 1.
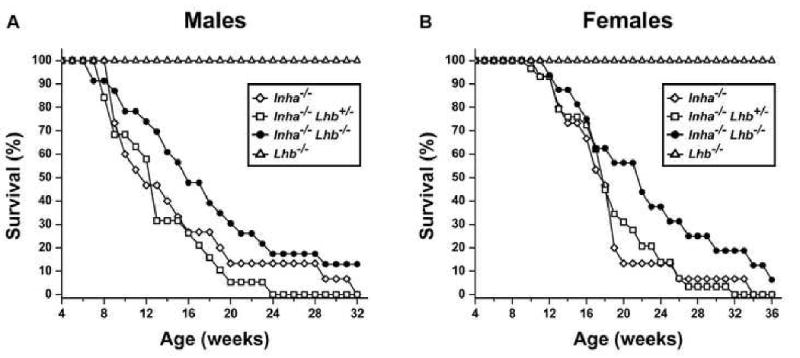
Survival curves for Inha-/-, Inha-/-Lhb+/-, Inha-/-Lhb-/-, and Lhb-/- males (A) and females (B). The increase in survival is significant for double knockout males, and from 17 weeks onward, significant for double knockout females (p<0.05, log-rank test). The following numbers of mice were used: Inha-/-, 15 males and 15 females; Inha-/-Lhb+/-, 19 males and 29 females; Inha-/-Lhb-/-, 23 males and 16 females; Lhb-/-, 20 males and 20 females.
End-stage double mutants have tumors that are histologically similar to Inha-/- tumors
All end-stage male and female Inha-/-Lhb-/- mice developed bilateral, multifocal, mixed, invasive sex cord-stromal tumors that were histologically similar to the tumors in Inha-/- controls (Figs. 2 and 3). The testes and ovaries of Inha-/- and Inha-/-Lhb-/- mutants were grossly enlarged and misshapen and often had multiple areas of hemorrhage. Microscopic analysis of testicular (Fig. 2) and ovarian (Fig. 3) tumors revealed multiple regions composed of poorly differentiated and mitotically active neoplastic cells that were circumscribed by a prominent neoplastic cellular stroma. In testicular tumors, seminiferous tubules were confined to the outer rim of tissue (Fig. 2, A and D) and often displayed architecture disrupted or invaded by adjacent nodules of tumor cells (Fig. 2, B and E). Ovarian tumors from both Inha-/-Lhb-/- mutants and Inha-/- controls demonstrated tubular structures lined with Sertoli-like cells (Fig. 3, B and E). Call-Exner bodies, consisting of an eosinophilic region surrounded by a rosette-like arrangement of granulosa cells, are characteristic of granulosa cell tumors and were also evident (Fig. 3, C and F). In contrast to the Inha-/- and Inha-/-Lhb-/- mutants, Lhb-/- mice had small gonads and did not develop gonadal tumors. Instead, Lhb-/- testes demonstrated sparse interstitium with few Leydig cells and spermatogenic arrest at the round spermatid stage (Fig. 2, G-I), while Lhb-/- ovaries contained many degenerating oocytes and a block in folliculogenesis at the antral follicle stage (Fig. 3, G-I). These observations are consistent with prior characterization of these mutants (Ma et al., 2004).
Fig. 2.
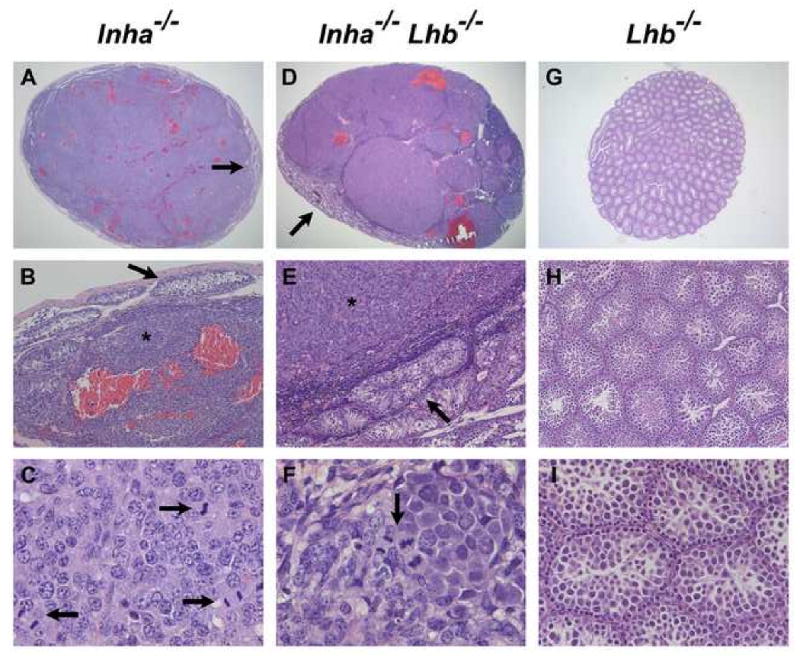
Histological analysis of testicular tumors from end-stage Inha-/- and Inha-/-Lhb-/- males as compared to testes from 12-week-old Lhb-/- males (n ≥ 5 mice of each sex and genotype). Multiple areas of hemorrhage are present in testicular tumors from Inha-/- (A) and Inha-/-Lhb-/- (D) mutants. The tumors display poorly differentiated neoplastic cells (asterisks in B, E) that are compressing the few remaining seminiferous tubules (arrows in A, B, D, E). Numerous mitotic figures are also present (arrows in C, F). In contrast, Lhb-/- testes are non-hemorrhagic and do not contain tumorigenic foci (G-I). The major histologic abnormalities in Lhb-/- testes are their small size (G), sparse interstitium with few Leydig cells (H), and spermatogenic arrest at the round spermatid stage (I), consistent with prior observations (Ma et al., 2004). Ages of mice: A-C: 9 wks; D-F: 18 wks; G-I: 12 wks. Magnification: A: ×15.6; B, E, H: ×100; C, F: ×640; D: ×12.5; G: ×20; I: ×250.
Fig. 3.
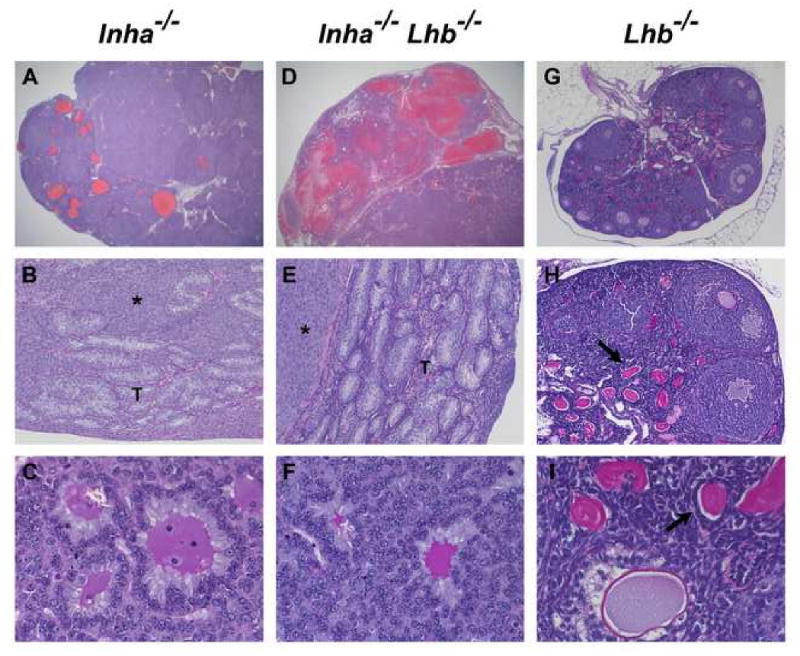
Histological analysis of ovarian tumors from end-stage Inha-/- and Inha-/-Lhb-/- females as compared to ovaries from 12-week-old Lhb-/- females (n ≥ 5 mice of each sex and genotype). The ovarian tumors from both Inha-/- and Inha-/-Lhb-/- mutants are hemorrhagic (A, D) and composed of poorly differentiated neoplastic cells (asterisks in B, E) along with tubular structures (T) lined with Sertoli-like cells and Call-Exner bodies (C, F) that are characteristic of granulosa cell tumors. In contrast, Lhb-/- ovaries are non-hemorrhagic and do not contain tumorigenic foci (G-I). These ovaries are small and contain numerous degenerating oocytes (arrows in H, I). Preovulatory follicles and corpora lutea are notably absent from Lhb-/- ovaries, indicating a block in folliculogenesis at the antral follicle stage, as previously reported (Ma et al., 2004). Ages of mice: A: 12 wks; B: 25 wks; C: 15 wks; D: 14 wks; E: 23 wks; F: 13 wks; G-I: 12 wks. Magnification: A, D: ×12.5; B, E, H: ×100; C, F, I: ×400; G: ×40.
Inha-/-Lhb-/- mice develop the cancer cachexia-like syndrome similar to Inha-/- mice
In contrast to Lhb-/- mutants, all Inha-/- and Inha-/-Lhb-/- males and females developed and died from the cancer cachexia-like syndrome secondary to gonadal tumor development. The wasting syndrome is caused by tumor-secreted activins signaling through the activin receptor type IIA, and is marked by progressive and severe weight loss, a hunchback and sunken-eye appearance, thoracic kyphoscoliosis, lethargy, and pathologic defects in the stomach and liver (Coerver et al., 1996; Matzuk et al., 1994). To investigate the influence of LH on the extent of the wasting syndrome in inhibin-deficient mice, we examined stomachs and livers from end-stage double mutants (Fig. 4). Similar to Inha-/- mice, Inha-/-Lhb-/- mice demonstrated mucosal atrophy with depletion of parietal cells in the glandular stomach (Fig. 4, A-C). These mice also displayed hepatocellular inflammation and necrosis around the central vein that resulted in liver weights comparable to those of cachectic end-stage Inha-/- controls and significantly decreased compared to Lhb-/- controls (Fig. 4, D-G).
Fig. 4.
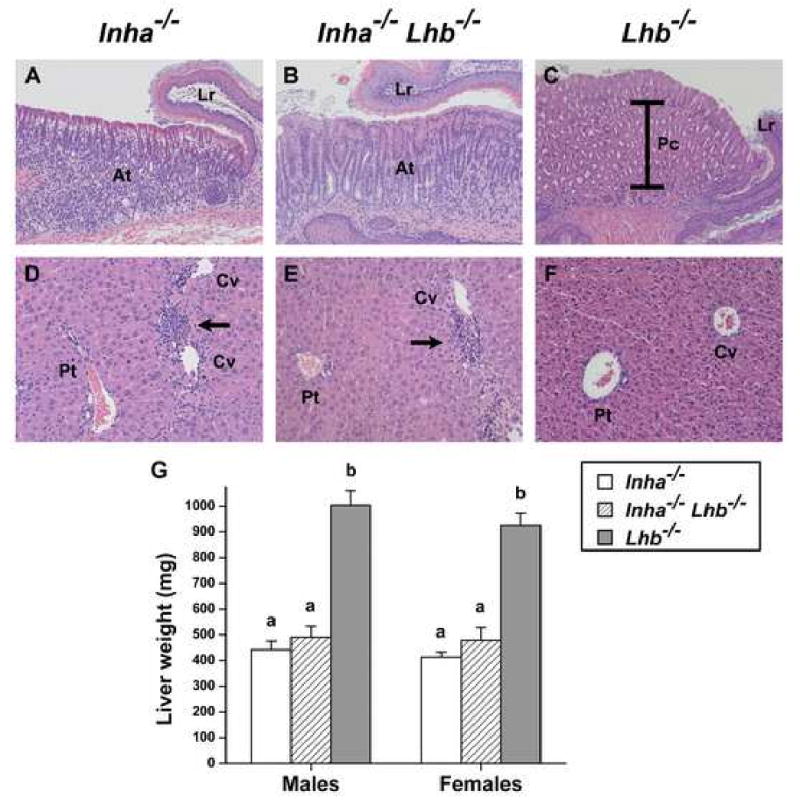
Histological analysis of stomachs (A-C) and livers (D-F) from end-stage Inha-/- and Inha-/-Lhb-/- mutants as compared to healthy Lhb-/- mice. (A-C) Glandular stomach photographed at the limiting ridge (Lr), the junction between the glandular stomach and the squamous epithelium of the forestomach. Multiple layers of large, eosinophilic parietal cells (Pc, brackets) comprise the majority of cells in the glandular region of Lhb-/- mice (C, 12 wks). In contrast, mucosal atrophy (At) and depletion of parietal cells are evident in the glandular epithelium of both Inha-/- (A, 14 wks) and Inha-/-Lhb-/- (B, 9 wks) mice. The magnification in panels A and B is 1.6-fold greater than the magnification in panel C in order to demonstrate the same region, thus indicating the severity of glandular atrophy. (D-F) Healthy liver from an Lhb-/- mouse (F, 17 wks) with portal tract (Pt) to the left and two central veins (Cv) to the right. In contrast, the livers from end-stage Inha-/- (D, 12 wks) and Inha-/-Lhb-/- (E, 27 wks) mice have lymphocytic infiltrates (arrows) around the central veins. (G) Liver weights (mean ± SEM) of end-stage Inha-/- and Inha-/-Lhb-/- males and females are similar to each other but significantly decreased compared to liver weights of Lhb-/- mice (p<0.0001, one-way ANOVA followed by Tukey's HSD test). The following numbers of end-stage mice were used to measure liver weights, with the range of ages indicated: Inha-/-, 7 males (8-28 wks) and 8 females (10-33 wks); Inha-/-Lhb-/-, 9 males (7-60 wks) and 11 females (13-35 wks); Lhb-/-, 6 males (12 wks) and 6 females (12 wks). Magnification: A, B: ×100; C: ×62.5; D, E, F: ×160.
Inhibin-deficient mice have delayed tumor progression in the absence of LH
In spite of the fact that no histologic differences were observed in end-stage testicular and ovarian tumors between Inha-/- and Inha-/-Lhb-/- mice, nor in the extent of the cancer cachexia-like syndrome, the increased survival of double mutant males and females suggested that tumor progression may be slower in these mice. To test this hypothesis, we examined the testes and ovaries of Inha-/-, Inha-/-Lhb-/-, and Lhb-/- mutants that were sacrificed at 6 weeks of age (Fig. 5). Lhb-/- gonads did not develop tumors (Fig. 5, C and E), while all of the testes from Inha-/- mice were more than half-filled with tumor foci that were usually hemorrhagic (Fig. 5A). In contrast, 4 of 6 testes from Inha-/-Lhb-/- mice were less hemorrhagic and had smaller tumor foci and larger regions containing seminiferous tubules (Fig. 5B). The ovaries of 6-week-old double mutants also demonstrated slower tumor progression, with 4 of 5 ovaries showing no hemorrhage (Fig. 5E), in contrast to all of the Inha-/- ovaries which had multiple hemorrhagic cysts (Fig. 5D). In addition, oocytes were rarely present in Inha-/- ovaries (Fig. 5D) and when observed they often appeared to be degenerating, while 2 of 5 Inha-/-Lhb-/- ovaries demonstrated many oocytes (Fig. 5E).
Fig. 5.
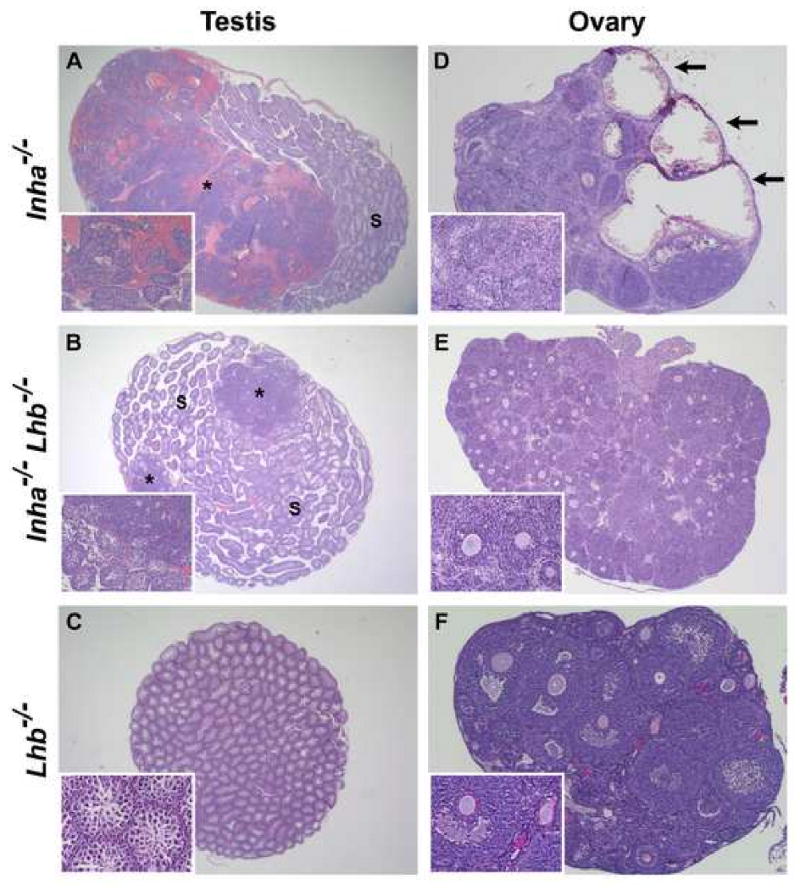
Histological analysis of testes (A-C) and ovaries (D-F) from 6-week-old Inha-/-, Inha-/-Lhb-/-, and Lhb-/- mice (n ≥ 5 gonads of each sex and genotype). (A) Inha-/- testis predominantly containing tumor cells and hemorrhage (asterisk), with a smaller region of normal seminiferous tubules (S). In contrast, the testis of an age-matched Inha-/-Lhb-/- male (B) is filled with seminiferous tubules, and only two small tumor foci (asterisks) are readily apparent. (C) No tumor foci are present in Lhb-/- testes. (D) Inha-/- ovary with architecture almost entirely disrupted by numerous hemorrhagic cysts (arrows) and nodules of poorly differentiated neoplastic cells. In contrast, the ovaries from age-matched Inha-/-Lhb-/- (E) and Lhb-/- (F) females are non-hemorrhagic, intact, and contain multiple oocytes. Magnification: A: ×12.5; B: ×15.6; C: ×20; D: ×31.3; E: ×25; F: ×62.5.
Inha-/-Lhb-/- mice have lower serum FSH and estradiol
Inhibin-deficient mice have elevated serum FSH and gonadal tumors that secrete large amounts of estradiol into the circulation (Matzuk et al., 1992; Matzuk et al., 1996). LH is critical for gonadal steroid biosynthesis, and Lhb-/- mice have decreased serum estradiol and testosterone (Ma et al., 2004). Accordingly, we measured serum hormone levels in Inha-/-, Inha-/-Lhb-/-, and Lhb-/- mutants (Fig. 6). At 6 weeks of age, double mutant females demonstrated serum FSH levels that were significantly lower than Inha-/- controls and higher than Lhb-/-controls (Fig. 6B), while 6-week-old double mutant males trended toward lower serum FSH as compared to Inha-/- controls (Fig. 6A). In addition, 6-week-old double mutant males had serum estradiol levels that were significantly lower than Inha-/- controls and higher than Lhb-/- controls (Fig. 6C). We did not detect any consistent differences in testosterone levels in males at 6 weeks of age (Fig. 6E).
Fig. 6.
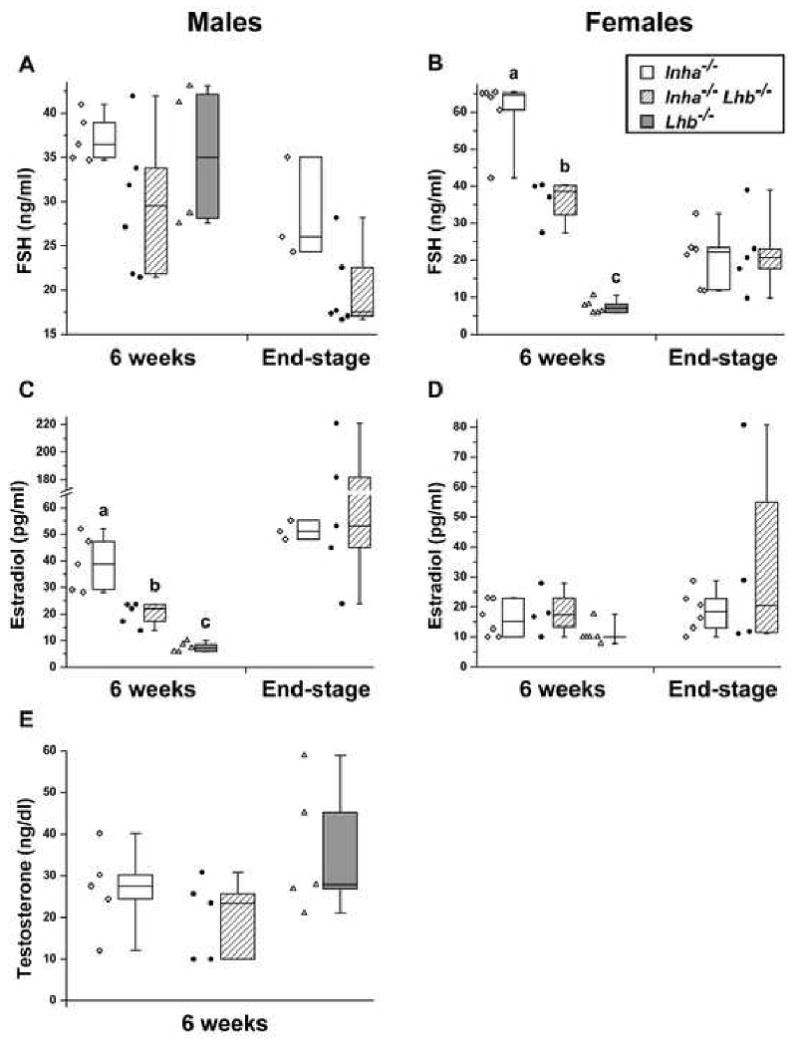
Serum FSH (A, B), estradiol (C, D), and testosterone (E) measurements for Inha-/-, Inha-/-Lhb-/-, and Lhb-/- males and females. The individual data points are shown and are also represented as standard box plots with whiskers extending to the minimum and maximum values. In 6-week-old females, FSH is significantly different between all three mutants, while in 6-week-old males, estradiol is significantly different between all three mutants (p<0.0001, oneway ANOVA followed by Tukey's HSD test). In addition, both 6-week-old and end-stage Inha-/-Lhb-/- males demonstrate a trend toward lower FSH compared to Inha-/- controls. Ages of end-stage Inha-/-mice: A, C: 9-11 wks; B, D: 10-25 wks. Ages of end-stage Inha-/- Lhb-/- mice: A, C: 7-60 wks; B, D: 13-33 wks.
Inha-/-Lhb-/- ovarian tumors have alterations in gene expression
We hypothesized that the absence of LH in inhibin-deficient mice would modify the gene expression pattern of gonadal tumors, perhaps explaining the differences in serum hormone levels and the slower progression of tumors in Inha-/-Lhb-/- mice compared to Inha-/- controls. To measure gene expression, we performed quantitative real-time PCR on mRNA from Inha-/- and Inha-/-Lhb-/- testicular and ovarian tumors (Fig. 7). Considering the role of LH in gonadal steroidogenesis, we examined genes involved in androgen and estrogen biosynthesis, namely Star (steroidogenic acute regulatory protein), Cyp11a1 (cytochrome P450 side chain cleavage), and Cyp17a1 (cytochrome P450 17α-hydroxylase/17,20-lyase). Because inhibin-deficient gonadal tumors are known to have aberrations in the expression of cell cycle regulatory genes (Cipriano et al., 2001), we determined the levels of Ccnd2 (cyclin D2), Cdkn1b (p27Kip1), and Cdkn2b (p15INK4b). Finally, we also measured the relative quantities of Inhba (inhibin/activin βA) and Inhbb (inhibin/activin βB), genes which encode the subunits responsible for activin signaling, the primary cause of the wasting syndrome in inhibin-deficient mice (Coerver et al., 1996). We found significantly decreased expression of Star, Cyp11a1, Ccnd2, Cdkn1b, Inhba, and Inhbb in testicular tumors from Inha-/-Lhb-/- males compared to Inha-/- controls (Fig. 7A). We also observed significantly decreased expression of Cyp17a1 and increased expression of Cdkn2b, as well as a trend toward increased expression of Cdkn1b, in double mutant ovarian tumors compared to Inha-/- controls (Fig. 7B).
Fig. 7.
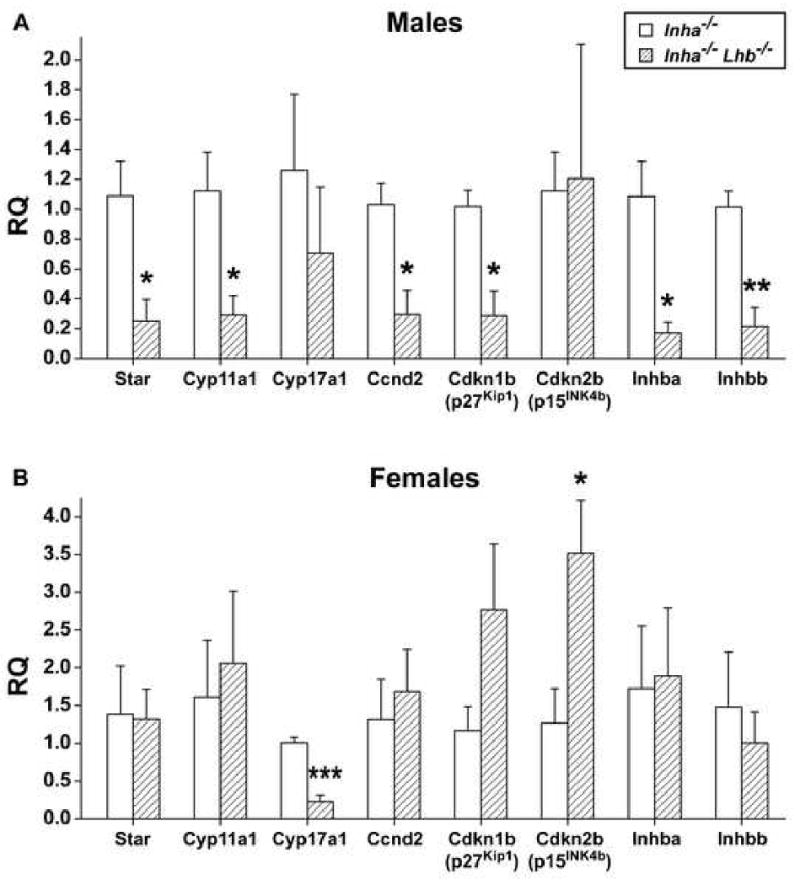
Quantitative real-time PCR analysis of end-stage Inha-/- and Inha-/-Lhb-/- gonadal tumors (mean ± SEM, n ≥ 4 tumors from independent mice of each sex and genotype). (A) Double mutant testicular tumors demonstrated a significant decrease in the relative quantity (RQ) of Star (4.3-fold), Cyp11a1 (3.9-fold), Ccnd2 (3.5-fold), Cdkn1b (3.6-fold), Inhba (6.4-fold), and Inhbb (4.7-fold). (B) Double mutant ovarian tumors demonstrated a significant decrease in the expression of Cyp17a1 (4.5-fold) and a significant increase in the expression of Cdkn2b (2.8-fold). We also noted a trend toward increased expression of Cdkn1b (2.4-fold) in double mutant ovarian tumors. Ages of end-stage mice: Inha-/- males (8-13 wks) and females (12-19 wks); Inha-/-Lhb-/- males (13-18 wks) and females (18-53 wks). *p<0.05, **p<0.005, ***p<0.001 (Student's t-test).
Discussion
The balance of growth and differentiation in the mammalian gonads depends on the coordinated actions of numerous signaling pathways that collectively comprise the hypothalamic-pituitary-gonadal axis. The inhibins are TGF-β superfamily members which are integral to this process by functioning as gonadal tumor suppressors (Matzuk et al., 1992). Mice lacking inhibins develop sex cord-stromal tumors (e.g., granulosa/Sertoli cell tumors) in a gonadotropin-dependent manner (Kumar et al., 1999; Kumar et al., 1996). In the present study, we hypothesized that LH contributes to the pathogenesis of ovarian and testicular cancers in the absence of inhibins.
To test our hypothesis, we generated mice deficient in both inhibins and LH and monitored for tumor development and the concomitant wasting syndrome. LH is not required for tumor formation in inhibin-deficient mice, as all Inha-/-Lhb-/- mice developed sex cord-stromal tumors that were histologically similar to the tumors in Inha-/- controls. Double mutants also demonstrated principal characteristics of the cancer cachexia-like syndrome, including progressive weight loss, a hunchback appearance, lethargy, mucosal atrophy with depletion of parietal cells in the glandular stomach, and hepatocellular inflammation and necrosis around the central vein. In spite of these pathologic defects, the loss of LH on the Inha-/- background prolonged median survival by 5 weeks and maximum lifespan by over 20 weeks in both males and females, suggesting that tumor progression may be slower in double mutants. To investigate this possibility further, we examined the testes and ovaries of 6-week-old Inha-/-Lhb-/- mice and compared them to gonads from age-matched Inha-/- controls. We observed smaller tumor foci and less hemorrhage in double mutant testes and ovaries, supporting the conclusion that LH promotes tumor progression in the absence of inhibins.
Inha-/- mice have elevated serum FSH levels, consistent with the fact that inhibins suppress FSH (Matzuk et al., 1992). Secretion of activins by the gonadal tumors in these mice probably also contributes to the increase in FSH. Under normal circumstances, activins produced locally within the pituitary stimulate Fshb transcription and FSH secretion in a paracrine fashion (Gregory and Kaiser, 2004), while circulating serum activins are neutralized by follistatin (Schneyer et al., 1994). Follistatin is a monomeric glycoprotein that irreversibly binds activins to prevent their interaction with activin receptors. Sex cord-stromal tumors in Inha-/- mice secrete substantial amounts of activins into the circulation, enough to overcome inhibition by follistatin and to bind activin receptors in multiple tissues, most notably the stomach and liver as evidenced by the resulting cancer cachexia-like syndrome (Matzuk et al., 1994). Elevated serum activins may also bind to the pituitary to increase FSH synthesis and secretion. Accordingly, overexpression of follistatin in inhibin-deficient males decreases serum FSH, presumably by interfering with the endocrine action of tumor-produced activins on the pituitary (Cipriano et al., 2000).
Interestingly, 6-week-old Inha-/-Lhb-/- females had lower serum FSH compared to Inha-/-controls, and 6-week-old double mutant males demonstrated a trend toward lower FSH. Given that Lhb-/- mice do not have alterations in serum FSH compared to wild type mice (Ma et al., 2004), the lower FSH in 6-week-old double mutants is most likely an effect of delayed tumor progression. Smaller tumors at this early age may produce less activins, thereby attenuating endocrine stimulation of pituitary FSH. The consequent decrease in FSH could in turn further slow tumor growth, perhaps by decreasing cyclin D2 (Ccnd2) expression. Ccnd2 is an FSH-induced gene (Robker and Richards, 1998; Sicinski et al., 1996) that is upregulated in inhibin-deficient testicular and ovarian cancers compared to wild type gonads (Cipriano et al., 2001) and known to enhance the progression of these tumors, especially in males (Burns et al., 2003b). Indeed, end-stage double mutant males trended toward lower FSH compared to Inha-/-males, and testicular tumors from Inha-/-Lhb-/- end-stage males had decreased expression of Ccnd2 compared to Inha-/- tumors. These observations may be secondary to decreased activin production, as both Inhba and Inhbb were lower in double mutant testicular tumors. Although our results indicate that Ccnd2 levels are similar between Inha-/- and Inha-/-Lhb-/- ovarian tumors, these data were collected from end-stage females in which FSH levels are similar. The expected decrease in Ccnd2 expression in double knockout females may only be apparent at earlier ages, when tumor growth is slower and serum FSH levels are lower.
One of the major functions of LH is the regulation of gonadal steroidogenesis. LH binds to receptors on testicular Leydig cells and ovarian theca cells to stimulate androgen production. In males, testosterone is required for spermatogenesis, while in females, theca-derived androgens are transported to granulosa cells and converted to estradiol. Accordingly, Lhb-/- males and females have reduced serum testosterone and estradiol, respectively, compared to wild type mice (Ma et al., 2004).
Prior studies have implicated the sex steroid hormones in the pathogenesis of gonadal cancers, and therefore we hypothesized that LH contributes to tumor development in Inha-/-mice by promoting steroidogenesis. Beamer and colleagues demonstrated that androgens are required for the initiation of spontaneous ovarian granulosa cell tumors in SWR/SWXJ recombinant inbred strains of mice (Beamer et al., 1988; Beamer et al., 1998; Beamer et al., 1993; Tennent et al., 1993). Importantly, treatment with hCG, but not FSH, can induce the tumors, supporting the link between LH, steroid biosynthesis, and cancer (Dorward et al., 2007). Treatment of Inha-/- females with flutamide, a nonsteroidal androgen antagonist, prolongs lifespan by slowing tumor progression (Burns et al., 2003a), and similar findings were observed in inhibin-deficient males carrying a loss-of-function mutation (tfm) at the androgen receptor locus (Shou et al., 1997). Contrary to our expectation, we did not observe a decrease in serum testosterone in Inha-/-Lhb-/- males compared to Inha-/- controls; instead, there was no difference. On the other hand, in double mutant ovarian tumors, we noted a significant decrease in the expression of Cyp17a1, encoding cytochrome P450 17α-hydroxylase/17,20-lyase, the LH-induced theca cell enzyme that converts pregnenolone and progesterone to dehydroepiandrosterone (DHEA) and androstenedione, respectively. The reduction in Cyp17a1 may lead to a parallel decrease in these androgens, resulting in slower tumor growth in Inha-/-Lhb-/- females.
Although both the testicular and ovarian tumors of inhibin α mutants secrete estradiol (Matzuk et al., 1996), the consequences of estrogen receptor signaling on tumor progression are distinct between males and females. While triple knockout males lacking inhibin α and estrogen receptors alpha (Esr1) and beta (Esr2) have prolonged survival compared to Inha-/-controls, triple knockout females suffer from more aggressive ovarian tumors and die earlier (Burns et al., 2003a). These data suggest that in the absence of inhibins, estrogen receptor signaling has more severe consequences in males but is protective in females. The detrimental effect in males may result from an increase in cyclin D2 within testicular tumors, as Ccnd2 is an estradiol-responsive gene (Robker and Richards, 1998) that promotes tumor growth (Burns et al., 2003b). The mechanism responsible for the protective effect in females is unclear, yet the observation is additionally supported by experiments in which estradiol treatment suppressed ovarian granulosa cell tumor incidence in genetically susceptible SWXJ mice, perhaps by decreasing serum DHEA (Beamer et al., 1988). Notably, we measured a reduction in estradiol in 6-week-old Inha-/-Lhb-/- males, but not in double mutant females, compared to Inha-/- controls. The decrease in estradiol is consistent with slower tumor progression in 6-week-old double mutant males, and may directly result from the absence of LH, or may be secondary to reduced serum FSH. FSH acts on granulosa cells and prepubertal Sertoli cells to stimulate the activity of cytochrome P450 aromatase (encoded by Cyp19a1), the enzyme responsible for the conversion of androgens to estrogens. The observation that 6-week-old double mutant females, in contrast to double mutant males, have estradiol levels similar to controls supports the notion that the regulation of tumor estradiol production may be sexually dimorphic. Furthermore, in comparison to tumors from end-stage Inha-/- males, Inha-/-Lhb-/- testicular tumors have decreased expression of steroidogenic acute regulatory protein (Star) and P450 side chain cleavage (Cyp11a1), two LH-dependent enzymes that catalyze the earliest steps of gonadal steroidogenesis. In spite of these observations, the comparable estradiol levels between end-stage Inha-/- and Inha-/-Lhb-/- mice suggest that estradiol synthesis from both testicular and ovarian tumors is independent of LH in later stages.
We previously reported that inhibin-deficient gonadal tumors have aberrant expression of cell cycle regulators compared to wild type testes and ovaries. Specifically, we demonstrated an increase in cyclin D2 and cyclin-dependent kinase 4 (encoded by Cdk4) mRNA, and a decrease in p27Kip1 (encoded by Cdkn1b) protein levels (Cipriano et al., 2001). D-type cyclins form a complex with CDK4/6 to promote cell proliferation, while p27Kip1 binds to cyclin-CDK complexes to inhibit cell cycle progression. In comparison to Inha-/- controls, mice deficient in both inhibins and cyclin D2 have prolonged survival because of delayed tumorigenesis (Burns et al., 2003b), while mice lacking both inhibins and p27Kip1 have more aggressive tumors leading to decreased survival (Cipriano et al., 2001). To determine if the slower tumor progression in Inha-/-Lhb-/- mutants was associated with alterations in cell cycle regulators, we measured the expression of cyclin D2 and p27Kip1 in gonadal tumors from end-stage double mutants and Inha-/- controls. We observed decreased levels of both cyclin D2 and p27Kip1 in testicular tumors from Inha-/-Lhb-/- males compared to Inha-/- males. Although these proteins have antagonistic effects in Inha-/- mice as mentioned above, previous experiments demonstrated that in inhibin-deficient males, the exacerbation of tumor progression by cyclin D2 (Burns et al., 2003b) was stronger than the mitigating influence of p27Kip1 (Cipriano et al., 2001). From the reverse angle, this suggests that the beneficial effects of decreased cyclin D2 in double mutant testicular tumors might outweigh the harmful effects of decreased p27Kip1. In contrast to our observations in males, we found similar levels of cyclin D2 in ovarian tumors from Inha-/-Lhb-/- females compared to Inha-/- females. However, we noted that double mutant ovarian tumors trend toward elevated expression of p27Kip1 and have significantly higher levels of p15INK4b (encoded by Cdkn2b), another cyclin-dependent kinase inhibitor that specifically targets CDK4/6 complexes. These data suggest that the absence of LH delays tumorigenesis in inhibin-deficient females through the upregulation of proteins that repress cell proliferation.
In summary, our current experiments extend upon prior lessons from LH/hCG gain-of-function models (Huhtaniemi et al., 2005; Kananen et al., 1995; Keri et al., 2000; Mikola et al., 2003; Neumann, 1991; Risma et al., 1995), suggesting that although LH may be sufficient for gonadal tumor formation, it may not be required. Furthermore, our present work expands upon numerous studies which indicate that inhibins genetically interact with multiple factors that influence testicular and ovarian growth and differentiation, including the pituitary gonadotropins (Kumar et al., 1999; Kumar et al., 1996). We have demonstrated that LH is not required for gonadal tumor formation in the absence of inhibins but promotes tumor progression. These findings provide additional perspective on the complex interplay between inhibins, gonadotropins, and cancer.
Acknowledgments
This research was supported by National Institutes of Health Grants CA60651 (to M.M.M) and T32GM008307 (to A.K.N.), the Joseph and Matilda Melnick Endowed Fund (to A.K.N.), and a scholarship from Baylor Research Advocates for Student Scientists (to A.K.N.). We thank Dr. Stephanie Pangas for critical reading of the manuscript, Roopa Nalam for help with figures, and the University of Virginia Ligand Assay and Analysis Core (Specialized Cooperative Centers Program in Reproduction Research NICHD/NIH U54 HD28934) for performing serum hormone assays.
Footnotes
Disclosure statement: A.K.N., J.E.A., T.R.K., and M.M.M. have nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beamer WG, Shultz KL, Tennent BJ. Induction of ovarian granulosa cell tumors in SWXJ-9 mice with dehydroepiandrosterone. Cancer Res. 1988;48:2788–92. [PubMed] [Google Scholar]
- Beamer WG, Shultz KL, Tennent BJ, Nadeau JH, Churchill GA, Eicher EM. Multigenic and imprinting control of ovarian granulosa cell tumorigenesis in mice. Cancer Res. 1998;58:3694–9. [PubMed] [Google Scholar]
- Beamer WG, Shultz KL, Tennent BJ, Shultz LD. Granulosa cell tumorigenesis in genetically hypogonadal-immunodeficient mice grafted with ovaries from tumor-susceptible donors. Cancer Res. 1993;53:3741–6. [PubMed] [Google Scholar]
- Burns KH, Agno JE, Chen L, Haupt B, Ogbonna SC, Korach KS, Matzuk MM. Sexually dimorphic roles of steroid hormone receptor signaling in gonadal tumorigenesis. Mol Endocrinol. 2003a;17:2039–52. doi: 10.1210/me.2003-0039. [DOI] [PubMed] [Google Scholar]
- Burns KH, Agno JE, Sicinski P, Matzuk MM. Cyclin D2 and p27 are tissue-specific regulators of tumorigenesis in inhibin alpha knockout mice. Mol Endocrinol. 2003b;17:2053–69. doi: 10.1210/me.2003-0038. [DOI] [PubMed] [Google Scholar]
- Cipriano SC, Chen L, Burns KH, Koff A, Matzuk MM. Inhibin and p27 interact to regulate gonadal tumorigenesis. Mol Endocrinol. 2001;15:985–96. doi: 10.1210/mend.15.6.0650. [DOI] [PubMed] [Google Scholar]
- Cipriano SC, Chen L, Kumar TR, Matzuk MM. Follistatin is a modulator of gonadal tumor progression and the activin-induced wasting syndrome in inhibin-deficient mice. Endocrinology. 2000;141:2319–27. doi: 10.1210/endo.141.7.7535. [DOI] [PubMed] [Google Scholar]
- Coerver KA, Woodruff TK, Finegold MJ, Mather J, Bradley A, Matzuk MM. Activin signaling through activin receptor type II causes the cachexia-like symptoms in inhibin-deficient mice. Mol Endocrinol. 1996;10:534–43. doi: 10.1210/mend.10.5.8732684. [DOI] [PubMed] [Google Scholar]
- Dorward AM, Shultz KL, Beamer WG. Luteinizing hormone analog and dietary isoflavones support ovarian granulosa cell tumor development in a spontaneous mouse model. Endocr Relat Cancer. 2007 doi: 10.1677/erc.1.01232. In press. [DOI] [PubMed] [Google Scholar]
- Gregory SJ, Kaiser UB. Regulation of gonadotropins by inhibin and activin. Semin Reprod Med. 2004;22:253–67. doi: 10.1055/s-2004-831901. [DOI] [PubMed] [Google Scholar]
- Huhtaniemi I, Rulli S, Ahtiainen P, Poutanen M. Multiple sites of tumorigenesis in transgenic mice overproducing hCG. Mol Cell Endocrinol. 2005;234:117–26. doi: 10.1016/j.mce.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Kananen K, Markkula M, Rainio E, Su JG, Hsueh AJ, Huhtaniemi IT. Gonadal tumorigenesis in transgenic mice bearing the mouse inhibin alpha-subunit promoter/simian virus T-antigen fusion gene: characterization of ovarian tumors and establishment of gonadotropin-responsive granulosa cell lines. Mol Endocrinol. 1995;9:616–27. doi: 10.1210/mend.9.5.7565808. [DOI] [PubMed] [Google Scholar]
- Keri RA, Lozada KL, Abdul-Karim FW, Nadeau JH, Nilson JH. Luteinizing hormone induction of ovarian tumors: oligogenic differences between mouse strains dictates tumor disposition. Proc Natl Acad Sci U S A. 2000;97:383–7. doi: 10.1073/pnas.97.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar TR, Palapattu G, Wang P, Woodruff TK, Boime I, Byrne MC, Matzuk MM. Transgenic models to study gonadotropin function: the role of follicle-stimulating hormone in gonadal growth and tumorigenesis. Mol Endocrinol. 1999;13:851–65. doi: 10.1210/mend.13.6.0297. [DOI] [PubMed] [Google Scholar]
- Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–4. doi: 10.1038/ng0297-201. [DOI] [PubMed] [Google Scholar]
- Kumar TR, Wang Y, Matzuk MM. Gonadotropins are essential modifier factors for gonadal tumor development in inhibin-deficient mice. Endocrinology. 1996;137:4210–6. doi: 10.1210/endo.137.10.8828479. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma X, Dong Y, Matzuk MM, Kumar TR. Targeted disruption of luteinizing hormone beta-subunit leads to hypogonadism, defects in gonadal steroidogenesis, and infertility. Proc Natl Acad Sci U S A. 2004;101:17294–9. doi: 10.1073/pnas.0404743101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci U S A. 1994;91:8817–21. doi: 10.1073/pnas.91.19.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk MM, Finegold MJ, Su JG, Hsueh AJ, Bradley A. Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature. 1992;360:313–9. doi: 10.1038/360313a0. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Kumar TR, Shou W, Coerver KA, Lau AL, Behringer RR, Finegold MJ. Transgenic models to study the roles of inhibins and activins in reproduction, oncogenesis, and development. Recent Prog Horm Res. 1996;51:123–54. discussion 155-7. [PubMed] [Google Scholar]
- Mikola M, Kero J, Nilson JH, Keri RA, Poutanen M, Huhtaniemi I. High levels of luteinizing hormone analog stimulate gonadal and adrenal tumorigenesis in mice transgenic for the mouse inhibin-alpha-subunit promoter/Simian virus 40 T-antigen fusion gene. Oncogene. 2003;22:3269–78. doi: 10.1038/sj.onc.1206518. [DOI] [PubMed] [Google Scholar]
- Neumann F. Early indicators for carcinogenesis in sex-hormone-sensitive organs. Mutat Res. 1991;248:341–56. doi: 10.1016/0027-5107(91)90067-x. [DOI] [PubMed] [Google Scholar]
- Pierson TM, Wang Y, DeMayo FJ, Matzuk MM, Tsai SY, Omalley BW. Regulable expression of inhibin A in wild-type and inhibin alpha null mice. Mol Endocrinol. 2000;14:1075–85. doi: 10.1210/mend.14.7.0478. [DOI] [PubMed] [Google Scholar]
- Risma KA, Clay CM, Nett TM, Wagner T, Yun J, Nilson JH. Targeted overexpression of luteinizing hormone in transgenic mice leads to infertility, polycystic ovaries, and ovarian tumors. Proc Natl Acad Sci U S A. 1995;92:1322–6. doi: 10.1073/pnas.92.5.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robker RL, Richards JS. Hormone-induced proliferation and differentiation of granulosa cells: a coordinated balance of the cell cycle regulators cyclin D2 and p27Kip1. Mol Endocrinol. 1998;12:924–40. doi: 10.1210/mend.12.7.0138. [DOI] [PubMed] [Google Scholar]
- Schneyer AL, Rzucidlo DA, Sluss PM, Crowley WF., Jr Characterization of unique binding kinetics of follistatin and activin or inhibin in serum. Endocrinology. 1994;135:667–74. doi: 10.1210/endo.135.2.8033815. [DOI] [PubMed] [Google Scholar]
- Shou W, Woodruff TK, Matzuk MM. Role of androgens in testicular tumor development in inhibin-deficient mice. Endocrinology. 1997;138:5000–5. doi: 10.1210/endo.138.11.5499. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature. 1996;384:470–4. doi: 10.1038/384470a0. [DOI] [PubMed] [Google Scholar]
- Tennent BJ, Shultz KL, Beamer WG. Genetic susceptibility for C19 androgen induction of ovarian granulosa cell tumorigenesis in SWXJ strains of mice. Cancer Res. 1993;53:1059–63. [PubMed] [Google Scholar]