

Abstract
Brain–derived neurotrophic factor (BDNF) was the first purified molecule identified to directly support the development of mesencephalic dopamine neurons. However, its physiologic role has remained unknown. Based on patterns of expression, it is unlikely to serve as a target-derived neurotrophic factor, but it may instead act locally in the mesencephalon, either released by afferent projections, or in autocrine fashion. To assess a possible local role, we blocked BDNF signaling in the substantia nigra (SN) of postnatal rats by injection of either neutralizing antibodies or a peptide antagonist. These treatments increased the magnitude of developmental cell death in the SN, indicating that endogenous local BDNF does play a regulatory role. However, we also find that elimination of BDNF in brain throughout postnatal development in BDNFfl/fl:Nestin-Cre mice has no effect on the adult number of SN dopamine neurons. We postulate that other forms of trophic support may compensate for the elimination of BDNF during early development. Although the number of SN dopamine neurons is unchanged, their organization is disrupted. We conclude that BDNF plays a physiologic role in the postnatal development of SN dopamine neurons.
Introduction
Disturbances of mesencephalic dopaminergic systems have been postulated to play a role in diverse and prevalent human psychiatric and behavioral disturbances, including schizophrenia (Jarskog et al. 2007), addiction (Kauer and Malenka 2007) and satiety disorders (Morton et al. 2006). The neurobiology of these neurons is also important for degenerative neurologic diseases, especially Parkinson's disease (PD) and many of the tauopathies (Lee et al. 2001), in which they are especially vulnerable. In view of the direct relevance of mesencephalic dopamine neurons to these disorders, it is important to understand the regulation of their development, and particularly the determinants of their final adult number and organization.
The first fully purified molecule reported to selectively and directly support the survival of mesencephalic dopamine neurons was brain-derived neurotrophic factor (BDNF) (Hyman et al. 1991). However, BDNF is not expressed in the striatum during development (Maisonpierre et al. 1990), and therefore it is unlikely to play a role as a target-derived neurotrophic factor, as envisioned by classic neurotrophic theory (Oppenheim 1991). Alternatively, it has been proposed that BDNF may serve as an afferent projection-derived factor (Altar et al. 1997;Altar and DiStefano 1998) for these neurons. This view is supported by patterns of BDNF mRNA and protein expression (Conner et al. 1997), and specifically by the observation that increased BDNF protein expression within the locus ceruleus afferent projection to the substantia nigra pars compacta (SNpc) results in a 50% increase in the adult number of dopamine neurons of the SNpc (Alonso-Vanegas et al. 1999). This effect was postulated to be due to a suppression of postnatal developmental cell death in dopamine neurons (Janec and Burke 1993;Oo and Burke 1997), resulting in an increase in their number in adulthood.
An alternative, or additional, possible role for BDNF in regulating the development of dopamine neurons of the SN is based on the observation that they express BDNF (Seroogy et al. 1994;Baquet et al. 2005), and it may therefore serve in an autocrine fashion (Acheson et al. 1995). Such a possibility is supported by the observations of Baquet et al (Baquet et al. 2005) who examined the effects of local mesencephalic-hindbrain deletion of BDNF by means of a Wnt1-Cre transgene expressed in BDNFfl/fl mice. These investigators observed a diminished number of tyrosine hydroxylase (TH)-positive neurons at birth. However, this effect appeared to be due principally to loss of phenotype, because there was no alteration in the number of SNpc neurons, based on NeuN staining.
The goal of these investigations therefore was to define the postnatal role of local BDNF, delivered either by afferent projections or autocrine release, in regulating the magnitude of the developmental cell death event in dopamine neurons of the SN, and, consequently, their final adult number. To achieve this goal we have made observations both acutely, by use of intranigral injection of inhibitors of BDNF, and chronically, in BDNFfl/fl: Nestin-Cre mice.
Results
To assess the regulation of the naturally occurring cell death event in SN by local BDNF signaling, we injected increasing amounts of anti-BDNF neutralizing antibody dorsal to the SNpc in PND5-6 rats, and determined the number of apoptotic profiles in the SNpc 24 hours later. These experiments revealed a trend for doses of 5.0 and 10.0 μm to induce apoptosis, and a significant induction of 2.5-fold over control IgG was achieved at 20.0 μg (Fig. 1A, B). To determine whether anti-BDNF neutralizing antibody could also induce apoptosis during the second phase of developmental cell death among SN dopamine neurons, we injected antibodies on PNDs 14 and 21 (Fig, 1C). These injections did not induce apoptosis, so we therefore conclude that local BDNF signaling regulates apoptosis in these neurons only during the first phase of developmental cell death.
Fig. 1.
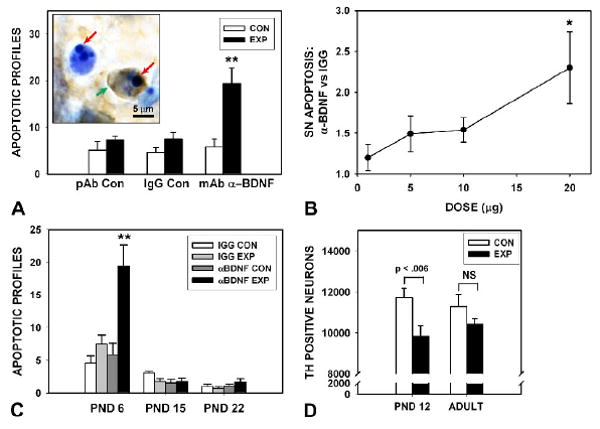
Postnatal neutralization of BDNF activity in the SN increases naturally occurring cell death. (A) Intranigral injection of a monoclonal, neutralizing anti-BDNF antibody (20.0 μg) at PND5-6, results in a 2.5-fold increase in the number of apoptotic profiles in the SN on post injection day 1 (N=7, anti-BDNF; N=7, mouse IgG control; **p < 0.001, ANOVA). For each condition, the side of the unilateral injection, Experimental (EXP), and the contralateral, non-injected Control (CON) side is shown. An additional control injection of a chicken polyclonal antibody is also shown (N=5). The insert shows two apoptotic profiles in the SNpc of a postnatal rat injected with anti-BDNF, and processed for TH immunostaining with thionin counterstain. The red arrows indicate the intensely basophilic nuclear chromatin clumps characteristic of apoptosis. One of the profiles retains TH expression in the cytoplasm (green arrow). (B) A dose-response analysis for neutralizing anti-BDNF antibody injections, expressed as fold-increase in comparison to the IgG control injection at each dose. A trend is observed at doses of 5.0 (N=5) and 10.0 μg (N=4) and a significant effect is observed at 20.0 μg (N=7) (*p < 0.02, ANOVA). (C) A developmental time course analysis of the effect of 20.0 μg anti-BDNF injections. The effect observed at PND6 (during the first phase of naturally occurring cell death in SN dopamine neurons) is not observed the second phase at PNDs13-15 (N=6, anti-BDNF and IgG control injections, Experimental (EXP) and non-injected Control (CON) sides), or later, at PND22 (N=6). (D) Stereologic counts of SN TH-positive neurons at PND12 following injection of 20.0 μg anti-BDNF at PND5 reveal a significant decrease in the number of surviving neurons (p < 0.006, paired t-test). However, this effect does not persist into adulthood (p = 0.19, NS) (N = 7, both age groups).
We have previously shown that intra-striatal injection of anti-GDNF antibodies during development induces apoptosis in SN dopamine neurons (Oo et al. 2003), in keeping with its possible role as a target-derived neurotrophic factor for these neurons. Since little BDNF mRNA is expressed in striatum (Maisonpierre et al. 1990), we anticipated that intrastriatal injection of neutralizing anti-BDNF antibodies, unlike anti-GDNF antibodies, and unlike intranigral injection, should not induce apoptosis in SN. We found that this was the case; intrastriatal injection of 20.0 μm of anti-BDNF had no effect on the amount of developmental apoptosis in SN (data not shown). Thus the ability of the BDNF neutralization to induce apoptosis depends on local SN injection.
To determine whether the increased amount of apoptosis induced by intranigral injection of neutralizing antibodies specifically affected the SN population of dopamine neurons, we determined their number after antibody injection. Stereologic counts of SN TH-positive neuron number on PND12, prior to the second phase of developmental cell death, demonstrated that their number was decreased by 16% (p = 0.006, paired t-test) following antibody injection at PND5-6 (Fig. 1D). However, this effect was no longer apparent after the second phase of developmental cell death, in adulthood. We have previously described a similar discordance between effects observed before and after the second phase, and have suggested that the magnitude of this event may be regulated to achieve a normal adult number of neurons (Kholodilov et al. 2004). This discordance notwithstanding, we may conclude that the administration of BDNF neutralizing antibodies induces apoptosis among SN dopamine neurons, resulting in a decrease in their number after the first phase of developmental cell death.
To confirm these observations made with neutralizing antibodies, we performed experiments using an alternative approach, based on a conformationally constrained small peptide analogue of Loop 2 of BDNF, L2-8 (Fig. 2A) (O'Leary and Hughes 1998). This peptide acts as a competitive antagonist of BDNF by mimicking the structure of Loop 2 of the BDNF molecule, which is required for interaction with the TrkB receptor (O'Leary and Hughes 1998). Intra-nigral injection of L2-8 in increasing doses achieved a significant induction of apoptosis (p = .001, ANOVA) in comparison to an identical, but non-conformationally restrained, peptide, reaching a maximal effect of a 1.5-fold induction at 20.0 μg (Fig. 2B).
Fig. 2.
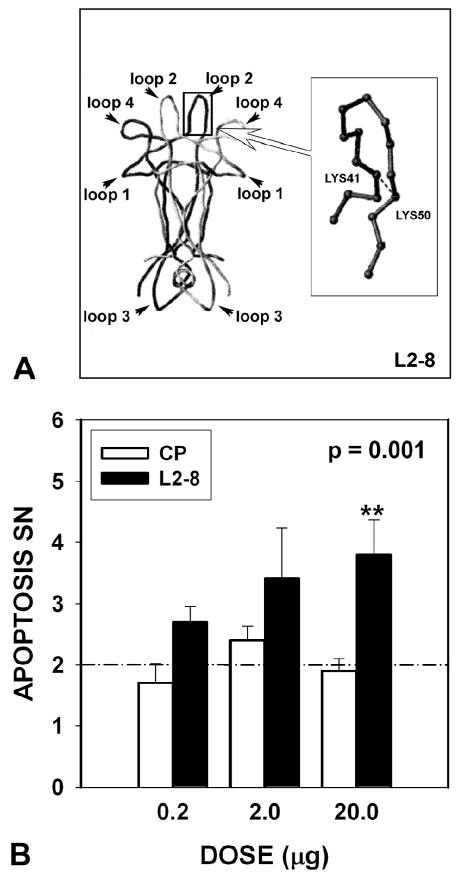
Postnatal inhibition of BDNF with a competitive antagonist augments naturally occurring cell death. (A) The constrained peptide L2-8 is an analogue of Loop 2 of BDNF, the region that mediates binding to the TrkB receptor. The position of the disulfide bond in L2-8, between Lys41 and Lys50, is shown in the inset as a dashed line. (B) L2-8 induces apoptosis in comparison to a control peptide (CP) (L28-Ala) (in which the terminal cysteines have been replaced by alanines to abrogate conformational constraint) (p= 0.001, ANOVA; for this analysis, there was no difference between the CP injected and non-injected sides, so their values have been combined at all doses to constitute the CP group) (0.2 μg: Control Peptide (CP) N = 5; L2-8 N = 5; 2.0 μg: CP N = 6, L2-8 N = 6; 20.0 μg: CP N = 13, L2-8 N = 14). There was no difference among the various control peptide conditions in the number of apoptotic profiles. The mean number for all control injections is indicated by a dashed line. Note that the x-axis is not to scale.
In view of a trend for a decrease in the adult number of dopamine neurons following injection of neutralizing antibodies at PND5-6 (Fig. 1D), we also considered the possibility that we had not successfully demonstrated the degree of dependence of postnatal dopamine neurons on BDNF by use of the neutralizing antibody technique. Intracerebral injection of neutralizing antibodies produces only a transient effect, and it is limited by the ability of antibody molecules to diffuse in the extracellular space. Furthermore, we sought to confirm at the gene level the observations made with the neutralizing and comparative peptide antagonist approaches. We achieved elimination of BDNF expression throughout brain by crossing BDNFfl/fl mice (Rios et al. 2001) with Nestin-Cre transgenic mice (Tronche et al. 1999). These latter mice have previously been reported to achieve Cre-mediated recombination throughout the central nervous system in newborn reporter mice (Tronche et al. 1999). In BDNFfl/fl:Nestin-Cre mice, there was elimination of BDNF mRNA in SN and other brain regions in PND2 and adult mice (Fig. 3A), and elimination of BDNF protein was confirmed in PND28 mice (Fig. 3B).
Fig. 3.
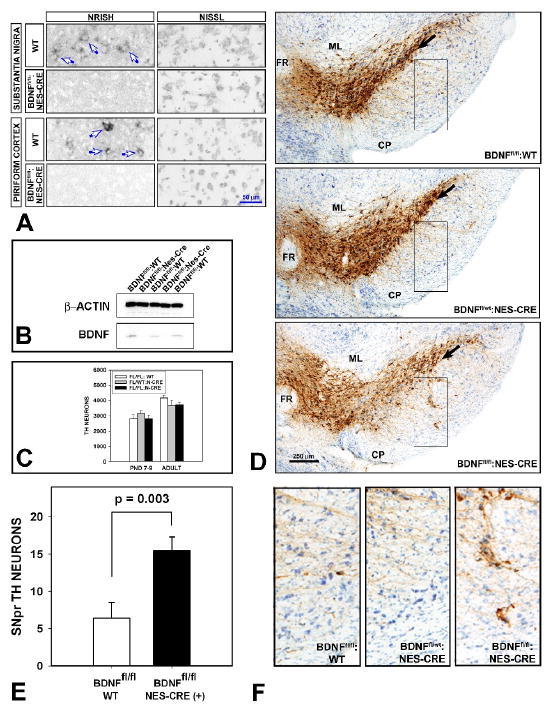
Elimination of BDNF expression in brain of BDNFfl/fl:Nestin-Cre mice disturbs the organization of SN dopamine neurons, but not their adult number. (A) Non-radioactive in-situ hybridization (NRISH) of BDNF mRNA reveals that expression in neurons (blue arrows) of the SNpc and piriform cortex in wildtype mice is absent in BDNFfl/f:Nestin-Cre adult mice. Nissl stain of adjacent sections demonstrates that the loss of BDNF mRNA expression is not due to neuron loss. Similar observations were made in PND2 mice (not shown). (B) Elimination of BDNF protein expression in whole forebrain is demonstrated by Western analysis at PND28. (C) Stereologic counts of TH-positive neurons in SN reveals no change in their number in BDNFfl/fl:Nestin-Cre mice in comparison to controls in both PND7-8 (BDNFfl/fl:WT, N = 6; BDNFfl/wt:Nestin-Cre, N = 6; BDNFfl/fl:Nestin-Cre, N = 7) and adult mice (BDNFfl/fl:WT, N = 12; BDNFfl/wt:Nestin-Cre, N = 6; BDNFfl/fl:Nestin-Cre, N = 12). (D) Two abnormalities are noted in the regional organization of TH-positive neurons in the SN of BDNFfl/fl:Nestin-Cre mice. First, whereas the ventral boundary of the SNpc, identified by TH immunostaining, is distinctly defined in both BDNFfl/fl:WT and BDNFfl/wt:Nestin-Cre controls (black arrows in two upper panels), it is not in the BDNFfl/fl:Nestin-Cre mice (black arrow lower panel. In addition, in anterior SN planes, clusters of TH-positive neurons are not usually observed ventrally in the SNpr. However, among BDNFfl/fl: Nestin-Cre mice, such TH-positive neurons were observed in significant numbers. Each of the representative sections from each of the three genotypes is matched for coronal plane -3.08 mm (relative to bregma) in the Paxinos-Franklin atlas (Paxinos and Franklin 2001) (ML: medial lemniscus; FR: fasciculus retroflexus; CP: cerebral peduncle). (E) The number of ectopic TH-positive neurons in the SNpr was determined in single sections within the -3.08 coronal plane for N = 12 BDNFfl/fl:WT and N = 12 BDNFfl/fl:Nestin-Cre mice. There was a 2.4-fold increase in the number of these ectopic neurons in the BDNFfl/fl:Nestin-Cre mice (p = 0.003, t-test). (F) An example of the SNpr in a BDNFfl/fl:Nestin-Cre mouse, containing multiple ectopic TH-positive neurons, is shown in comparison to identical SNpr regions in representative examples of BDNFfl/fl:WT and BDNFfl/wt:Nestin-Cre mice. Each of the panels represents the regions demarcated by black rectangles in (D).
Surprisingly, based on the observations made in studies of acute BDNF inhibition, and given that SNpc dopamine neurons are post-mitotic in the postnatal period, we found that elimination of BDNF expression in brain of BDNFfl/fl: Nestin-Cre mice did not affect the total number of SN dopamine neurons either before (at PND7-9) or after (at adulthood) the second phase of naturally occurring cell death (Fig. 3C). However, elimination of BDNF expression in brain throughout postnatal development did result in two abnormalities in the regional anatomy of SN dopamine neurons in adulthood. In wildtype adult mice, and in the BDNFfl/fl:WT and BDNFfl/wt:Nestin-Cre controls, there is a distinct ventral border of the SNpc, defined by TH immunostaining, that demarcates its separation from the SN pars reticulata (SNpr) (Fig. 3D). In the BDNFfl/fl:Nestin-Cre mice this distinct boundary was obscured due to the presence of a substantial number of TH-positive neurons in the SNpr, just ventral to the SNpc in both adult (Fig. 3D) and PND 7-9 pups (not shown). A second, and possibly related, abnormality was the presence of islands of TH-positive neurons throughout the SNpr (Fig. 3D). While clusters of TH-positive neurons are normally observed in the SNpr in caudal SN planes in mice and rats, where they constitute a component of the ventral tier (Gerfen et al. 1987), they are not normally observed in rostral planes. These alterations in the regional anatomy of dopamine neurons in the SN did not, however, result in a change in the organization of dopaminergic efferent projections to the forebrain. Analysis of TH-immunostained sections of the forebrain, encompassing the striatum, the nucleus accumbens, cingulate cortex and piriform cortex did not reveal alterations at the regional level.
Discussion
A novel neurotrophic activity, distinct from NGF, was first identified in the conditioned medium of a glioma cell line on the basis of effects on the viability of chick dorsal root ganglion neurons (Barde et al. 1980). Using this bioassay, BDNF was purified (Barde et al. 1982), and subsequently cloned by Barde and colleagues (Leibrock et al. 1989) (reviewed by Lindsay (Lindsay 1993)). BDNF was the first purified molecule demonstrated to directly support the viability of embryonic dopamine neurons (Hyman et al. 1991). Subsequently, it was shown to provide neuroprotection for embryonic mesencephalic dopamine neurons against neurotoxins (Spina et al. 1992). Nevertheless, the physiologic role of endogenous BDNF in regulating the development of dopamine neurons has remained unknown, in part because conventional null mutations of either BDNF or its receptor TrkB are incompatible with long-term survival (Ernfors et al. 1994;Jones et al. 1994;Klein et al. 1993). The possibility of a physiologically relevant role for BDNF in the development of these neurons is supported by the expression of BDNF and TrkB mRNA in SNpc neurons (Seroogy et al. 1994;Numan et al. 2005;Numan and Seroogy 1999).
To examine the possible role of endogenous BDNF in dopamine neuron development, we initially used approaches based on acute blockade to circumvent the occurrence of compensatory changes that may occur, even in regionally selective nulls, to obscure a relevant phenotype. Intranigral injection of a neutralizing antibody to BDNF induced apoptosis in SNpc, in keeping with a role for endogenous local BDNF in regulating this postnatal developmental cell death event. Although the antibody used lacks cross-reactivity with other neurotrophins, we cannot formally exclude the possibility of interaction with other brain proteins that might regulate developmental cell death. We therefore sought to confirm these observations by an independent approach using a conformationally constrained, synthetic peptide competitive antagonist (O'Leary and Hughes 1998). By the incorporation of cysteines into the Lys41 and Lys50 positions of the amino acid sequence of the second β-hairpin loop (Loop 2) of BDNF, and the formation of a disulfide bond between them, it is possible to constrain the Loop 2 peptide sequence and thereby mimic its native structure. Since Loop 2 is essential for interaction with the TrkB receptor, it would be predicted that the constrained peptide would serve as a competitive antagonist, and this is the case (O'Leary and Hughes 1998). This peptide, L2-8, has a maximal inhibitory effect of only 50% in vitro, that may be due either to the presence of TrkB isoforms, or a tendency of the peptide to self-aggregate (O'Leary and Hughes 1998). In spite of this limitation of L2-8, it did induce apoptosis, like the neutralizing antibody, albeit with a less robust effect, as anticipated. Given the similar results obtained with these two different approaches, we conclude that local, endogenous BDNF regulates developmental cell death in postnatal dopamine neurons. A developmental time course analysis revealed that BDNF appears to play a role only during the first phase of naturally occurring cell death in these neurons, and not during the second phase.
To confirm that the induction of apoptosis by neutralizing antibodies was affecting dopamine neurons, we determined their number 6-7 days after injection (at PND12), before the occurrence of the second phase of developmental cell death at PND14. As expected, induction of apoptosis decreased the number of post-mitotic dopamine neurons. Given that this loss of TH-positive neurons is associated with an induction of apoptosis by injection of neutralizing antibody at PND5-6, it is exceedingly unlikely that it is due to diminished expression of TH protein rather than an actual decrease in the neuronal population. However, this decrease in the number of neurons did not persist into adulthood. This ability of the developing nigrostriatal system to compensate for an augmented loss of their numbers early in development is unlikely to be due to the birth of new neurons. Mitosis stops in this population at E15 in rat (Marchand and Poirer 1983;Lauder and Bloom 1974), and there is no evidence for a resurgence in the postnatal period (El-Khodor and Burke 2002). We have previously described a similar ability of the nigrostriatal system to normalize its adult numbers, but in the opposite direction, in transgenic mice over-expressing GDNF in striatal target (Kholodilov et al. 2004). These mice have a greater than normal number of dopamine neurons prior to the second phase of developmental cell death, but in adulthood their number has returned to normal. We postulated that during the second phase of developmental cell death, a compensatory increase in the magnitude of apoptosis could account for this normalization. Similarly, a compensatory decrease in apoptosis could account for a normalization in the postnatal rats treated with anti-BDNF. Alternatively, it is also possible that during late development, the nigrostriatal system is capable of modifying the phenotype of existing SNpc neurons to either increase or decrease the number of dopaminergic neurons after the developmental cell death event, to achieve a normal final adult number. In any case, our observations with acute, early developmental BDNF neutralization provide a second example of the ability of the nigrostriatal system to “fine tune” the number of dopamine neurons between the termination of the first phase of developmental cell death and adulthood.
In spite of this evidence from acute studies that local BDNF regulates the first phase of developmental cell death in dopamine neurons, we found that elimination of BDNF in brain during the postnatal period did not affect the number of dopamine neurons surviving after this phase or in adulthood. To reconcile these disparate observations, we propose that an effect is not observed in BDNFfl/fl:Nestin-Cre mice because alternate sources of trophic support have compensated for its prenatal loss. A precedent exists in the investigation of these neurons for a discordance between studies utilizing acute ablation and those using long-term ablation of neurotrophic molecules. In spite of evidence suggesting a role for GDNF in supporting the developmental viability in SN dopamine neurons (Oo et al. 2003;Kholodilov et al. 2004) two recent studies found, surprisingly, that embryonic, regionally selective knockout of Ret, the receptor tyrosine kinase that mediates GDNF signaling (Jain et al. 2006;Kramer et al. 2007) had no effect on young adult SN dopamine neuron number. However, it has recently been shown that acute knockout of GDNF, utilizing a tamoxifen-inducible form of Cre recombinase, results in extensive degeneration of dopamine neurons of the SN (Pascual et al. 2008). These authors propose, as we do, that embryonic disruption of trophic signaling induces developmental compensatory mechanisms that obscure a relevant phenotype.
Given that our acute studies, for methodological reasons, were performed in postnatal rats, and the chronic studies were performed in transgenic mice, we cannot formally exclude the possibility that the disparate results observed are due to a species difference. However, there is no known basis for this possibility; the developmental cell death events between the two species are highly similar in time course and morphology (Oo and Burke 1997;Jackson-Lewis et al. 2000).
Although we postulate that there are alternative trophic mechanisms to compensate for the prenatal loss of brain BDNF expression in the regulation of the final adult number of dopamine neurons, such does not appear to be the case for BDNF regulation of SN dopaminergic anatomical organization. In adulthood, BDNFfl/f:Nestin-Cre mice showed a loss of definition of the SNpc-SNpr boundary, and the appearance of ectopic dopaminergic neurons in the SNpr. Similar abnormalities were depicted in the study by Baquet and colleagues in BDNFfl/fl:Wnt1-Cre mice (Baquet et al. 2005), and by Baker et al in BDNF-/- mice at PND14 (Baker et al. 2005).
Although these investigations identify a role for local BDNF in SN in regulating the first phase of naturally occurring cell death, they do not identify its source. It may be provided by an afferent projection from the locus ceruleus (Alonso-Vanegas et al. 1999) or on an autocrine basis from SNpc neurons themselves (Baquet et al. 2005).
Experimental methods
Experimental animals
Timed, multiple pregnancy Sprague Dawley female rats were obtained from Charles River Laboratories (Wilmington, MA) 1 week before delivery. The day of delivery was defined as P1 (Janec and Burke 1993;Oo and Burke 1997). BDNFfl/fl mice (on a mixed background) were obtained from Jackson Laboratory (Bar Harbor, ME; Stock Number 004339) and maintained as homozygotes (Rios et al. 2001). Nestin-Cre mice (on a C57BL/6J background) were also obtained from Jackson Laboratory (Stock Number 003771) (Tronche et al. 1999). These two strains were crossed to obtain BDNFfl/wt:Nestin-Cre offspring, which were then crossed with BDNFfl/fl mice. Genotypes of the offspring were the expected Mendelian ratio. BDNFfl/fl:Nestin-Cre mice comprised the Experimental group, and BDNFfl/wt:Nestin-Cre and BDNFfl/fl:WT were used as littermate Controls. BDNFfl/wt:WT mice in these litters were not used. Mice were genotyped according to protocols recommended by Jackson Labs. For these studies all adult mice were 8 to 10 weeks of age.
Animal surgery
Postnatal rat pups were anesthetized either by hypothermia (postnatal day (PND) 5-6), or by ketamine/xylazine injection (PND 14 and older). Animals were positioned in a stereotaxic apparatus (Kopf Instruments, Tujunga, CA) to conform with the neonatal brain atlas described by Heller et al (Heller et al. 1979). The apparatus is equipped with a hypothermic miniaturized stereotaxic adapter for neonatal rats (Stoelting Co., Wood Dale, IL). The needle of a Hamilton microliter syringe was lowered to a point just dorsal to the substantia nigra (SN): AP: -0.32 LAT: +0.15 DV: -0.40 (cm) (relative to bregma). A solution containing anti-BDNF neutralizing antibodies (or control IgG) or BDNF antagonist (or control peptide) was injected at 0.1 μl/30 sec for 10 min, for a total volume of 2.0 μl. The needle was held in place for 2 min, and then slowly withdrawn. Intra-striatal injections were performed in similar fashion at AP: +0.10 LAT: +0.25 DV: -0.25 (cm) (relative to bregma) as described (Oo et al. 2003). All animal surgery procedures were approved by the Columbia University Animal Care and Use Committee.
Anti-BDNF neutralizing antibodies and BDNF peptide antagonist
For these studies a mouse monoclonal anti-BDNF neutralizing antibody (#GF35L; Calbiochem, La Jolla, CA) was used. This antibody was selected by the manufacturer for its ability to neutralize human recombinant BDNF. It has less than 2% cross-reactivity with human β-NGF, NT-3, and NT-4. This antibody has previously been demonstrated to achieve specific blockade of BDNF signaling in neurons (Kerr et al. 2003). Antibody (or immunoglobulin control) was first dialyzed in 0.1 M phosphate-buffered saline (PBS) (pH 7.1), and the concentration adjusted for the required dose. Two immunoglobulin controls were used in these studies: mouse IgG (Calbiochem) and chicken IgY (Promega). A conformationally constrained peptide analogue of Loop 2 of BDNF, which has been demonstrated to act as a competitive antagonist of BDNF (O'Leary and Hughes 1998), was used for these studies. This peptide consists of the sequence of Loop 2 of BDNF in which the lysines at positions 41 and 50 have been replaced by cysteine, and a disufide bond formed, thus mimicking the three dimensional structure of Loop 2 of BDNF. A control peptide contained alanine substitutions at lysine positions 41 and 50. These peptides were diluted in PBS at varying concentrations and injected dorsal to the SN, as described for neutralizing antibodies.
Immunohistochemistry for tyrosine hydroxylase (TH)
Pups were anesthetized with either hypothermia (within one postnatal week) or ketamine/xylazine (older than one postnatal week) and then perfused intracardially with 0.9% saline for 5 min by gravity, followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB) for 10 min at room temperature. The brain was removed and the SN was blocked. The SN was post-fixed in 4% paraformaldehyde in 0.1 M PB for one week at 4°C and then placed in 20% sucrose in 0.1 M PB for 24 h prior to sectioning. Each SN was rapidly frozen by immersion in isopentane on dry ice and then sectioned in a cryostat at 30 μm. For stereology, a complete set of serial sections was obtained, and every fourth section was processed free-floating. After washes with PBS, sections were incubated in primary antibody (MAB5280; Chemicon, Temecula, CA, USA), a mouse monoclonal anti-tyrosine hydroxylase (TH) at 1 : 40 in PBS/10% normal horse serum for 24 h at 4°C. Sections were then washed with PBS and incubated with biotinylated horse anti-mouse IgG (Vector Laboratories) at 1:50 in PBS/10% normal horse serum at 4°C. Sections were washed in PBS at room temperature and then incubated with avidin-biotinylated horseradish peroxidase complexes (ABC, Vector Labs, Burlingame, CA) at 1:600 for 1 h at room temperature. Following washes in PBS, sections were incubated in a solution of diaminobenzidine (50 mg in 100 mL Tris buffer, pH 7.6) containing glucose oxidase, ammonium chloride and D(+) glucose to generate H2O2. Sections were then mounted on glass slides, left to dry overnight at room temperature and thionin counterstained. SN and forebrain sections from adult mice were processed in a similar fashion, except that sections were incubated with rabbit anti-TH (Calbiochem, La Jolla, CA) primary antibody at 1:750. Sections were then treated with biotinylated protein A and ABC (Vector Labs, Burlingame, CA).
Quantitative morphology
(1) Determination of SN neuron numbers by stereologic analysis. SN dopamine neuron numbers were determined by stereologic analysis of TH-immunostained sections for both postnatal rats and adult mice. For the experiments performed on postnatal rats, the SN on both sides of the brain was analyzed; for mice, a single side of the SN was counted for each animal. For each section the entire SN, including the pars compacta, reticulata and lateralis, was identified as the region of interest. Using StereoInvestigator software (MicroBright Field, Inc, Williston, VT), a fractionator probe was established for each section. The number of TH-positive neurons in each counting frame was then determined by focusing down through the section, using 100× objective under oil, as required by the optical disector method. Our criterion for counting an individual TH-positive neuron was the presence of its nucleus either within the counting frame, or touching the right or top frame lines (green), but not touching the left or bottom lines (red). The total number of TH-positive neurons for each side of the SN was then determined by the StereoInvestigator program. (2) Qualitative morphologic analysis of apoptosis in SN. In the TH immunoperoxidase-stained sections, apoptosis was identified at the light microscope level by performing a thionin counterstain and visualizing intranuclear chromatin clumps at 600× as one or more intensely basophilic, homogeneously stained, round and distinctly bounded structures. We have previously shown that, for naturally occurring cell death in dopamine neurons and for induction of this death event, apoptotic profiles so identified are confirmed to be apoptotic by electron microscopy (Macaya et al. 1994;Jackson-Lewis et al. 2000;El-Khodor and Burke 2002), TUNEL labeling (Macaya et al. 1994;Jackson-Lewis et al. 2000;El-Khodor and Burke 2002), and by immunostaining for activated caspase-3 and its cleavage products (Jeon et al. 1999;El-Khodor and Burke 2002) and for activated caspase-9 (Ganguly et al. 2004). Apoptotic profiles in each SN were quantified by scanning the SN at 600× in its entirety on three sections in each of three planes through the SN corresponding to Paxinos-Watson planes 4.2, 3.7 and 3.2 (Paxinos and Watson 1982), for a total of 9 sections per brain. The total number of profiles for each plane was averaged, and the sum of these averages provided a measure of the magnitude of apoptosis in SN for that brain (Oo et al. 2003). All quantitative morphologic assessments were performed on coded slides, blind to experimental condition.
Non-radioactive in situ hybridization for BDNF mRNA
A probe for in situ hybridization was designed based on the published mouse BDNF sequence (Accession no.: BC034862). Oligonucleotide primers for probe isolation were synthesized by Gene Link (Tarrytown, NY) corresponding to sequence as follows: Forward (nucleotides 11-20)- ACCAGGTGAGAAGAGTGATG; reverse(nucleotides 321-340) – CCAAAGGCACTTGACTGCTG. The PCR product derived from the cDNA created from SN mRNA was subcloned in pGEM-T vector (Promega, Madison) and used for making sense or antisense riboprobe. Brains were rapidly removed from PND2 and adult mice, and rapidly frozen in OCT (Tissue-Tek) on dry ice. Sections (14 μm) were thaw-mounted on glass slides (Superfrost Plus, Fisher). For hybridization, sections were warmed to room temperature and fixed by immersion in 4% paraformaldehyde in 0.1 M PBS. Sections were then treated with a pre-hybridization solution as previously described (Burke et al. 1994) for 2 h at room temperature. Sections were then covered with hybridization solution and incubated overnight at 68°C. Hybridization solution contained either anti-sense or sense BDNF probe labeled with digoxigenin-UTP, prepared as per the manufacturer's instructions (Roche Diagnostics, Penzberg, Germany). The size and integrity of labeled probe were confirmed by gel electrophoresis. After washes in 0.2×SSC at 68°C, sections were incubated with an anti-digoxigenin antibody (Roche) at 1:5000 overnight at 4°C. After additional washes, sections were then incubated with a developing solution containing BCIP/NBT (Promega) overnight at room temperature in the dark. Sections were washed and coverslipped with DAKO aqueous mounting medium.
Western analysis of BDNF protein expression. Western analysis was performed as previously described (Neystat et al. 2001) with a monospecific polyclonal rabbit anti-BDNF antibody (sc-546; Santa Cruz Biotechnology, Santa Cruz,CA;).
Statistical analysis
For comparisons of two groups, data were analyzed by Student's t-test. For multiple group comparisons, one way analysis of variance was performed. If a significant difference was found, then a post-hoc analysis was performed by use of a Tukey all pairwise multiple comparison procedure. All statistical analyses were performed with SigmaStat version 3.0.
Acknowledgments
This work was supported by NS26836, NS38370, and the Parkinson's Disease Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- Alonso-Vanegas MA, Fawcett JP, Causing CG, Miller FD, Sadikot AF. Characterization of dopaminergic midbrain neurons in a DBH:BDNF transgenic mouse. J Comp Neurol. 1999;413:449–462. doi: 10.1002/(sici)1096-9861(19991025)413:3<449::aid-cne7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Altar CA, DiStefano PS. Neurotrophin trafficking by anterograde transport. Trends Neurosci. 1998;21:433–437. doi: 10.1016/s0166-2236(98)01273-9. [DOI] [PubMed] [Google Scholar]
- Baker SA, Stanford LE, Brown RE, Hagg T. Maturation but not survival of dopaminergic nigrostriatal neurons is affected in developing and aging BDNF-deficient mice. Brain Res. 2005;1039:177–188. doi: 10.1016/j.brainres.2005.01.052. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Bickford PC, Jones KR. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–6259. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA, Edgar D, Thoenen H. Sensory neurons in culture: changing requirements for survival factors during embryonic development. Proc Natl Acad Sci U S A. 1980;77:1199–1203. doi: 10.1073/pnas.77.2.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982;1:549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, Franklin SO, Inturrisi CE. Acute and persistent suppression of preproenkephalin mRNA expression in the striatum following developmental hypoxic- ischemic injury. J Neurochem. 1994;62:1878–1886. doi: 10.1046/j.1471-4159.1994.62051878.x. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khodor BF, Burke RE. Medial forebrain bundle axotomy during development induces apoptosis in dopamine neurons of the substantia nigra and activation of caspases in their degenerating axons. J Comp Neurol. 2002;452:65–79. doi: 10.1002/cne.10367. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Kuo-Fen L, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Oo TF, Rzhetskaya M, Pratt R, Yarygina O, Momoi T, Kholodilov N, Burke RE. CEP11004, a novel inhibitor of the mixed lineage kinases, suppresses apoptotic death in dopamine neurons of the substantia nigra induced by 6-hydroxydopamine. J Neurochem. 2004;88:469–480. doi: 10.1046/j.1471-4159.2003.02176.x. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Herkenham M, Thibault J. The neostriatal mosaic: II. Patch- and matrix-directed mesostriatal dopaminergic and non-dopaminergic systems. J Neurosci. 1987;7:3915–3934. doi: 10.1523/JNEUROSCI.07-12-03915.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller A, Hutchens JO, Kirby ML, Karapas F, Fernandez C. Stereotaxic electrode placement in the neonatal rat. J Neurosci Methods. 1979;1:41–76. doi: 10.1016/0165-0270(79)90006-2. [DOI] [PubMed] [Google Scholar]
- Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, Lindsay RM. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. 1991;350:230–232. doi: 10.1038/350230a0. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Vila M, Djaldetti R, Guegan C, Liberatore G, Liu J, O'Malley KL, Burke RE, Przedborski S. Developmental cell death in dopaminergic neurons of the substantia nigra of mice. J Comp Neurol. 2000;424:476–488. doi: 10.1002/1096-9861(20000828)424:3<476::aid-cne6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Jain S, Golden JP, Wozniak D, Pehek E, Johnson EM, Jr, Milbrandt J. RET is dispensable for maintenance of midbrain dopaminergic neurons in adult mice. J Neurosci. 2006;26:11230–11238. doi: 10.1523/JNEUROSCI.1876-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janec E, Burke RE. Naturally occurring cell death during postnatal development of the substantia nigra of the rat. Mol Cell Neurosci. 1993;4:30–35. doi: 10.1006/mcne.1993.1004. [DOI] [PubMed] [Google Scholar]
- Jarskog LF, Miyamoto S, Lieberman JA. Schizophrenia: new pathological insights and therapies. Annu Rev Med. 2007;58:49–61. doi: 10.1146/annurev.med.58.060904.084114. [DOI] [PubMed] [Google Scholar]
- Jeon BS, Kholodilov NG, Oo TF, Kim S, Tomaselli KJ, Srinivasan A, Stefanis L, Burke RE. Activation of caspase-3 in developmental models of programmed cell death in neurons of the substantia nigra. J Neurochem. 1999;73:322–333. doi: 10.1046/j.1471-4159.1999.0730322.x. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kerr DA, Llado J, Shamblott MJ, Maragakis NJ, Irani DN, Crawford TO, Krishnan C, Dike S, Gearhart JD, Rothstein JD. Human embryonic germ cell derivatives facilitate motor recovery of rats with diffuse motor neuron injury. J Neurosci. 2003;23:5131–5140. doi: 10.1523/JNEUROSCI.23-12-05131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodilov N, Yarygina O, Oo TF, Zhang H, Sulzer D, Dauer WT, Burke RE. Regulation of the development of mesencephalic dopaminergic systems by the selective expression of glial cell line-derived neurotrophic factor in their targets. J Neurosci. 2004;24:3136–3146. doi: 10.1523/JNEUROSCI.4506-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- Kramer ER, Aron L, Ramakers GM, Seitz S, Zhuang X, Beyer K, Smidt MP, Klein R. Absence of Ret signaling in mice causes progressive and late degeneration of the nigrostriatal system. PLoS Biol. 2007;5:e39. doi: 10.1371/journal.pbio.0050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauder JM, Bloom FE. Ontogeny of monoamine neurons in the locus coeruleus, raphe nuclei and substantia nigra of the rat. J Comp Neurol. 1974;155:469–482. doi: 10.1002/cne.901550407. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde YA. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- Lindsay RM. Brain-derived neurotrophic factor: An NGF-related neurotrophin. In: Loughlin SE, Fallon JH, editors. Neurotrophic Factors. Academic Press; San Diego: 1993. pp. 257–284. [Google Scholar]
- Macaya A, Munell F, Gubits RM, Burke RE. Apoptosis in substantia nigra following developmental striatal excitotoxic injury. Proc Natl Acad Sci USA. 1994;91:8117–8121. doi: 10.1073/pnas.91.17.8117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: Parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Marchand R, Poirer LJ. Isthmic origin of neurons of the rat substantia nigra. Neuroscience. 1983;9:373–381. doi: 10.1016/0306-4522(83)90300-7. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Neystat M, Rzhetskaya M, Oo TF, Kholodilov N, Yarygina O, Wilson A, El-Khodor BF, Burke RE. Expression of cyclin-dependent kinase 5 and its activator p35 in models of induced apoptotic death in neurons of the substantia nigra in vivo. J Neurochem. 2001;77:1611–1625. doi: 10.1046/j.1471-4159.2001.00376.x. [DOI] [PubMed] [Google Scholar]
- Numan S, Gall CM, Seroogy KB. Developmental expression of neurotrophins and their receptors in postnatal rat ventral midbrain. J Mol Neurosci. 2005;27:245–260. doi: 10.1385/JMN:27:2:245. [DOI] [PubMed] [Google Scholar]
- Numan S, Seroogy KB. Expression of trkB and trkC mRNAs by adult midbrain dopamine neurons: a double-label in situ hybridization study. J Comp Neurol. 1999;403:295–308. doi: 10.1002/(sici)1096-9861(19990118)403:3<295::aid-cne2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- O'Leary PD, Hughes RA. Structure-activity relationships of conformationally constrained peptide analogues of loop 2 of brain-derived neurotrophic factor. J Neurochem. 1998;70:1712–1721. doi: 10.1046/j.1471-4159.1998.70041712.x. [DOI] [PubMed] [Google Scholar]
- Oo TF, Burke RE. The time course of developmental cell death in phenotypically defined dopaminergic neurons of the substantia nigra. Dev Brain Res. 1997;98:191–196. doi: 10.1016/s0165-3806(96)00173-3. [DOI] [PubMed] [Google Scholar]
- Oo TF, Kholodilov N, Burke RE. Regulation of natural cell death in dopaminergic neurons of the substantia nigra by striatal GDNF in vivo. J Neurosci. 2003;23:5141–5148. doi: 10.1523/JNEUROSCI.23-12-05141.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Ann Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pascual A, Hidalgo-Figueroa M, Piruat JI, Pintado CO, Gomez-Diaz R, Lopez-Barneo J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat Neurosci. 2008;11:755–761. doi: 10.1038/nn.2136. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Academic Press; New York: 2001. [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego, CA: 1982. [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Seroogy KB, Lundgren KH, Tran TM, Guthrie KM, Isackson PJ, Gall CM. Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J Comp Neurol. 1994;342:321–334. doi: 10.1002/cne.903420302. [DOI] [PubMed] [Google Scholar]
- Spina MB, Squinto SP, Miller J, Lindsay RM, Hyman C. Brain-derived neurotrophic factor protects dopamine neurons against 6-hydroxydopamine and N-methyl-4-phenylpyridinium ion toxicity: involvement of the glutathione system. J Neurochem. 1992;59:99–106. doi: 10.1111/j.1471-4159.1992.tb08880.x. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]