

Abstract
Antithrombin, a plasma glycoprotein serpin, requires conformational activation by heparin to induce an anticoagulant effect, which is mediated through accelerated factor Xa inhibition. Heparin, a highly charged polymer and an allosteric activator of the serpin, is associated with major adverse effects. To design better, but radically different activators of antithrombin from heparin, we utilized a pharmacophore-based approach. A tetrahydroisoquinoline-based scaffold was designed to mimic four critical anionic groups of the key trisaccharide DEF constituting the sequence-specific pentasaccharide DEFGH in heparin. Activator IAS5 containing 5,6-disulfated tetrahydroisoquinoline and 3,4,5-trisulfated phenyl rings was found to bind antithrombin at pH 7.4 with an affinity comparable to the reference trisaccharide DEF. IAS5 activated the inhibitor nearly 30-fold, nearly 2 to 3-fold higher than our first generation flavan-based designs. This work advances the concept of antithrombin activation through non-saccharide, organic molecules and pinpoints a direction for the design of more potent molecules.
Introduction
Antithrombin, a member of the serpin (serine proteinase inhibitor) family of proteins, is a major regulator of several proteinases, especially thrombin, factor Xa, and factor IX, which play pivotal roles in the coagulation cascade. Yet, antithrombin alone is a rather poor inhibitor of all three proteinases under physiologic conditions.1–3 A naturally occurring polymer called heparin accelerates this weak inhibition several thousand-fold,2–4 which is the reason for heparin’s clinical use as an anticoagulant.
Heparin is a linear polysaccharide of varying chain length and is composed of uronic acid and glucosamine residues that are variably sulfated and acetylated.3,5 Both these features introduce major structural complexity in heparin, which is known to be associated with multiple adverse effects including hemorrhage, thrombocytopenia, osteoporosis and variable patient response.6 These side-effects are reduced by using homogeneous heparin preparations such as fondaparinux, which is a synthetic five-residue sequence based on the naturally occurring DEFGH# sequence (Fig. 1).
Figure 1. Structures of pentasaccharide DEFGH and organic activators.

Historically, D, E, F, G, and H refer to residue labels from the non-reducing end of the pentasaccharide. Sulfate groups in DEFGH highlighted as filled ovals ( ) are critical for its high-affinity interaction with antithrombin. (+)-CS is a flavanoid-based first generation non-saccharide activator designed earlier,12,14 while isoquinoline-based molecules labeled as either IAS or IES type are being studied in this work.
) are critical for its high-affinity interaction with antithrombin. (+)-CS is a flavanoid-based first generation non-saccharide activator designed earlier,12,14 while isoquinoline-based molecules labeled as either IAS or IES type are being studied in this work.
The DEFGH sequence binds antithrombin with high affinity and induces an allosteric conformational change in the serpin that allows the inhibitor to better recognize its target proteinases, especially factor Xa, resulting in acceleration of inactivation.4 DEFGH binds antithrombin in the pentasaccharide-binding site (PBS),7 while in addition to interacting with PBS, full-length UFH interacts with an adjacent region called the extended heparin–binding site (EHBS).8,9 Detailed structure-activity studies show that several anionic groups of DEFGH are critical for its high-affinity interaction with antithrombin (Fig. 1). More importantly, studies with truncated variants of DEFGH indicate that trisaccharide sequence DEF is the minimum structure that retains the functional role of heparin, i.e., antithrombin activation, albeit with significant loss of affinity under physiological conditions.10
Numerous attempts have been made to design or discover new molecules that activate antithrombin,3,11 however each of these searches has relied on utilizing a saccharide scaffold as a mimic of heparin. Implicit in these designs was the assumption that a saccharide scaffold was critical to induce antithrombin activation. We questioned this assumption and undertook the challenge of designing non-saccharide activators of the serpin. Non-saccharide, organic mimics of heparin are likely to offer special advantages such as greater non-ionic binding energy in antithrombin recognition,12 greater hydrophobicity for possible oral delivery, greater synthetic accessibility, and higher specificity of action in comparison to heparin’s numerous activities.13 However, designing non-saccharide, antithrombin activators is intrinsically challenging. Three major hurdles compound this challenge. One, antithrombin activation is a two-step, induced-fit process in which the activator is expected to recognize both the native state and the activated state such that key interactions could be ‘turned on’ for conformational transformation.4,10 Secondly, heparin mimics invariably have to possess several negative charges, which can induce recognition of many electropositive domains on protein surfaces. Finally, large organic scaffolds that can contain multiple sulfate and carboxylate groups capable of mimicking heparin, or pentasaccharide DEFGH (~20–25 Å long), are difficult to design.
In our first attempt on designing non-saccharide antithrombin activators, we designed sulfated flavan-3-ol molecules (Fig. 1) based on a receptor-based approach involving computerized hydropathic interaction (HINT) analyses.12,14 The sulfated flavan-3-ol molecules were found to bind antithrombin with an affinity comparable to DEF and accelerated factor Xa inhibition approximately 8–10-fold. We have now utilized a pharmacophore-based approach of mimicking trisaccharide DEF to design tetrahydroisoquinoline-based sulfated activator IAS5 (Fig. 1). Our studies suggest that IAS5 activates antithrombin nearly 30-fold, an increase of nearly 2 to 3-fold higher than the first generation rationally designed agents.12,14 This work advances the concept of antithrombin activation through non-saccharide, organic molecules and pinpoints a direction for the design of more potent molecules.
Experimental Procedures
Proteins and Chemicals
Antithrombin and factor Xa (human forms) were purchased from Haematologic Technologies (Essex Junction, VT) and used as received. Molar concentrations of antithrombin were calculated from absorbance measurements at 280 nm using a εMAX of 37,700 M−1cm−1. Antithrombin was stored in 20 mM sodium phosphate buffer, pH 7.4, containing 100 mM NaCl, 0.1 mM EDTA and 0.1% (w/v) PEG8000 at −78 °C, while factor Xa was stored in 5 mM MES buffer, pH 6.0, containing 25 mM NaCl at −78 °C until use. Factor Xa substrate Spectrozyme FXa was obtained from American Diagnostics, Greenwich, CT. Trimethylamine-sulfur trioxide complex was purchased from Alfa-Aesar (Ward Hill, MA). All other reagents/chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Experimental Conditions
Antithrombin interaction and activation studies were performed at 25 °C and in 20 mM sodium phosphate buffer, containing 100 mM NaCl, 0.1 mM EDTA and 0.1% (w/v) PEG 8000, adjusted to 7.4. The ionic strength of this buffer is 0.135 and the buffer is labeled as pH 7.4, I 0.15, 25 °C buffer.
Synthesis and Characterization of Organic Activators
IES4 and IES5 were synthesized from IEM4 and IEM5 (Fig. 1) in two steps, respectively (Scheme I), as described in our earlier work.15 The carboxylate derivatives IAS4 and IAS5 were synthesized from IES4 and IES5, respectively. Briefly, each ester (0.03 – 0.08 mmol) was stirred with potassium t-butoxide and H2O (2:1 molar equivalent, 3 equiv. KBuO-t) in DMSO (0.1 M) under nitrogen at room temperature until the starting material was consumed (HPLC analysis, 1–3 h),16 after which sodium dibasic phosphate (10 equiv) in H2O (2 mL) was added to the reaction mixture. Following 15 min of stirring, the reaction mixture was loaded onto a Sephadex G10 column (160 cm) and chromatographed using water as an eluent. Fractions were combined, concentrated under vacuum and lyophilized to obtain the sodium salt of the carboxylate as a white solid. Capillary electrophoresis of both products using a fused silica capillary in 20 mM sodium phosphate buffer, pH 2.3, at 10 kV showed a single peak with greater than 95% purity (see Supplementary Information). IAS4: 1H-NMR (400 MHz, 2H2O): δ 6.99–7.51 (m, 5H), 4.72 – 4.85 (m, 2H), 4.42 – 4.52 (m, 1H), 3.01–3.16 (m, 2H); ESI (-ve) m/z calcd for C17H10NNa5O19S4 [(M-Na)−] 751.83, found 751.91; IAS5: 1H NMR (400 MHz, 2H2O) δ 7.05 – 7.46 (m, 5H), 4.75–4.99 (m, 2H), 4.47–4.53 (m, 1H), 3.03–3.11 (m, 2 H); ESI (-ve) m/z calcd for C17H9NNa6O23S5 [(M+H)+] 869.77, found 869.83.
Scheme I.

Fluorescence Spectroscopy and Equilibrium Binding Studies
Fluorescence experiments were performed using a QM4 fluorometer (Photon Technology International, Birmingham, NJ) in pH 7.4, I 0.15, 25 °C buffer. Equilibrium dissociation constants (KD) for the interaction of organic activators with plasma antithrombin were determined by titrating the activator into a solution of plasma antithrombin and monitoring the decrease in the fluorescence at 340 nm (λEX = 290 nm). The slit widths on the excitation and emission side were 1 and 2 mm, respectively. The decrease in fluorescence signal was fit to the quadratic equilibrium binding equation I to obtain the KD of interaction, wherein ΔF represents the change in fluorescence following each addition of the activator ([ACT]O) from the initial fluorescence FO and ΔFMAX represents the maximal change in fluorescence observed on saturation of antithrombin ([AT]O).
| I |
Factor Xa Inhibition Studies
The kinetics of inhibition of factor Xa by antithrombin in the presence of organic activators under pseudo-first order conditions was measured spectrophotometrically in a manner similar to our earlier work.10,12 A fixed 10 nM concentration of factor Xa was incubated with fixed concentrations of plasma antithrombin (0.1 to 0.5 μM) and the sulfated activator (0 to 100 μM) in pH 7.4, I 0.15 buffer at 25 °C. At regular time intervals, an aliquot of the inhibition reaction (100 μL) was diluted with 900 μL of 100 μM Spectrozyme FXa in pH 7.4, I 0.15 buffer and the initial rate of substrate hydrolysis was measured from the increase in absorbance at 405 nm. The exponential decrease in the initial rate of substrate hydrolysis as a function of time was used to determine the observed pseudo-first rate constant of factor Xa inhibition (kOBS). A plot of kOBS values measured as a function of different concentrations of an organic activator could be described by equation II, in which kUNCAT is the second-order rate constant of factor Xa inhibition by antithrombin alone, i.e., 2300 M−1s−1 at pH 7.4, I 0.15, 25 °C,10 and kACT is the second-order rate constant of factor Xa inhibition by antithrombin – organic activator complex.
| II |
Results and Discussion
Rational design of tetrahydroisoquinoline-based activator
Our first generation agents, the flavanoid-based activators, were designed on the basis of a receptor-based approach involving computerized docking and scoring.14 In this work, we utilized a pharmacophore-based approach to design potential heparin mimics. Trisaccharide DEF, rather than pentasaccharide DEFGH, was chosen as the saccharide scaffold to mimic because of the expectation that the smaller de novo organic design would be easier to synthesize. In addition, it is known that both DEF and DEFGH conformationally activate antithrombin fully and equally (~300-fold).10 Structure-activity studies show that three sulfate groups, i.e., the 6-O-sulfate on residue D and 3-O-sulfate and 2-N-sulfate on residue F, and the 6-carboxylate group of residue E are critical for conformational activation of antithrombin, while the trisaccharide scaffold only serves to position these groups for optimal interaction.11,17 Thus, the pharmacophore was extracted and the four critical groups were connected in three-dimensional space using a linear carbon linker to arrive at a first ‘blueprint’ of an activator (Fig. 2). The blueprint was transformed into a potential organic activator by introducing rigidity and simplifying the structure for rapid synthesis. In this process, several scaffolds were modeled and their 3D similarity with the pharmacophore assessed. IAS5, the tetrahydroisoquinoline-based activator containing an acid functionality and five sulfate groups, was selected because it retained the three-dimensional configuration of the four critical groups (Fig. 3). In addition, the 2-N-sulfate of residue D was also effectively mimicked by a sulfate group of the isoquinoline ring.
Figure 2. Rationale used in the design of isoquinoline-based organic activators.

Four critical anionic groups (highlighted as filled ovals) of trisaccharide DEF formed the pharmacophore. Connecting the groups using a carbon framework followed by engineering of rigidity that matches their orientation gave rise to a ‘blueprint’, which was transformed into a synthetically plausible target, IAS5.
Figure 3. Overlay of DEF and IAS5 showing superposition of five anionic groups.
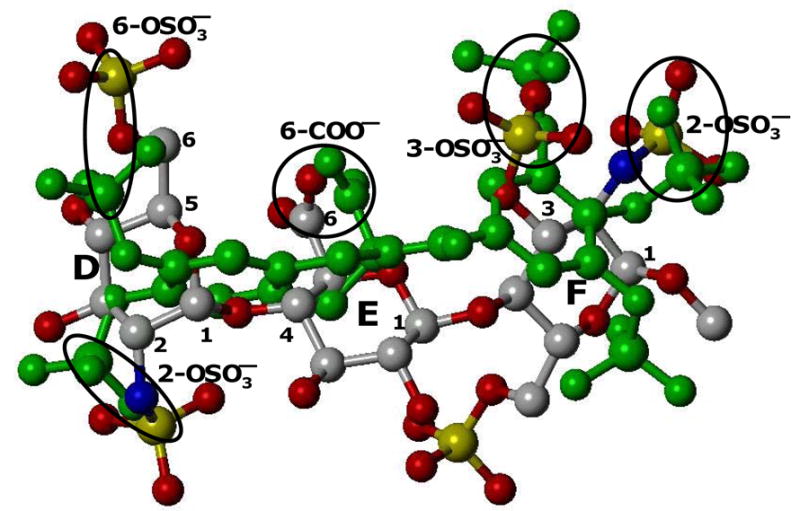
The three critical sulfate groups – 6-OSO3− on residue D and 3-OSO3− and 2-OSO3− on residue F – as well as the 6-COO− group of residue E overlay well on three sulfate and one carboxylate groups of IAS5 in three dimensional space. In addition, the 2-OSO3− group of residue D matches one of the remaining two sulfates on IAS5. The overall RMSD for the five anions was 2.0 Å. See text for details.
Calculation of the root mean square deviation between the five corresponding groups gave a value of 2.0 Å (range 1.1 to 2.7 Å), which reduced to 1.6 Å when only the groups of the pharmacophore were considered. In this design, it is suggested that the bicyclic ring mimics the D and E rings, the unicyclic ring mimics ring F, and the carboxylate groups match each other. To assess the importance of selected groups, we chose to study ester analogs, IES5 and IES4, as well as an acid derivative with one less sulfate group, IAS4.
Synthesis of isoquinoline-based sulfated activators
Nearly all small organic sulfates reported in the literature are mono- or di-sulfated molecules, typically prepared using sulfur trioxide complexes with amines in a highly polar solvent (DMF or DMA).18–20 Yet, as the number of –OH groups increase on a small scaffold, sulfation becomes progressively difficult because of anion crowding. We recently developed a rapid and high-yielding microwave-based synthesis of polysulfated organic molecules using trimethylamine– or pyridine– sulfur trioxide complexes in the presence of excess amine in anhydrous acetonitrile at 100 – 120 °C.15 Thus, ester analogs IES4 and IES5 were prepared in two steps, debromination and sulfation, in good yields from the corresponding methoxy-protected compounds IEM4 and IEM5, respectively (Scheme I, see Supplementary Information for details on the synthesis of IEM4 and IEM5). However, the microwave-assisted sulfation failed to yield any sulfated products when the substrate was the polyphenolic molecule containing an acid group. It is possible that the presence of naked carboxylic acid functionality under microwave conditions induces degradation of organic sulfates resulting in multiple products. To circumvent this problem, the carboxylic acid ester in IES4 and IES5 was hydrolyzed in excellent yields by simply stirring with KBuO-t/H2O (2:1) in anhydrous DMSO at RT.16 It is worthwhile to mention that this hydrolysis fails miserably when standard saponification conditions (NaOH/water) are used.
Equilibrium Dissociation Constant of Antithrombin Interaction with Sulfated Isoquinoline-based Activators
Previously we have used fluorescence spectroscopy for measuring the affinity of ligands for antithrombin. For saccharide ligands, e.g., DEFGH and DEF, the ~30% increase in intrinsic tryptophan fluorescence affords a convenient signal for the determination of the KD of the interaction,4,10 while for our first generation agents, i.e., (+)-CS and others, we used an external probe, TNS, to determine binding affinity.12,14 In this study, we found that interaction of our designed organic ligands with antithrombin resulted in a decrease in intrinsic tryptophan fluorescence that reached a plateau at high ligand concentrations (Fig. 4). An equivalent limiting decrease of ~100% was obtained for IAS5, IES4 and IES5, which could be fitted with the standard quadratic binding equation I to obtain the KD of interaction at pH 7.4, I 0.15, 25 °C. KD values of 320, 330 and 805 μM were measured for IAS5, IES4 and IES5, respectively, corresponding to a similar free energy of binding between 4.2 and 4.8 kcal/mol (Table 1). For IAS4, the fluorescence titration could not be made to reach an endpoint and, hence, a significantly weaker affinity (KD > 1mM) is estimated. The affinity of reference trisaccharide DEF for antithrombin under similar conditions (pH 7.4, I 0.15, 25 °C), has not been reported, however a KD value of 66 ± 4 μM has been measured at pH 7.4 in the absence of any added salt (pH 7.4, I 0.05, 25 °C).10 The affinity at pH 7.4, I 0.15, 25 °C can be estimated to be ~500–600 μM based on salt dependence studies of tetrasaccharide DEFG, which an increase of 9.4-fold at higher salt concentration.10 Likewise, the affinities of IAS5, IES4 and IES5 at pH 7.4, I 0.05, 25 °C were found to be 37, 50, and 130 μM (data not shown), respectively, suggesting that the designed activators compare favorably with trisaccharide DEF in antithrombin affinity. In comparison, the affinities of our flavanoid-based designs were found to be ~130 ± 20 μM at pH 7.4, I 0.15, 25 °C,12 which is ~0.5 to 1 kcal/mol better than the tetrahydroisoquinoline molecules investigated here.
Figure 4. Fluorescence-based measurement of the equilibrium dissociation constant of organic activator – antithrombin complex at pH 7.4, I 0.15, 25°C.

Interaction of organic activators (○ = IES5; □ = IES4; ◇ = IAS5, IAS4 not shown) with antithrombin resulted in a saturable decrease in intrinsic tryptophans fluorescence at 340 nm (λEX = 290 nm), which was fitted to the quadratic binding equation I to calculate the observed KD. Solid lines represent the non-linear regressional fit.
Table 1.
Thermodynamic and kinetic parameters for the interaction of the organic activators with plasma antithrombin at pH 7.4, I 0.15, 25 °C.
| KD,OBS(μM)a | ΔFMAX(%)a | ΔG° (kcal/mol) | kACT(M−1s−1) | Accelerationb | |
|---|---|---|---|---|---|
| IAS5 | 320±10c | −1.3±0.2 | 4.8±0.2 | 69,780±580 | 30 |
| IES5 | 805±70 | −1.2±0.2 | 4.2±0.4 | 6160±240 | 2.7 |
| IES4 | 330±20 | −1.4±0.2 | 4.8±0.3 | 10860±480 | 4.7 |
| IAS4 | >1000d | na | na | 5,370e | 2.3e |
Measured spectrofluorometrically using a saturable decrease in intrinsic tryptophan fluorescence (λEM = 340 nm) due to the binding of the organic activator to plasma antithrombin. See ‘Experimental Procedures’ for details.
Represents the ratio of second-order rate constant of antithrombin inhibition of factor Xa in the presence of organic activator (kACT) to that in its absence (kUNCAT). The kUNCAT value for pH 7.4 buffer used in calculations was 2300 M−1s−1, as reported previously.10
Errors represent ±1 S.E.
This value was not possible to measure accurately because of insufficient saturation of antithrombin.
Estimated on the assumption that KD is 1,000 μM.
Antithrombin Activation by Tetrahydroisoquinoline-based Activators
A distinguishing characteristic of antithrombin activation is the acceleration in rate of factor Xa inhibition. The second–order rate constant for antithrombin inhibition of factor Xa in the presence of tetrahydroisoquinoline-based activators (kACT) was determined at pH 7.4, I 0.15, 25 °C, as previously described.10 The concentration dependence of the observed rate constant of factor Xa inhibition for each activator is shown in Figure 5. The concentration dependence profile is linear and can be analyzed on the basis of equation II, which considers the observed rate of reaction as consisting of the uncatalyzed (kUNCAT) and catalyzed (kACT) components. A comparison of the linear profiles indicates that the slope for IAS5 is significantly greater than that for IES4 or IES5 suggesting that IAS5 is a better activator of antithrombin. Linear regression of the data using equation II gives kACT of 69780, 6160 and 10860 M−1s−1 for IAS5, IES5 and IES4, respectively (Table 1). Using the ill-defined KD value for IAS4, the kACT was estimated to be 5370 M−1s−1 under identical conditions. Thus, the acceleration in antithrombin inhibition of factor Xa induced by IAS5 was found to be 30-fold, while IES5, IES4, and IAS4 induce a 2.7-, 4.7-, and 2.3-fold acceleration, respectively (Table 1). The results suggests that nearly all sulfate and carboxylate groups of IAS5 are important for activation of antithrombin as even a unit change in functional group modification appears to be not tolerated well. This highlights the fidelity of pharmacophore-based design approach used in the study.
Figure 5. Antithrombin activation for acceleration inhibition of factor Xa by organic activators.

The observed rate constant (kOBS) of factor Xa inhibition by antithrombin in the presence of varying concentrations of each organic activator (○ = IES5; □ = IES4; ◇ = IAS5, IAS4 not shown) was measured in a spectrophotometric assay at pH 7.4, I 0.15 and 25 °C. Solid lines represent linear regressional fits to the data using equation II to calculate the second-order rate constant of factor Xa inhibition by antithrombin – organic activator complex (kACT).
In comparison to the first generation flavan-based activators,12,14,21 the new molecules were found to be ~1.5–3-fold better at antithrombin activation suggesting reasonable improvement in design. This implies that appropriate modifications to the scaffold, e.g., extension of the linker between the tetrahydroisoquinoline and phenyl rings to more closely mimic pentasaccharide DEFGH, may yield a more potent activator. A key consideration in the future design would be interactions with antithrombin’s pentasaccharide binding site residues, specifically Lys114, Lys125 and Arg129, which are possibly not engaged well with IAS5 resulting in significantly less efficacy as compared to trisaccharide DEF.10
Conclusions and Significance
Antithrombin activation is a complex process in which the energy of heparin binding at an allosteric site is transferred to the proteinase recognition domain. Heparin, nature’s highly efficient activator, is a design pinnacle and emulating it through a radically novel, as-yet-undeciphered scaffold is an engineering challenge. The gain in activation observed with our new pharmacophore-based approach is a small, but important, step in this direction. The results suggest that the tetrahydroisoquinoline-linker-phenyl-based scaffold possesses the potential to mimic a small fragment of heparin. Thus, appropriately modified molecules in which the linker consists of multiple atoms may lead to the design of a DEFGH mimic. This implies that antithrombin activation through non-saccharide molecules appears feasible. Finally, the design of a robust algorithm for the design of heparin mimics will likely open up many protein–heparin systems for non-saccharide modulation.
Supplementary Material
Acknowledgments
This work was supported by the National Heart, Lung and Blood Institute (RO1 HL069975 and R41 HL081972), the American Heart Association National Center (EIA 0640053N), the A. D. Williams Foundation (6-48708), and the Mizutani Foundation for Glycoscience.
Footnotes
Abbreviations used: DEFGH, heparin pentasaccharide DEFGH; DEF, trisaccharide DEF of DEFGH; (+)-CS, (+)-catechin sulfate; PBS, pentasaccharide–binding site; EHBS, extended heparin–binging site; PEG, polyethylene glycol; MES, 2-(N-morpholino)ethanesulfonic acid; EDTA, ethylene diamine tetraacetic acid
Supplementary Information Available: A description of experimental procedures used to synthesize IES4 and IES5, and their spectral characterization.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Björk I, Olson ST. Adv Exp Med Biol. 1997;425:17–33. [PubMed] [Google Scholar]
- 2.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, Herault JP, Herbert JM, Björk I. Thromb Haemost. 2004;92:929–939. doi: 10.1160/TH04-06-0384. [DOI] [PubMed] [Google Scholar]
- 3.Desai UR. Med Res Rev. 2004;24:151–181. doi: 10.1002/med.10058. [DOI] [PubMed] [Google Scholar]
- 4.Olson ST, Björk I, Sheffer R, Craig PA, Shore JD, Choay J. J Biol Chem. 1992;267:12528–12538. [PubMed] [Google Scholar]
- 5.Rabenstein DL. Nat Prod Rep. 2002;19:312–331. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 6.Hirsh J, Anand SS, Halperin JL, Fuster V. Circulation. 2001;103:2994–3018. doi: 10.1161/01.cir.103.24.2994. [DOI] [PubMed] [Google Scholar]
- 7.Jin L, Abrahams J-P, Skinner R, Petitou M, Pike RN, Carrell RW. Proc Natl Acad Sci. 1997;94:14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W, Johnson DJD, Esmon CT, Huntington JA. Nature Struct Mol Biol. 2004;11:857–862. doi: 10.1038/nsmb811. [DOI] [PubMed] [Google Scholar]
- 9.Meagher JL, Huntington JA, Fan B, Gettins PG. J Biol Chem. 1996;271:29353–29358. doi: 10.1074/jbc.271.46.29353. [DOI] [PubMed] [Google Scholar]
- 10.Desai UR, Petitou M, Björk I, Olson ST. J Biol Chem. 1998;273:7478–7487. doi: 10.1074/jbc.273.13.7478. [DOI] [PubMed] [Google Scholar]
- 11.Petitou M, van Boeckel CA. Angew Chem Int Ed Engl. 2004;43:3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]
- 12.Gunnarsson GT, Desai UR. J Med Chem. 2002;45:4460–4470. doi: 10.1021/jm020132y. [DOI] [PubMed] [Google Scholar]
- 13.Capila I, Linhardt RJ. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 14.Gunnarsson GT, Desai UR. J Med Chem. 2002;45:1233–1243. doi: 10.1021/jm020012q. [DOI] [PubMed] [Google Scholar]
- 15.Raghuraman A, Riaz M, Hindle M, Desai UR. Tetrahedron Lett. 2007;48:6754–6758. doi: 10.1016/j.tetlet.2007.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gassman PG, Schenk WN. J Org Chem. 1977;42:918–920. [Google Scholar]
- 17.Desai UR, Gunnarsson GT. Med Chem Res. 1999;9:643–655. [Google Scholar]
- 18.Santos GA, Murray AP, Pujol CA, Damonte EB, Maier MS. Steroids. 2003;68:125–132. doi: 10.1016/S0039-128X(02)00166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Lien IFF, Ruttgaizer S, Dove P, Taylor SD. Org Lett. 2004;6:209–212. doi: 10.1021/ol036157o. [DOI] [PubMed] [Google Scholar]
- 20.Simpson LS, Widlanski TS. J Am Chem Soc. 2006;128:1605–1610. doi: 10.1021/ja056086j. [DOI] [PubMed] [Google Scholar]
- 21.Gunnarsson GT, Desai UR. Bioorg Med Chem Lett. 2003;13:579–583. doi: 10.1016/s0960-894x(02)01055-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.