

Abstract
Deoxyinosine (dI) is produced in DNA by the hydrolytic or nitrosative deamination of deoxyadenosine. It is excised in a repair pathway that is initiated by endonuclease V, the product of the nfi gene. The repair was studied in vivo using high-efficiency oligonucleotide transformation mediated by the Beta protein of bacteriophage λ in a mismatch repair-deficient host. Escherichia coli was transformed with oligonucleotides containing a selectable A to G base substitution mutation. When the mutagenic dG was replaced by a dI in the oligonucleotide, it lost 93 to 99% of its transforming ability in an nfi+ cell, but it remained fully functional in an nfi mutant. Therefore, endonuclease V is responsible for most of the removal of deoxyinosine from DNA. New nfi mutants were isolated based on the strong selection provided by their tolerance for transformation by dI-containing DNA. The repair patch for dI was then measured by determining how close to the transforming dG residue a dI could be placed in the oligonucleotide before it interferes with transformation. At the endonuclease V cleavage site, three nucleotides were preferentially removed from the 3′ end and two nucleotides were removed from the 5′ end. dI:dT and dI:dC base pairs gave the same results. Caveats include possible interference by Beta protein and by mispaired bases. Thus, oligonucleotide transformation can be used to determine the relative importance of redundant repair pathways, to isolate new DNA repair mutants, and to determine with high precision the sizes of repair tracts in intact cells.
1. Introduction
Most base lesions in DNA are removed by excision-repair pathways (reviewed in ref. [1]. The excision may be limited to the damaged nucleotide, but it often extends beyond it. The resulting gaps in the DNA are filled in by a DNA polymerase and sealed by a DNA ligase to produce a repair patch, that is, a tract of one or more newly inserted nucleotides that replace the ones that were excised. Several types of enzymes may participate in the excision steps, which are usually unique to the lesion. Endonucleases may hydrolyze phosphodiester bonds on one or both sides of the lesion. Sometimes, excision is initiated by a glycosylase, which releases the damaged base leaving an abasic site. In that case the C-O bonds on one or both sides of the base-free sugar may be disrupted by an elimination reaction catalyzed by an endolyase activity, which in some cases belongs to the glycosylase itself. The strand break produced by an endonuclease or endolyase provides a site of entry for an exonuclease that may remove the damaged nucleotide or the base-free sugar phosphate. In addition, a helicase may be required, either to release a lesion-containing oligodeoxyribonucleotide (oligo) from the DNA or to unwind a lesion-containing end so that it may be degraded by single-strand specific exonucleases.
The size of the repair patch for a given lesion is specific to that lesion and may provide some insight into the repair pathway. A patch length as short as one nucleotide has been reported for the repair of DNA uracil in prokaryotic and eukaryotic systems in vitro [2]. In the VSR (very short patch repair) system of E. coli, which removes deaminated 5-methylcytosine from DNA methylation sites, a patch length of between two and ten nucleotides was measured by genetic experiments [3]. The UvrABC system of E. coli, which removes bulky adducts, has a patch size of 12 nucleotides in vitro [4]. A single base mismatch in E. coli or in mammalian DNA may cause the replacement of as many as 1000 nucleotides [5]. Most patch sizes have been measured in vitro using cell extracts or mixtures of purified enzymes. Working with cell extracts introduces possible artifacts: component proteins may be denatured on isolation, protein complexes may dissociate, repair is measured in the absence of transcription and DNA replication, and effective levels of some enzymes are not reduced by competing lesions as they may be in the cell. Moreover, some system components such as deoxynucleoside triphosphates, metal ions, salts, enzymes and DNA substrates may be added at levels that do not mimic the intracellular conditions. Studies with purified enzymes have many of the same drawbacks and cannot take into account enzymes and accessory proteins that have yet to be discovered.
In only a few cases have measurements been made in vivo. Methods based on the co-conversion of linked genetic markers are usually crude; they can be no more accurate than the frequency of available markers in any particular region. A promising approach examined the removal of mismatches at various distances from lesions in transfected plasmids [6,7]. It may have general applicability, but the physical nature of the plasmid might affect the results.
In this current study, we present a general method for measuring patch sizes in situ in chromosomal DNA. It is precise, unlike other in vivo methods, and it avoids most of the tacit assumptions of the in vitro methods. The technique involves transformation of chromosomal DNA by single-stranded oligos containing a DNA lesion. This type of transformation is quite efficient in E. coli that express the Beta protein of bacteriophage λ. This single-stranded binding protein promotes the annealing of oligos to single-stranded chromosomal regions [8] such as those near the replication fork. We can gauge the size of a repair patch by examining the inheritance of a mutagenic base substitution situated at various distances from a DNA lesion in the transforming oligo. The lesion to which this method is applied is deoxyinosine (hypoxanthine deoxyribonucleoside), which arises normally from the hydrolytic [9] or nitrosative [10] deamination of deoxyadenosine in DNA and would lead to an A:T to G:C transition mutation if it were not repaired [11]. Its repair is initiated by endonuclease V (Endo V), which is encoded by the nfi gene. The enzyme cleaves the second phosphodiester bond 3′ to the deoxyinosine [12]. The lesion must then be removed by a second DNase cleaving on its 5′ side. This unidentified enzyme could either be an endonuclease or a 3′→5′ exonuclease.
The results will be presented in three major parts. In the first, evidence will be presented that Endo V is the major endonuclease that destroys the transforming activity of dI-containing DNA. This will provide the basis of a selection procedure for nfi mutants. In the second part, the patch size produced during Endo V-mediated repair will be measured. In the third part, control experiments will demonstrate that in a wild type cell, dI-containing oligos are not cleaved before they are incorporated into the chromosome; thus, we are examining repair rather than restriction of dI-containing DNA.
2. Materials and methods
2.1. Bacterial strain construction
The strains used are listed in Table 1. The mutS::Gm insertion in BW1945 and its derivatives is a PCR-mediated replacement [13] of nt 1 to nt 2522 of mutS by a gentamycin resistance cassette. The template was plasmid pKD::Gm, in which an 855-bp SacI Gm cassette from plasmid pUCGm [14] replaced a 360-bp PvuII segment of the kanamycin resistance gene in plasmid pKD4 [15]. The primers were ATCACACCCCATTTAATATCAGGGAACCGGACATAACCCCGTGTAGGCTGGAGCTGCTTC and TTACACCAGGCTCTTCAAGCGATAAATCCACTCCAGCGCCCATATGAATATCCTCCTTAG. Disruptions of mutS nfi, and λ exo genes were verified by PCR. lacZ mutations were introduced by transformations [16] with 70-mers. Lac− colonies were detected on X-Gal/IPTG plates [17], and the mutations were verified by DNA sequencing of PCR products. Transductions with phage P1 dam rev6 were as previously described [18].
Table 1. E. coli strains useda.
| Strain | Description | Source or reference |
|---|---|---|
| BW1185 | Hfr KL16 nfi-1::cat thi-1 spoT1 relA1 | [46] |
| BW1892 | mutS::Gm λcI857 Δ(cro-bio) lacZ [nt 1384 G→A; ΔGG (nt 1370-71)] | From BW1945 by oligo-mediated gene replacement |
| BW1904 | mutS::Gm λcI857 Δ(cro-bio) lacZ [nt 1384 G→A; ΔGG (nt 1370-71)] nfi-1::cat | Transduction: P1(BW1185) × BW1892 |
| BW1945 | mutS::Gm λcI857 Δ(cro-bio) | From DY378 by PCR-mediated gene replacement |
| BW1946 | mutS::Gm λcI857 Δ(cro-bio) lacZ [Δ(nt 1370-71)] | From BW1945 by oligo-mediated gene replacement |
| BW1947 | mutS::Gm λcI857 Δ(cro-bio) lacZ (nt 1384 G→A) | From BW1945 by oligo-mediated gene replacement |
| BW1948 | mutS::Gm λcI857 Δ(cro-bio) lacZ(Am) (nt 50 G→A) | From BW1945 by oligo-mediated gene replacement |
| BW1950 | mutS::Gm λcI857 Δ(cro-bio) Δ(exo)::cat lacZ [nt 1384 G→A; Δ(nt 1370-71)] | Transduction: P1(HME27) × BW1892 |
| DY378 | λcI857 Δ(cro-bio) | [13] |
| HME27 | Δ(argF-lac)169 galK λcI857 Δ(cro-bio) Δ(exo)::cat | [22] |
All strains except BW1185 were derivatives of E. coli W3110 [E. coli K-12 F− λ− IN(rrnD-rrnE)1].
2.2. Bacteriological media
LB media [19] were used for routine growth. They were supplemented with gentamycin at 20 μg/ml, streptomycin at 200 μg/ml, or chloramphenicol at 25 μg/ml for genetic selections. Strr transformants were plated after growth in broth for at least ten generations. Cells containing the EZ-Tn5™ <DHFR-1> transposon were selected on Mueller-Hinton agar (Difco) containing 10 μg/ml of trimethoprim. Lac+ recombinants were selected on 1.5% agar plates containing Vogel-Bonner Medium E [20] supplemented with lactose (0.4%), thiamine (10 μg/ml), and biotin (0.8 μg/ml). A low-salt growth medium, YENB [21], was used for the preparation of electrocompetent cells.
2.3. Transformation
Transformations with single-stranded oligos were performed as previously described [22], using 100 ng of oligonucleotide per 50 μl of electrocompetent cells. Transformations with the EZ-Tn5™ <DHFR-1>Tnp transposome complex (Epicentre Biotechnologies, Madison, WI) were performed as described by the manufacturer.
2.4. Other methods and materials
Oligonucleotides were synthesized by Operon Biotechnologies (Huntsville, AL). DNA sequencing of PCR products was performed by Agencourt Bioscience Corp. (Beverly, MA).
3. Results
3.1. The transformation indicator strain BW1892
E. coli strain BW1892 was constructed to serve as a transformational recipient for oligos and as a parental strain for the isolation of dI-tolerant mutants. It contains a partially deleted λ prophage that expresses the Beta and exonuclease recombination proteins of λ under the thermoinducible control of the λcI857 repressor. The Beta protein enables the efficient incorporation of electroporated oligos into DNA [23]. It binds specifically to single-stranded DNA [24], probably protecting oligos against DNases in the cytoplasm [25,26]. It should also promote annealing of an oligo to a single-stranded region of chromosomal DNA and displacement of regions of adjacent strands [24], such as Okazaki fragments. The mutS mutation in the host enhances the frequency of transformation by blocking mismatch repair [16], which would otherwise degrade the newly formed heteroduplex. Mismatch repair would also interfere with the measurement of endonuclease V-mediated repair if the dI residue is opposite anything other than dC. The strain is also a lacZ mutant, and it is streptomycin sensitive (Strs, rpsL+). It may be transformed to Lac+ or to streptomycin resistance (Strr) with an appropriate oligo that substitutes a dG for a dA in the corresponding gene (Fig. 1). An additional 2-nucleotide (nt) deletion in the lacZ gene prevents spontaneous reversion to Lac+.
Fig. 1.

Transformation indicator regions. Numbers are the nucleotide positions in the ORFs of the lacZ and rpsL genes. Highlighted bases are those that will be replaced by guanines during transformation, yielding Lac+ or Strr (rpsL mutant) recombinants. In BW1892, the base substitution is that of mutation indicator strain CC106 of Cupples and Miller [44], which reverts to Lac+ only by an A→G transition at nt 1384. BW1892 also has a deletion of two guanines at nt 1370 to 1371 in lacZ. An A→G mutation at nt 253 in rpsL produces the Strr phenotype associated with the rpsL40 (strA40) mutation [45]. In strain BW1948, replacing A with G at nt 50 changes the amber codon to the original TGG (tryptophan).
3.2. Transformations with dI-containing oligodeoxyribonucleotides
Because dI pairs with dC, it may be used instead of dG in the transforming oligos, but transformation should occur only if the cell is deficient in the excision of dI from DNA. This principle might provide the basis for methods to isolate or to detect DNA repair mutants and to evaluate the relative roles of individual repair enzymes like Endo V. However, the method has to be tested because even a repair-proficient cell could produce transformants if replication were to occur faster than repair. In that case, the dI-containing oligo would serve as a template for Lac+ or Strr progeny. Accordingly, strains BW1892 (nfi+) and an nfi mutant derivative, BW1904, were electroporated with dG- and dI-containing oligos. The 70-mers either spanned nt 1350 to 1419 of the lacZ gene and had dG or a dI at nt 1384, or they covered nt 229 to 278 of rpsL and had a dG or dI at nt 263 (see Fig. 1). The sequences of the oligos were derived from the coding strands, which were the lagging strand of lacZ and the leading strand of rpsL. The efficiency of transformation by dG-containing oligos was 40-fold better for the lacZ gene than the rpsL gene, which is consistent with known preference for the lagging strand in oligo-mediated transformation [16,27].
For the nfi+ strain BW1892, 120 times as many Lac+ transformants were produced by the dG-containing oligo than with the dI-containing one. Transformations to Strr demonstrated a 14-fold preference for dG DNA. For the nfi mutant, BW1904, the transformation efficiencies were about the same (±50%) for dG and dI-containing DNAs. No spontaneous revertants were seen (<0.05%). This is the first evidence that in vivo, 93 to 99% of the repair of dI is dependent on Endo V. This strong selectivity may provide the basis for mutant isolation. Using dI-containing oligos, a selection for Lac+ transformants followed by one for Strr transformants should produce about a 1700-fold enrichment for repair-deficient mutants.
3.3. Mutant selection with dI-containing oligos
Strain BW1892 was mutagenized by random insertions of EZ-Tn5™ <DHFR-1>, a synthetic transposon specifying trimethoprim resistance. About 5 × 105 trimethoprim-resistant cells were pooled and transformed to Lac+ by a 70-mer containing dI at nt 1384 in the lacZ ORF (Fig. 1). Greater than 2 × 104 transformants were pooled and transformed to Strr by a 70-mer containing dI at nt 263 of rpsL (Fig. 1). Then, greater than 300 transformants were pooled, and their transposons were transferred back into strain BW1892 by generalized transduction with phage P1. The backcross had two functions. First, it placed the insertion mutations back into a Lac− Strs strain to facilitate testing of dI tolerance by using the same transformational selections that were used for the selective enrichment. Second, it excluded from the final pool any spontaneous mutation that was not closely linked to a transposon. Spontaneous mutations should be common in BW1892, which is a mutator (mutS) strain. In the backcrossed transductants, any mutation to dI tolerance would most likely be caused by the transposon insertion itself and therefore would be easily mapped.
Of 64 transductants that were tested, 22 had transposons that were closely linked to nfi. These were tested by PCR using primers ATGGATCTCGCGTCATTAC and CAGTTTACCTGAATTAGGG and confirmed to have insertions directly within the nfi gene. Seven of the 22 putative nfi mutant strains were tested for dI tolerance by transformation to Lac+ with a 70-mer containing dI at position 1384 (Fig. 1). Each had a high frequency of transformation comparable to that of BW1904, the nfi-1::cat derivative. On the other hand, none of the 42 non-nfi mutants had an elevated efficiency of transformation with the dI-containing 70-mer; they may have merely been cells in which, during the enrichment procedure, replication of the incorporated oligo preceded repair. Thus the procedure of selective enrichment by oligo transformation was successful only for the isolation of new nfi mutants. It failed to uncover mutations affecting additional steps in nfi-mediated repair or affecting parallel pathways for dI repair (see Discussion).
3.5 Measuring the dI repair patch
The results so far indicate that the dI-intolerance of a wild type cell is mostly due to Endo V and that a dI-tolerant cell is invariably an nfi mutant. Therefore, the study of dI repair during transformation in a wild type cell is a study of Endo V-mediated repair. To measure the size of the repair patch after Endo V scission, we may study the transformation of an nfi+ cell by a dI-containing oligo. The principle is that if a dI in the oligo prevents the acquisition of a nearby mismatched nucleotide, the dI repair patch extends through that nucleotide. An oligo is used that contains a mutagenic base substitution, such as the G at position 1384 in the lacZ gene, which can transform BW1892 to Lac+ (Fig. 1). A dI residue is placed nearby. Because 99% of it will be excised, it should not be mutagenic even if it is mispaired. After incorporation of the oligo, if excision repair of the dI encompasses the dG, then there will be no Lac+ transformants (Fig. 2A). If the excision of dI spares the dG, then there will be Lac+ transformants (Fig. 2B). In this example, the dG is located −5 nucleotides from the Endo V cleavage site, which occurs at the second phosphodiester bond 3′ to the dI. If the dI residue markedly reduces the number of Lac+ transformants, then at least 5 nucleotides are removed from the 3′ end created by Endo V cleavage. Strain BW1892 was transformed with a series of similar oligos containing dI at various positions. The results (Fig. 3A) indicate a sharply demarcated minimum size of 5 nucleotides for the repair patch, with three nucleotides removed from the 3′ end and two from the 5′ end. Those dI-containing oligos that produced a high number of transformants produced from 0.75 to 1.7 times as many transformants as the dG-containing oligo. Therefore, there cannot be a large fraction of DNA molecules with patch sizes greater than five nucleotides. These are the first results of which the author is aware for the measurement of the Endo V repair patch either in vivo or in vitro.
Fig. 2.

Principle of repair patch measurement. Oligonucleotide transformation produces a heteroduplex region of which 20 bp are shown (nt 1377 to 1396 of lacZ in BW1892). The oligo contains a mutagenic dG and a nearby dI. The production of Lac+ transformants will depend on whether or not the excision of dI encompasses the dG (A versus B). See text for details.
Fig. 3.

Extent of the dI repair patch. As outlined in Fig. 2, lacZ mutants were transformed by a set of 70-mers, each of which contained a mutagenic dG at a fixed position and a dI at a variable position. A position of −1 indicates that the dI is placed so that the dG is at the 3′ end of the Endo V cleavage site, whereas a position of +1 indicates that it is at the 5′ end. Each bar represents the mean ± SEM for four transformations, normalized to the values obtained for dI-free oligos. (A) Strain BW1892 transformed by 70-mers covering 1350 to nt 1419 of lacZ. (B) Strain BW1945 transformed with 70-mers covering nt 15 to nt 84 of lacZ. (See Fig. 1 for sequences). The data in B were corrected for the spontaneous reversion frequency of BW1945 (<1% of peak values).
Similar results (Fig. 3B) were obtained with the lacZ(Am) strain BW1948 (Fig. 1), indicating that the measurements were not largely affected by sequence context. The lacZ(Am) transformations employed different oligos, a different region of the gene, and a chromosomal mutation that did not include a deletion. Although not shown, similar results were also obtained when 40-mers were used instead of 70mers to transform strain BW1948.
In addition to producing the λ Beta protein, the strains used in this study also produce λ exonuclease. This condition was ignored initially because this double-strand-specific 5′→3′ exonuclease has no measurable activity at nicks or gaps in DNA [28,29], although it might be able to excise a short 5′ flap displaced by DNA synthesis [30]. However, results similar to those of Fig. 3A were obtained when the experiment was repeated with BW1950, an isogenic strain with a λ exo mutation (results not shown).
3.6. A dI-containing oligo Is not cleaved before incorporation into DNA
Endo V will cleave near dI residues in single-stranded as well as double-stranded DNA. It was possible that we have been observing restriction rather than repair of dI-containing DNA, i.e., events before incorporation into the chromosome rather than events occurring afterward. Thus, the apparent “repair patch” might have been due to Endo V cleavage of the oligos in the cytoplasm followed by nibbling at the new ends by exonucleases before the fragments annealed to the chromosome. To rule this out, an experiment was devised that relied on the great difference between the transformation efficiency of a 40-mer and that of a 20-mer [22]. It also made use of the finding, in the previous section, that the repair patch is limited. Two lacZ recipients were used. BW1946 (Fig. 4A) had only the 2-nt deletion of strain BW1892 (Fig. 1), whereas BW1947 had only its G→A point mutation (Fig. 4B). The 40-mers used covered both mutations and were capable of transforming either strain to Lac+. One of the 40-mers had a dI at position 19. If cleaved in the cytoplasm by Endo V, it would produce two 20-mers. To prevent dI repair from removing the transforming dG residue, the dI was placed five nucleotides 5′ to it. The dI-containing 40-mers transformed about as well as the dG-containing ones (Fig. 4). However, neither of the predicted cleavage products, i.e., the 20-mers, was effective. Because the dI-containing 40-mer could transform the cells but the predicted cleavage products could not, the 40-mer could not have been cleaved before incorporation into the chromosome.
Fig. 4.
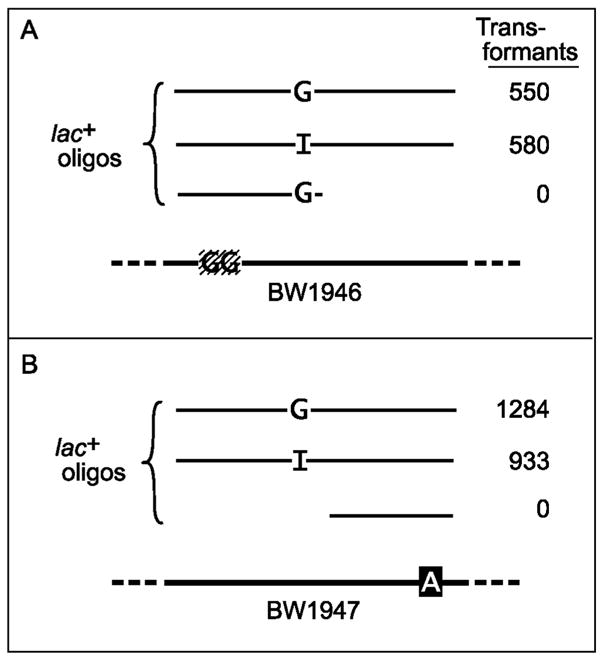
dI-containing DNA is not split in the cytoplasm. Strains BW1946 and BW1947 were transformed to Lac+ with the indicated 40-mers or 20-mers. BW1946 has a 2-nt (GG) deletion. Strain BW1947 has a G→A mutation. The 40-mers, which were the same for parts A and B, spanned nt 1361 to 1400 of the wild type lacZ gene and contained either dG or dI at position 1379 (see Fig. 1 for sequence). The 20-mers were the length of the predicted cleavage products of the dI-containing 40-mer and covered either nt 1361 to 1380 (A) or nt 1381 to 1400 (B). In duplicate experiments, 10 μl (A) or 0.1 μl (B) of the saturated cultures were plated.
4. Discussion
4.1. Screening for mutants by oligo transformation
Before the discovery of Endo V, it was believed that dI in DNA was repaired through a base excision pathway, i.e., a pathway initiated by a glycosylase. There were reports of a hypoxanthine-DNA glycosylase activity in E. coli [31], of a similar (probably the same) activity associated with 3-methyladenine DNA glycosylase II (AlkA) [32], and of an unconfirmed discovery (discussed in ref. [33] of a hypoxanthine DNA glycosylase with different physical properties from the others [34]. There might also be other repair enzymes that are too labile to be assayed in crude extracts and for which our selection procedure offered the best hope of discovery. This study indicates that compared to Endo V, other activities are too small to matter under our growth conditions, which include recovery from electroshock. However, Endo V catalyzes only the first step of dI repair, an incision near the lesion; it does not actually remove the dI. Consequently, the selective enrichment procedure should also have yielded excision-deficient mutants. Its failure to do so suggests that either there are redundant DNases for this step, that the loss of such an enzyme would reduce viability or transformability of the cell, or that there was a strongly biased distribution of insertion sites for the transposon used to generate the mutants.
Before using oligo transformation as an enrichment procedure, plasmid transformation was attempted. In pilot experiments in our lab, synthetic plasmids were either heavily substituted with dI or dU or had several of these residues in nearly opposite locations. A repair-proficient cell should produce lethal double-strand breaks that would block the inheritance of an antibiotic marker on the plasmid. The procedure did not work. The transformation efficiencies for the modified plasmids were equally high in wild type cells and in mutants lacking Endo V or uracil-DNA glycosylase. It is possible that the replication of a small plasmid is just too fast; theoretically, it might be complete in just a few seconds. If the modified DNA is replicated before the first cleavages occur, subsequent repair will only generate single-strand breaks. Oligo transformation has a clear advantage in this respect. It has been postulated that transforming single-stranded oligos bind to the template for the lagging strand, in the gaps between the Okazaki fragments [16,27]. This assumption is based on the requirement for a single-strand binding protein (Beta protein), the preference for incorporation into the lagging over the leading strand, and the preference for single-stranded over double-stranded oligos. If the oligo is indeed incorporated into the chromosome just behind a replication fork, then it has twenty minutes to be repaired before it is traversed by the next fork [35]. This may be the reason for the success of our methods. This principle may also explain why transformations to Strr did not select as strongly against dI-containing DNA as transformations to Lac+. Each of the oligos used in this study had the sequence of the coding strand. However, the lacZ oligos were incorporated into the lagging strand, whereas the rpsL oligos were incorporated into the leading strand. Thus, in many cells, an rpsL oligo may have been integrated in front of a replication fork and therefore had little time in which to be repaired before it was replicated. Nevertheless, other explanations are possible. For example, repair might have been affected by different rates of transcription of the lacZ and rpsL genes or by the different sequence contexts of the lesions.
4.2. Precision of dI repair
After cleavage by Endo V, excision of the dI from DNA requires that only two nucleotides be removed from the 3′ end of the cut and none from the 5′ end. Instead, three are removed from the 3′ end and two from the 5′ end. Another unexpected finding was the striking apparent precision of the repair patch (Fig. 3), with a sharp drop-off below five nucleotides. It is hard to imagine that this sharp demarcation results from a purely stochastic process. It suggests that the enzymes act in concert, perhaps in a complex, but it must involve at least three enzymes that are not known to act in a concerted fashion: Endo V, DNA polymerase I, and DNA ligase. The mechanism of excision of dI after Endo V cleavage is not known. Endo V from Thermotoga maritima has an associated 3′→5′ exonucleolytic activity that appears to be triggered by the endonucleolytic cut [36], but this activity has not been found in the E. coli enzyme, which remains tightly bound to the nick after cleavage [37]. The removal of nucleotides from the 5′ terminus at the nick could be caused by nick translation, i.e., the 5′→3′ exonuclease activity of DNA polymerase I that accompanies polymerization. This cannot be tested directly because mutations for this activity are conditionally lethal [38]. However, we might be able to use oligonucleotide transformation to screen for mutants for another enzyme that might perform this excision. For example, suppose that a dI at nt 1381 in the lacZ gene produces a Lac− phenotype. Then we could transform BW1892 (Fig. 1) with an oligo containing a dI at position 1381 and the wild type dG at position 1384 (2 nt from the prospective 5′ terminus). Lac+ transformants would be produced only if the dI were removed and the dG were not. This general approach should be widely applicable in cases where the repair patch is known with some precision.
A precise repair patch that brackets the lesion is reminiscent of that produced by the UvrABC protein complex, which removes 12-mers containing bulky adducts such as UV pyrimidine dimers [4]. It raises the additional possibility of a similar complex involved in dI repair, perhaps one containing one or two additional endonucleases. Thus, Endo V might act in concert with other DNases (endo- or exonucleases) that define the ends of the repair patch.
4.3. Other applications
These procedures may be used to study other lesions that are repaired by base or nucleotide excision. dI was ideal because its base pairing is fairly consistent, enabling us to measure the efficiency of repair directly when it was used as the transforming nucleotide. However, when a lesion displays no pairing or degenerate pairing, its excision may still be measured, although indirectly. We would just determine the decline in transformation frequency of a normal nucleotide when it is situated within the repair patch. One problem is that even if the lesion is not repaired, it may preclude transformation to Lac+ by producing a mutation. To prevent this outcome, the lesion could be positioned as the third nucleotide of a triplet that encodes the same amino acid regardless of which base is in that position. There are eight sets of such codons. Of course, this procedure will only work if the repair patch is longer than one nucleotide and if the enzyme that attacks the lesion recognizes it when it is opposite the given base.
4.4. Caveats
A matter of concern was that during transformation, the heteroduplex region near the dI contains a base mismatch that might destabilize it and thereby affect the size of the repair patch. For this reason, transformations were chosen that pair a dG in the oligo with a dT in the chromosome. G:T is the most stable of the non-canonical base pairs. It stacks well with other base pairs and produces relatively little distortion of the double helix [39]. However, the G:T wobble pair has a shorter half life and a longer open-state lifetime than the normal base pairs. It also destabilizes adjacent base pairs, although not its second nearest neighbors [40,41]. This relative instability could affect the patch size. For example, it is possible that normally only two nucleotides are excised at the 3′ end of the scission (the minimum required to remove the dI). Our conclusion that a third nucleotide is removed is based on the use of oligos in which the dI is immediately 3′ to a dG in a G:T pair. Perhaps the third nucleotide is removed only because it is in the unstable G:T pair that is attacked, for example, by a single-strand-specific exonuclease. Thus, its removal may be an artifact of our experimental conditions.
However, the patch size does not appear to be influenced by mispairing of the dI. Two different mutated regions were examined so that dI would be paired with different deoxynucleosides at some of the same relative positions. In various oligos, the dI replaced dC, dA, dT, or dG. There were two experiments in which the dI was placed immediately 3′ to the transforming dG, causing the removal of dGMP as the third nucleotide from the end. In the transformation of BW1892, the dI (at nt 1385, Fig. 1) was paired with dT, as it would be if it had arisen in vivo from the deamination of dA. In the transformation of BW1948, however, the dI (at nt 51, Fig. 1) was paired with dC, as it would be if it had arisen from the misincorporation into DNA of dITP that had escaped dITP pyrophosphohydrolase [42]. The results (Fig. 3A versus Fig. 3B) were similar, suggesting that the mechanism of dI excision is the same whether it is mispaired with dT or paired normally with dC.
Another possible artifact may be introduced by the expression in our cells of the Beta protein of bacteriophage λ. It may be the Beta protein that is responsible for protecting our dI-containing oligos from cleavage in the cytoplasm. Although the protein binds specifically to single-stranded DNA to promote annealing, it remains bound to the duplex after it is formed [24], raising the question of possible interference with repair. Several findings suggest that the Beta protein might not affect our repair patch measurements. (i) It is obviously not blocking the removal of dI from the DNA. (ii) Although single-stranded DNA is initially protected by Beta protein against digestion by pancreatic DNase, it loses that protection during renaturation [25]. (iii) The apparent size of the Endo V repair patch is greater than the minimal size we may have expected (i.e., greater than 2 nucleotides). Nevertheless, we cannot rule out the possibility that Beta protein is constraining the excision.
Although this system for the study of DNA repair lacks many of the shortcomings of the in vitro systems (as discussed in the introduction), it evidently comes with caveats of its own and therefore cannot replace them but merely complement them.
Nevertheless this approach requires fewer tacit assumptions than in vitro studies, and by using intact cells and by studying chromosomal events, it may be closer to reality. Moreover, it may be extended to the study of other DNA lesions and to other organisms, such as Saccharomyces cerevisiae [43], for which efficient oligo transformation systems exist.
Acknowledgments
I am grateful to Dawit Seyfe for his capable technical assistance. This work was supported by a NIH grant ES11163.
Abbreviations
- oligo(s)
oligodeoxyribonucleotide(s)
- Endo V
endonuclease V
- nt
nucleotide
- Strr
streptomycin resistant (or resistance)
- Strs
streptomycin sensitive (or sensitivity)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, D.C: 2006. [Google Scholar]
- 2.Dianov G, Price A, Lindahl T. Generation of single-nucleotide repair patches following excision of uracil residues from DNA. Mol Cell Biol. 1992;12:1605–1612. doi: 10.1128/mcb.12.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieb M, Allen E, Read D. Very short patch mismatch repair in phage lambda: repair sites and length of repair tracts. Genetics. 1986;114:1041–1060. doi: 10.1093/genetics/114.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sibghat U, Sancar A, Hearst JE. The repair patch of E. coli (A)BC excinuclease. Nucleic Acids Res. 1990;18:5051–5053. doi: 10.1093/nar/18.17.5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 6.Sattler U, Frit P, Salles B, Calsou P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Reports. 2003;4:363–367. doi: 10.1038/sj.embor.embor796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muheim-Lenz R, Buterin T, Marra G, Naegeli H. Short-patch correction of C/C mismatches in human cells. Nucleic Acids Res. 2004;32:6696–6705. doi: 10.1093/nar/gkh990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kmiec E, Holloman WK. β protein of bacteriophage λ promotes renaturation of DNA. J Biol Chem. 1981;256:12636–12639. [PubMed] [Google Scholar]
- 9.Karran P, Lindahl T. Hypoxanthine in deoxyribonucleic acid: generation by heat-induced hydrolysis of adenine residues and release in free form by a deoxyribonucleic acid glycosylase from calf thymus. Biochemistry. 1980;19:6005–6011. doi: 10.1021/bi00567a010. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro R, Pohl SH. The reaction of ribonucleosides with nitrous acid. Side products and kinetics. Biochemistry. 1968;7:448–455. doi: 10.1021/bi00841a057. [DOI] [PubMed] [Google Scholar]
- 11.Schouten KA, Weiss B. Endonuclease V protects Escherichia coli against specific mutations caused by nitrous acid. Mutat Res. 1999;435:245–254. doi: 10.1016/s0921-8777(99)00049-x. [DOI] [PubMed] [Google Scholar]
- 12.Yao M, Hatahet Z, Melamede RJ, Kow YW. Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3′ endonuclease, from Escherichia coli. J Biol Chem. 1994;269:16260–16268. [PubMed] [Google Scholar]
- 13.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Nat Acad USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schweizer HD. Small broad-host-range gentamycin resistance gene cassettes for site-specific insertion and deletion mutagenesis. Biotechniques. 1993;15:831–834. [PubMed] [Google Scholar]
- 15.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costantino N, Court DL. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc Natl Acad Sci USA. 2003;100:15748–15753. doi: 10.1073/pnas.2434959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1989. [Google Scholar]
- 18.Sternberg NL, Maurer R. Bacteriophage-mediated generalized transduction in Escherichia coli and Salmonella typhimurium. Methods Enzymol. 1991;204:18–24. doi: 10.1016/0076-6879(91)04004-8. [DOI] [PubMed] [Google Scholar]
- 19.Miller JH. A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1992. A short course in bacterial genetics. [Google Scholar]
- 20.Vogel HJ, Bonner DM. Acetylornithinase of Escherichia coli: partial purification and some properties. J Biol Chem. 1956;218:97–106. [PubMed] [Google Scholar]
- 21.Sharma RC, Schimke RT. Preparation of electrocompetent E. coli using salt-free growth medium. Biotechniques. 1996;20:42–44. doi: 10.2144/96201bm08. [DOI] [PubMed] [Google Scholar]
- 22.Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci USA. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu Rev Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 24.Li Z, Karakousis G, Chiu SK, Reddy G, Radding CM. The Beta protein of phage λ promotes strand exchange. J Mol Biol. 1998;276:733–744. doi: 10.1006/jmbi.1997.1572. [DOI] [PubMed] [Google Scholar]
- 25.Muniyappa K, Radding CM. The homologous recombination system of phage lambda. Pairing activities of beta protein. J Biol Chem. 1986;261:7472–7478. [PubMed] [Google Scholar]
- 26.Dutra BE, Sutera VA, Jr, Lovett ST. RecA-independent recombination is efficient but limited by exonucleases. Proc Natl Acad Sci USA. 2007;104:216–221. doi: 10.1073/pnas.0608293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XT, Costantino N, Lu LY, Liu DP, Watt RM, Cheah KS, Court DL, Huang JD. Identification of factors influencing strand bias in oligonucleotide-mediated recombination in Escherichia coli. Nucleic Acids Res. 2003;31:6674–6687. doi: 10.1093/nar/gkg844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masamune Y, Fleischman RA, Richardson CC. Enzymatic removal and replacement of nucleotides at single strand breaks in deoxyribonucleic acid. J Biol Chem. 1971;246:2680–2691. [PubMed] [Google Scholar]
- 29.Carter DM, Radding CM. The role of exonuclease and beta protein of phage lambda in genetic recombination. II. Substrate specificity and the mode of action of lambda exonuclease. J Biol Chem. 1971;246:2502–2512. [PubMed] [Google Scholar]
- 30.Sriprakash KS, Lundh N, Huh MO, Radding CM. The specificity of lambda exonuclease. Interactions with single-atranded DNA. J Biol Chem. 1975;250:5438–5445. [PubMed] [Google Scholar]
- 31.Karran P, Lindahl T. Enzymatic excision of free hypoxanthine from polydeoxynucleotides and DNA containing deoxyinosine monophosphate residues. J Biol Chem. 1978;253:5877–5879. [PubMed] [Google Scholar]
- 32.Saparbaev M, Laval J. Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc Natl Acad Sci USA. 1994;91:5873–5877. doi: 10.1073/pnas.91.13.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dianov G, Lindahl T. Preferential recognition of I.T base-pairs in the initiation of excision-repair by hypoxanthine-DNA glycosylase. Nucleic Acids Res. 1991;19:3829–3833. doi: 10.1093/nar/19.14.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harosh I, Sperling J. Hypoxanthine-DNA glycosylase from Escherichia coli. Partial purification and properties. J Biol Chem. 1988;263:3328–3334. [PubMed] [Google Scholar]
- 35.Helmstetter C, Cooper S, Pierucci O, Revelas O. On the bacterial life sequence. Cold Spr Hrbr Symp Quant Biol. 1960;33:809–822. doi: 10.1101/sqb.1968.033.01.093. [DOI] [PubMed] [Google Scholar]
- 36.Feng H, Dong L, Klutz AM, Aghaebrahim N, Cao W. Defining amino acid residues involved in DNA-protein interactions and revelation of 3′-exonuclease activity in endonuclease V. Biochemistry. 2005;44:11486–11495. doi: 10.1021/bi050837c. [DOI] [PubMed] [Google Scholar]
- 37.Yao M, Kow YW. Interaction of deoxyinosine 3′-endonuclease from Escherichia coli with DNA containing deoxyinosine. J Biol Chem. 1995;270:28609–28616. doi: 10.1074/jbc.270.48.28609. [DOI] [PubMed] [Google Scholar]
- 38.Kornberg A, Baker TA. DNA Replication. W.H. Freeman and Company; New York: 1992. pp. 162–163. [Google Scholar]
- 39.Brown T, Kennard O, Kneale G, Rabinovich D. High-resolution structure of a DNA helix containing mismatched base pairs. Nature. 1985;315:604–606. doi: 10.1038/315604a0. [DOI] [PubMed] [Google Scholar]
- 40.Patel DJ, Kozlowski SA, Ikuta S, Itakura K. Dynamics of DNA duplexes containing internal G·T, G·A, A·C, and T·C pairs: hydrogen exchange at and adjacent to mismatch sites. Fed Proc. 1984;43:2663–2670. [PubMed] [Google Scholar]
- 41.Varnai P, Canalia M, Leroy JL. Opening mechanism of G·T/U pairs in DNA and RNA duplexes: a combined study of imino proton exchange and molecular dynamics simulation. J Am Chem Soc. 2004;126:14659–14667. doi: 10.1021/ja0470721. [DOI] [PubMed] [Google Scholar]
- 42.Bradshaw JS, Kuzminov A. RdgB acts to avoid chromosome fragmentation in Escherichia coli. Mol Microbiol. 2003;48:1711–1725. doi: 10.1046/j.1365-2958.2003.03540.x. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto T, Moerschell RP, Wakem LP, Ferguson D, Sherman F. Parameters affecting the frequencies of transformation and co-transformation with synthetic oligonucleotides in yeast. Yeast. 1992;8:935–948. doi: 10.1002/yea.320081104. [DOI] [PubMed] [Google Scholar]
- 44.Cupples CG, Miller JH. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci USA. 1989;86:5345–5349. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Funatsu G, Wittmann HG. Ribosomal proteins. XXIII. Location of amino-acid replacements in protein S12 isolated from Escherichia coli mutants resistant to streptomycin. J Mol Biol. 1972;68:547–550. doi: 10.1016/0022-2836(72)90108-8. [DOI] [PubMed] [Google Scholar]
- 46.Guo G, Weiss B. Endonuclease V (nfi) mutant of Escherichia coli K-12. J Bacteriol. 1998;180:46–51. doi: 10.1128/jb.180.1.46-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]