

Abstract
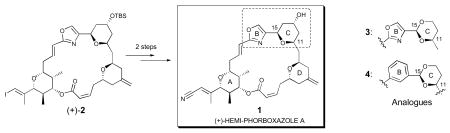
A synthesis providing totally synthetic (+)-hemi-phorboxazole A (1), proceeding in two steps (85% yield) from known vinyl iodide precursor (+)-2, has been achieved in conjunction with the design, synthesis and biological evaluation of two hemi-phorboxazole analogues [(+)-3 and (−)-4] featuring ring replacements inscribed within the macrolide. Although hemi-phorboxazole A (1) displayed no activity when tested against Candida albicans and two human cancer cell lines, analogue (−)-4 exhibited significant tumor cell growth inhibitory activity in the nanomolar range against HCT-116 (colon) and SK-BR-3 (breast), while (+)-3 displayed promising antifungal activity against C. albicans.
In April 2009 Dalisay and Molinski reported1 the isolation of hemi-phorboxazole A (1), a diminutive form of (+)-phorboxazole A (5), derived from the same marine sponge (Phorbas sp.) that not only provides phorboxazoles A and B (5 and 6),2 but also the unrelated phorbasides A–F (7–12)3 and very recently muironolide A (13)4 (Figure 1). Remarkably, the complete structure including absolute configuration was assigned with only 16.5 μg (!) of natural material, exploiting state-of-the-art microcryoprobe NMR, in conjunction with HRMS and circular dichromism (CD) studies. The truncated hemi-phorboxazole A (1), the first reported example of a natural phorboxazole variant since the original Molinski et al. disclosure of the phorboxazoles in 1995,2 was found to be ~10,000 times less abundant than phorboxazoles A and B (cf. 0.07 vs. 400 ppm, respectively). Not surprisingly the limited availability of the natural material precluded any significant evaluation of the biological activity. Thus, to confirm the assigned structure of 1, as well as to provide material to define the biological properties recourse to total synthesis was clearly required for forward progress.
Figure 1.

Natural Products Isolated from Phorbas sp.
Given that our Laboratory has had a long-standing interest in the phorboxazole area, with first- and second-generation total syntheses of (+)-phorboxazole A (5) recorded in 20015 and 2005,6 we were intrigued with the diminutive form of hemi-phorboxazole A (1). Indeed, a synthetic program directed at hemi-phorboxazole A could take ready advantage of the Petasis–Ferrier union/rearrangement,7 an effective tactic specifically designed and developed for the stereocontrolled construction of the A andC cis-tetrahydropyran rings inscribed within the parent phorboxazole macrolide. Equally exciting was the potential for the design, synthesis and biological evaluation of hemi-phorboxazole A analogues, a program that would fit nicely with our ongoing phorboxazole analogue venture.8
With this as background, we envisioned that (+)-hemi-phorboxazole A (1) could be readily elaborated by exploiting the same late-stage vinyl iodide macrolide (+)-2, which was constructed via a longest linear sequence of 20 steps (20% yield) exploiting our now scalable second-generation phorboxazole A synthesis.6 Indeed, removal of the TBS group at C(13), by treatment with tetrabutylammonium fluoride (TBAF), followed by a palladium-catalyzed cyanation,9 employing tributyltin cyanide, provided totally synthetic (+)-hemi-phorboxazole A (1) in 84 % yield for the two steps (Scheme 1). Importantly, the spectral data of synthetic (+)-1 [cf. 1D and 2D NMR, UV, circular dichromism (CD) and HRMS] were identical in all respects with the data derived from natural hemi-phorboxazole A (1), thereby confirming both the complete relative stereochemistry and assigned absolute configuration.
Scheme 1.

With synthetic (+)-hemi-phorboxazole A (1) in hand, we turned to the design, elaboration and biological evaluation of analogues, with the important goal of introducing synthetic simplicity, while maintaining the potentially important macrolide ring conformation of the natural congener. At the outset, we selected two analogues featuring macrolide ring replacements, based on our earlier analogue studies of (+)-phoroboxazole A (5).8 In particular, we proposed: (i) replacement of the C-ring tetrahydropyran with a cyclic acetal, and (ii) a bis-ring replacement comprising a similar acetal for the C-ring tetrahydropyran and a phenyl ring for the B-ring oxazole (Figure 2). Replacement of the tetrahydropyran ring with a synthetically simplified, but geometrically similar cyclic acetal (i.e., 1,3-dioxane) comprises a tactic that was introduced and elegantly employed by Wender et al. in their bryostatin analogue program.10 This concept was also utilized with some success in our (+)-phorboxazole A (5) SAR study, to furnish a novel, fully endowed phorboxazole analogue (14) possessing the macrolide ring, complete with an acetylenic sidechain. Importantly, only moderate loss (ca. 10 fold) in tumor cell growth inhibition activity was observed, when compared to (+)-phorboxazole A (5).8b
Figure 2.

Phorboxazole Analogues
Towards this end, the C-ring replacement congener of (+)-hemi-phorboxazole A (cf. 3) was constructed via a palladium-catalyzed cyanation of vinyl iodide (+)-15 (Scheme 2). Vinyl iodide (+)-15 was prepared in accordance with our reported synthetic route to (+)-14.8b
Scheme 2.

The second analogue, hemi-phorboxazole congener 4, possessing B- and C-ring replacements, was envisioned to be constructured in an analogous manner to that of (+)-1, exploiting two late-stage intermediates, vinyl iodide (+)-178a and diol (−)-18,8b readily available from our second- generation phorboxazole A (5) total synthesis (Scheme 3). Construction of the key C(11–15) acetal would then result via union of diol (−)-18 and aldehyde 16, the latter prepared in two steps from commercially available 1,3-benzenedimethanol (see Supporting Information).
Scheme 3.

We began with the condensation of diol (−)-18 and aldehyde 16 (Scheme 5), employing dehydrating conditions, to provide cis-acetal (−)-19 in 94% yield (dr > 20:1). Removal of the PMB group at C(19) and exposure of the resultant primary alcohol to CCl4 and PPh3 furnished chloride (−)-20 in 88% yield for the two steps.5 Next, conversion of chloride (−)-20 to the corresponding tributyl phosphonium salt, followed by Wittig reaction with aldehyde (+)-17 provided the C(3–28) carbon skeleton of analogue 4. The Wittig reaction, which employed a stablized phosphonium ylide, proceeded in 94% yield with modest selectivity (ca. 4:1) favoring the E-isomer. Hydroxy-aldehyde (−)-22 was then obtained via a three-step sequence: removal of the silyl ether at C(3), Dess–Martin oxidation of the resultant primary alcohol to the corresponding aldehyde, and DDQ mediated generation of the secondary hydroxyl at C(24). Macrocyclization of (−)-22 to furnish the desired Z-macrolide (−)-24 was then achieved employing a Still–Genari modified-Horner–Emmons olefination11 similar to that employed for the construction of (+)-2. Thus, generation of the phosphonate ester by reaction at the C(24) hydroxyl of (−)-22 with 2-[bis-(2,2,2-trifluoroethoxy)phosphoryl]acetic acid 23, and subsequent exposure to K2CO3/18-crown-6 in toluene afforded (−)-24 in 44% overall yield for the two steps. The E-macrolide was also obtained in 15% yield. Finally, palladium-catalyzed cyanation at C(28) of vinyl iodide (−)-24 provided hemi-phorboxazole congener (−)-4 in 93% yield.
Synthetic (+)-hemi-phorboxazole A (1) and congeners (+)-3 and (−)-4 were assayed for tumor cell growth inhibitory activity, in parallel with (+)-phorboxazole A (5), against two human cancer cell lines (HCT-116, colon and SK-BR-3, breast). Neither (+)-hemi-phorboxazole A (1) nor congener (+)-3, involving replacement of the C-ring tetrahydropyran with an acetal, displayed activity against either cell line. This result is perhaps not surprising given earlier reports that truncation of the phorboxazole side chain results in complete loss of potency.12 Pleasingly however, analogue (−)-4, that entailed replacement of two rings, displayed activity at the nanomolar level against both HCT-116 and SK-BR-3 cells.
Hemi-phorboxazole A (+)-1 and analogue (+)-3 were also assayed for antifungal activity against a panel of C. albicans strains. Although (+)-hemi-phorboxazole A (1) was again inactive, analogue (+)-3 recorded a miniminum inhibitory concentration (MIC) of 16 μg/mL.
In summary, the first synthesis of (+)-hemi-phorboxazole A (1) has been achieved in two steps from our previously reported macrolide (+)-2, thereby confirming the assigned structure and complete stereochemistry. Two analogues of (+)-hemi-phorboxazole A, featuring macrolide ring replacements, were also designed and constructed. Biological evaluations of (+)-hemi-phorboxazole A (1) and analogues (+)-3 and (−)-4 against two human cancer cell lines (HCT-116, colon and SK-BR-3, breast) and C. albicans strains were conducted with the synthetic material. Although (+)-hemi-phorboxazole A (1) proved to be inactive in all cases, two analogues, (+)-3 and (−)-4, respectively displayed promising antifungal activity and significant anticancer activity against the two human cell lines. Although disappointed with our initial biological evaluation of (+)-hemi-phorboxazole A (1), the biological results revealed by analogues (+)-3 and (−)-4 warrant further investigation, and thus will constitute part of our continuing phorboxazole synthetic program.
Supplementary Material
Scheme 4.

Table 1.
IC50 values for (+)-hemi-phorboxazole A and analogues against human cancer cell lines.13
| compound | HCT-116 IC50 (ng/mL) | SKBR-3 IC50 (ng/mL) |
|---|---|---|
| (+)-Phorboxazole A (5) | 0.71 | 2.0 |
| (+)-Hemi-phorboxazole A (1) | >6200 | >6200 |
| 3 | >6200 | >6200 |
| 4 | 207 | 258 |
Table 2.
Minimium Inhibitory Concentration (MIC, μg/mL) of (+)-Phorboxazole A and analogues against pathogenic C. albicans strains.14
|
C. albicans (μg/mL) |
|||
|---|---|---|---|
| compound | ATCC14503 | UCD-FR1a | 96-489a |
| (+)-Phorboxazole A (5) | 1.0 | 2.0 | 1.0 |
| (+)-Hemi-phorboxazole A (1) | >64 | >64 | >64 |
| 3 | 16.0 | 16.0 | >64 |
Clinical isolate, Fluconazole-resistant (MIC > 64 μg/mL).
Acknowledgments
Support was provided by the National Institute of Health (National Cancer Institute) through grants CA-19033 (to ABS) and CA-122256 (to TFM), and the University of Pennsylvania. We thank Dr. Thomas M. Razler and Ms. Regina M. Meis (University of Pennsylvania) for some of the advanced intermediates prepared in connection with our phorboxazole synthetic program. We also thank Dr. Furst, Dr. Gu and Dr. Kohli at the University of Pennsylvania for assistance in obtaining NMR and high-resolution mass spectra, respectively.
Footnotes
Supporting Information Available: Experimental procedures and full spectroscopic data are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dalisay DS, Molinski TF. Org Lett. 2009;11:1967. doi: 10.1021/ol9004189. [DOI] [PubMed] [Google Scholar]
- 2.(a) Searle PA, Molinski TF. J Am Chem Soc. 1995;117:8126. [Google Scholar]; (b) Searle PA, Molinski TF, Brzezinski LJ, Leahy JW. J Am Chem Soc. 1996;118:9422. [Google Scholar]; (c) Molinski TF. Tetrahedron Lett. 1996;37:7879. [Google Scholar]
- 3.(a) MacMillan JB, Xiong-Zhou G, Skepper CK, Molinski TF. J Org Chem. 2008;73:3699. doi: 10.1021/jo702307t. [DOI] [PubMed] [Google Scholar]; (b) Skepper CK, MacMillan JB, Zhou GX, Masuno MN, Molinski TF. J Am Chem Soc. 2007;129:4150. doi: 10.1021/ja0703978. [DOI] [PubMed] [Google Scholar]; (c) Dalisay DS, Molinski TF. J Nat Prod. 2009;72:739. doi: 10.1021/np900009b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalisay DS, Morinaka BI, Skepper CK, Molinski TF. J Am Chem Soc. 2009;131:7552. doi: 10.1021/ja9024929. [DOI] [PubMed] [Google Scholar]
- 5.Smith AB, III, Verhoest PR, Minbiole KP, Schelhaas M. J Am Chem Soc. 2001;123:4834. doi: 10.1021/ja0105055. [DOI] [PubMed] [Google Scholar]
- 6.(a) Smith AB, III, Razler TM, Ciavarri JP, Hirose T, Ishikawa T. Org Lett. 2005;7:4399. doi: 10.1021/ol051584i. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Razler TM, Ciavarri JP, Hirose T, Ishikawa T, Meis RM. J Org Chem. 2008;73:1192. doi: 10.1021/jo7018152. [DOI] [PubMed] [Google Scholar]
- 7.Smith AB, III, Fox RJ, Razler TM. Acc Chem Res. 2008;41:675. doi: 10.1021/ar700234r. [DOI] [PubMed] [Google Scholar]
- 8.(a) Smith AB, III, Razler TM, Pettit GR, Chapuis J-C. Org Lett. 2005;7:4403. doi: 10.1021/ol051585a. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Razler TM, Meis RM, Pettit GR. Org Lett. 2006;8:797. doi: 10.1021/ol060014v. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Razler TM, Meis RM, Pettit GR. J Org Chem. 2008;73:1201. doi: 10.1021/jo701816h. [DOI] [PubMed] [Google Scholar]
- 9.For related transformations see: Duffey MO, LeTiran A, Morken JP. J Am Chem Soc. 2003;125:1458. doi: 10.1021/ja028941u.Nicolaou KC, Pratt BA, Arseniyadis S, Wartmann M, O’Brate A, Giannakakou P. ChemMedChem. 2006;1:41. doi: 10.1002/cmdc.200500056.
- 10.Wender PA, Baryza JL, Bennett CE, Bi FC, Brenner SE, Clarke MO, Horan JC, Kan C, Lacôte E, Lippa B, Nell PG, Turner TM. J Am Chem Soc. 2002;124:13648. doi: 10.1021/ja027509+. [DOI] [PubMed] [Google Scholar]
- 11.Still WC, Gennari C. Tetrahedron Lett. 1983;24:4405. [Google Scholar]
- 12.Uckan FM, Forsyth CJ. Bioorg Med Chem Lett. 2001;11:1181. doi: 10.1016/s0960-894x(01)00191-3. [DOI] [PubMed] [Google Scholar]
- 13.Cory AH, Owen TC, Barltrop JA, Cory JG. Cancer Commun. 1991;7:207. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 14.National Committee for Clinical Laboratory Standards. Approved standard M27-A2. 2. National Committee for Clinical Laboratory Standards; Wayne, PA: 2002. Reference method for broth dilution antifungal susceptibility testing of yeast. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.