

Abstract
Under near-physiological pH, temperature, and ionic strength, a kinetics constant composition (CC) method was used to examine the roles of phosphorylation of a 14 amino acid segment (DDVDDTDDSHQSDE) corresponding to potential crystal binding domains within the osteopontin (OPN) sequence. The phosphorylated 14-mer OPN peptide segment significantly inhibits both the nucleation and growth of calcium oxalate monohydrate (COM), inhibiting nucleation by markedly increasing induction times and delaying subsequent growth by at least 50% at concentrations less than 44 nM. Molecular modeling predicts that the doubly phosphorylated peptide binds much more strongly to both (−101) and (010) faces of COM. The estimated binding energies are, in part, consistent with the CC experimental observations. Circular dichroism spectroscopy indicates that phosphorylation does not result in conformational changes in the secondary peptide structure, suggesting that the local binding of negatively charged phosphate side chains to crystal faces controls growth inhibition. These in vitro results reveal that the interactions between phosphorylated peptide and COM crystal faces are predominantly electrostatic, further supporting the importance of macromolecules rich in anionic side chains in the inhibition of kidney stone formation. In addition, the phosphorylation-deficient form of this segment fails to inhibit COM crystal growth up to concentrations of 1450 nM. However, at sufficiently high concentrations, this nonphosphorylated segment promotes COM nucleation. Dynamic light scattering (DLS) and small-angle X-ray scattering (SAXS) results confirm that aggregation of the nonphosphorylated peptide segment takes place in solution above 900 nM when the aggregated peptide particles may exceed a well-defined minimum size to be effective crystallization promoters.
Introduction
Calcium oxalate monohydrate (COM, CaC2O4 · H2O) is the primary constituent of most human kidney stones, and osteopontin (OPN) present in urine1,2 and prototype of the aspartic acid (D)-rich proteins (AARP), has been identified as an inhibitory molecule, against kidney stones3 and in vitro COM crystallization.4–6 Additionally, kinetic constant composition (CC) studies revealed that a linear aspartic acid-rich peptide with serine spacers (DDDS) was more than 30 times more effective as a COM crystal growth inhibitor than the corresponding peptide with glycine spacers (DDDG).7 This result clearly demonstrates that the number of acidic amino acids in a peptide is not the sole stone inhibitory determinant, and the contributions of other factors such as peptide sequence modifications and their extended conformations are also important for the inhibition of COM nucleation and growth. Prior structural studies have indicated that native OPN undergoes extensive post-translation modifications such as phosphorylation and glycosylation.8–11 It has been established that phosphate groups present in OPN are required for efficient inhibition of hydroxyapatite (HAP)12–15 or COM growth.16,17 This suggests that phosphorylation of full OPN molecules containing negatively charged phosphate group side chains might also have a functional role in COM crystallization. Although OPN inhibits several aspects of COM crystallization, specific structural features and the individual active domains whereby OPN exerts its inhibitory effects were not previously known. Moreover, the quantitative studies of bulk COM crystal nucleation and growth in the presence of phosphorylated or nonphosphorylated OPN are lacking. Thus, we selected from the full OPN sequence a 14 amino acid segment with highly conserved aspartic acid residues (DDVDDTDDSHQSDE) (Figure 1) (sequence of amino acids from 93 to 106 of OPN with two phosphorylation serine sites) rather than a complex protein in an attempt to evaluate the relative contributions of aspartic acid residues and phosphorylation to the functional activities of this small OPN peptide in modulating COM crystallization. In addition, we chose a relatively low supersaturation to attain a slow kinetic crystallization process similar to that within the kidney in order to define molecular features and the active domains causing inhibition of crystallization through interactions with COM crystals in vitro under near-physiological pH (7.0), temperature (37 °C), and ionic strength (0.15 M). A CC method sensitive to concentration changes at the nanomolar level was used to provide reproducible induction times and growth rates for COM crystallization kinetics, and the results show that the phosphorylated 14-mer OPN peptide segment significantly inhibits both the nucleation and growth of COM; the phosphorylation-deficient form of this segment failed to inhibit COM crystal growth. Conversely, increasing the concentration of this non-phosphorylated segment promoted COM nucleation.
Figure 1.

Sequences of amino acids from 62 to 116 of OPN in the four pairs of peptides. Peptide 93–106 used for the CC crystallization studies has three phosphorylation sites (two serine (S) and one threonine (T)), but only serine sites are phosphorylated. Reproduced from ref 16.
Experimental Section
OPN Peptide Fragments
Peptide fragments were purchased from CPC Scientific, San Jose, CA.
Constant Composition COM Crystal Growth and Nucleation
a. COM Seeded Growth
Solutions and seed crystals were prepared as described previously.7 The relative supersaturation, σ, for COM crystal growth in supersaturated solutions is defined by eq 1
| (1) |
where (Ca2+) and (C2O42−) are the ionic activities calculated by successive approximation for the ionic strength using the Davies extended form of the Debye–Hückel equation and mass balance expressions for total calcium and total oxalate with appropriate equilibrium constants. The solubility activity product, Ksp, of COM at 37.0 °C is 2.20 × 10−9 mol2 L−2.7 Supersaturated solutions (σ =0.266, ionic strength I = 0.15 mol L−1, pH 7.0) were prepared by slowly mixing filtered (0.22 μm Millipore filter) calcium chloride, potassium oxalate, and sodium chloride. To exclude carbon dioxide, the reaction vessels were purged with nitrogen saturated with water vapor at 37 °C. Crystallization experiments, initiated by the introduction of known amounts of COM seed crystals (specific surface area (SA) of 3.3 m2/g) were conducted in magnetically stirred (450 rpm) double-jacketed vessels thermostatically controlled at 37.0 ± 0.1 °C. In the CC method, lattice ions were simultaneously added to the reaction solution to compensate for changes due to crystal growth. When growth was initiated, changes in electrode potential triggered the addition of two titrant solutions, which were designed to maintain a constant activity of all ionic species in the reaction solution. The titrant concentrations having COM stoichiometries were calculated using eqs 2–4:
| (2) |
| (3) |
| (4) |
in which W and V are the total concentrations in the reaction solutions and titrants, respectively, and Ceff is the effective titrant concentration (5.00 × 10−3 mol L−1 in this study).7 Titrant addition was triggered by a potentiometer (Orion 720A, U.K.) incorporating a calcium ion selective electrode (Orion 93–20) and a reference electrode (Orion 900100, U.K.). During the experiment, the output of the potentiometer was constantly compared with a preset value, and a difference in output (error signal) activated motor-driven titrant burets, thereby maintaining a constant thermodynamic driving force.7
In these COM growth experiments, peptides were added to supersaturated solutions to achieve concentrations ranging from 10.1 to 108.0 nM for PP and from 108.0 to 1450 nM for NPP. The reaction solutions were maintained at pH 7.0 using 0.010 mol L−1 tris(hydroxymethyl)aminomethane (Tris) buffer. The crystal growth rates were determined from the slopes of the plots of titrant volume added as a function of time during the first 15 min of reaction. The overall growth rate, R, is defined by eq 5
| (5) |
where dV/dt is the titrant curve gradient and ms is the initial seed mass. Growth rates are expressed as the mean ± SD for a series of six experiments at each peptide concentration.
b. COM Nucleation
Nucleation experiments were performed at a higher relative supersaturation, σCOM = 1.53, pH = 7.0, T = 37.0 ± 0.1 °C, and an ionic strength of 0.15 mol L−1, adjusted by the addition of sodium chloride.7 These unseeded experiments were initiated by the slow mixing of solutions to create reaction solution supersaturation.1 The peptide concentration used in nucleation studies ranged from 10.1 to 40.4 nM for PP and from 20.2 to 1120 nM for NPP. Induction times are reported as mean ± SD.
Circular Dichroism (CD) Spectroscopy of Peptides
CD measurements were made on a JASCO J-715 spectropolarimeter. Experiments were performed at room temperature using a quartz cuvette with a path length of 2 mm. The final concentration of the samples was 15.0 μM in triply distilled deionzed (TDW) water or in 10 mM Tris buffer containing 0.147 M NaCl and 5.2 × 10−4 M CaCl2 (I ≈ 0.15 M) at pH 7.0 (37 °C). The baseline spectrum obtained under identical conditions using a TDW/buffer blank was subtracted from the sample spectra. To improve the signal-to-noise ratio, an average of three scans was used to plot the spectra.
Dynamic Light Scattering (DLS) Analysis
The particle size distribution of the two peptides at concentrations of 1118 nM–30 μM in Tris buffer solutions under similar COM nucleation experimental conditions (pH 7.00, 37 °C, and 0.147 M NaCl, and 5.2 × 10−4 M CaCl2, I ≈ 0.15 M), was analyzed using a Brookhaven BI-200SM goniometer equipped with a Lexel model 95 argon ion laser as a light source at 514 nm. The detection angle was 90°, and the temperature was controlled at 37 °C. The scattered intensity was recorded by means of a multichannel digital correlator and converted to the correlation function.
Small-Angle X-ray Scattering (SAXS) Analysis
SAXS measurements of both peptides in Tris buffer solutions containing 0.147 M NaCl and 5.2 × 10−4 M CaCl2 (I ≈ 0.15 M) at pH 7.0 were made at room temperature using a Bruker AXS NanoStar. This instrument employed a pinhole SAXS camera with a sealed X-ray tube (Cu anode, Kα wavelength of 1.54 Å operating at 40 kV and 35 mA). The cross-coupled Göbel mirrors reflected a parallel beam of high-intensity X-rays on the sample. The sample-to-detector distance was adjustable and was maintained at about 1060 mm to achieve a small angle of scattering. The detector was a Bruker HI-STAR position-sensitive area detector with very low dark current (4–5 counts/s over 1024 × 1024 pixels). The whole beam path was under vacuum to avoid air scattering effects. SAXS NT software was used for data acquisition and evaluation. The rapidly mixed solutions with precise peptide and salt concentrations were transferred to a thin-walled quartz capillary with a diameter of 1.8 mm that was then tightly vacuum-sealed for SAXS measurements.
Molecular Modeling
Molecular modeling of phosphorylated and nonphosphorylated 14-mer (DDVDDTDDSHQSDE) OPN peptide fragments was performed using Materials Studio v4.0 modeling software (Accelrys Inc., San Diego, CA, 2005). For the purpose of this study, we assumed that the phosphate groups were singly deprotonated (charge = −1) and that the histidine residue was in a protonated state (charge = +1). The COMPASS force field, as implemented in Materials Studio v4.0 modeling software, was used with a dielectric of 10. The molecular geometries shown in Figure 3 were obtained by docking the peptides on the surface followed by minimization, dynamics at 600K for 5000 fs, and then final minimization. The Poisson–Boltzmann electrostatic potential maps were produced using MOE v2005.06 software (Chemical Computing Group Inc., Montreal, Canada, 2006).
Figure 3.

Molecular modeling of (a) PP and (b) NPP to straight (−101) step riser on the (010) face of COM, and binding of (c) PP and (d) NPP to flat (−101) face of a COM crystal. In the Poisson–Boltzmann electrostatic potential maps red = negative, yellow = neutral, blue = positive.
Results and Discussion
Figure 2 shows typical CC curves for supersaturation maintenance in the seeded growth of COM in the absence and presence of 14-mer OPN segments with (Figure 2a) and without (Figure 2b) phosphorylation. The normalized COM growth rate R in the absence of added peptide segments as control was (1.38 ± 0.07) × 10−5 mol m−2 min−1 (n = 6). However, the rate was reduced by about 60% [R = (5.40 ± 0.06) × 10−6 mol m−2 min−1 (n = 6)] in the presence of 43.9 nM PP, and the degree of inhibition increased with increasing peptide concentration to (1.04 ± 0.07) × 10−6 mol m−2 min−1 (n = 6) or about 93% inhibition at a concentration of 108.0 nM. Thus, PP is an effective COM crystal growth retardant. In striking contrast, nonphosphorylated 14-mer OPN peptide segment (NPP) has little influence on COM growth; rates are not significantly changed in the presence of additive concentration up to 1450 nM NPP (Figure 2b).
Figure 2.

CC growth curves of COM in the presence of (a) phosphorylated (PP) and (b) nonphosphorylated 14-mer OPN peptide (NPP). The relative supersaturation (σ) with respect to COM was 0.266; the pH and ionic strength were 7.00 and 0.15 M, respectively; the curves have all been normalized to a seed mass of 10.0 mg. Growth rates are calculated from the slopes of the plots of titrant volume added as a function of time during the first 15 min of reaction.
To further investigate the differences in the COM growth inhibition by these two peptides, molecular modeling was used to calculate binding energies of PP and NPP bound to steps and faces of COM. COM crystals typically express three major families of crystallographically distinct faces, i.e., the {−101}, {010}, and {120}, although under certain conditions others are also possible.4 On the basis of their relative sizes and the previous atomic force microscopy (AFM)4 and CC5,6 studies, the motion velocities of atomic steps generated at dislocation hillocks on the (−101) and (010) faces dominate the overall macroscopic growth of the crystal, and additive molecules such as citrate and OPN control growth habit and kinetics by pinning step motion on the (−101) and (010) faces. Therefore, we chose both the (−101) and (010) faces as the main objects of the molecular modeling investigation. The calculations predict that the doubly phosphorylated peptide binds much more strongly to both flat and stepped surfaces of COM (Figure 3); PP has an estimated binding energy of −590 kcal/mol for the straight −101 step riser on a (010) face, whereas for NPP, the binding energy is about −520 kcal/mol. When compared to the binding of PP or NPP to the flat (−101) face, the average binding energies are −779 and −708 kcal/mol, respectively. These estimated binding energies are, in part, consistent with the CC experimental observations that PP significantly influences COM growth, and similar effects have been observed in comparing the binding of citrate and phosphocitrate to COM.18
CC kinetics and molecular modeling results suggest that growth inhibition is associated with peptide binding to crystal faces or pinning of the steps by peptides adsorbed on growth hillocks. Circular dichroism spectra (Figure 4) of PP or NPP in TDW water or in 10 mM Tris buffer containing 0.147 M NaCl and 5.2 × 10−4 M CaCl2 at 37 °C revealed that these two peptide segments (93–106) are present in solution in the same state of unordered structure at about 199 nm (Figure 4). In the absence of calcium ion, both peptides in either Tris buffer or water with NaCl are also largely unordered (not shown). The presence of 5.2 × 10−4 M CaCl2 did not result in a spectrum consistent with any secondary structures such as α-helix or β-sheet, clearly indicating that both peptides are unable to undergo calcium-dependent conformational changes regardless of the presence of calcium ions. However, very small changes in peak heights rather than peak positions for the mean residue ellipticity in the presence of 5.2 × 10−4 M CaCl2 in both NPP and PP solutions may result from the binding of Ca2+ to the carboxylate groups of all the aspartic acid residues within this peptide sequence domain (Figure 4). The present studies of the influence of phosphorylation on the structure of OPN peptide segment did not show comparable conformational changes when analyzed using identical conditions. The structure of the phosphorylated peptide segment in water or in Tris buffer was completely unordered. The CD results are fully consistent with the previous analysis of this OPN peptide with less ordered structure in water.16 Thus, the different CC growth inhibition observed for each peptide further suggests that local binding of negatively charged phosphate side chains to crystal faces controls growth inhibition rather that any secondary peptide structure. These results reveal that the phosphorylated peptide–COM crystal face interactions are probably dominated by electrostatics, further supporting an important role for macromolecules rich in anionic side chains in the regulation of kidney stone formation.19–21
Figure 4.

Circular dichroism (CD) spectra of both peptides at pH 7.0 (37 °C) in TDW water or in 10 mM Tris buffer with 0.147 M NaCl and 5.2 × 10−4 M CaCl2.
To further test the binding (adsorption) behavior of PP or NPP at crystal faces, CC nucleation studies were conducted. The results showed that PP dramatically increased the induction times, retarding nucleation with increasing PP concentration (Figure 5). The nucleation initiation induction time in pure supersaturated solution was 43 ± 3 min (n = 4), whereas in the presence of 10.1, 20.2, 30.3, or 40.4 nM PP, the induction times increased to 52 ± 5 (n = 4), 65 ± 3 (n = 4), 109 ± 4 (n = 4), and 157 ± 5 (n = 4) min, respectively. In contrast, NPP at concentrations less than 609 nM did not show any influence on COM nucleation; the induction times were not significantly changed in the presence of NPP at concentrations ranging from 20.2 to 609 nM (Figure 5). Surprisingly, however, nucleation was promoted when NPP concentrations were further increased. The presence of 812, 966, or 1120 nM concentrations of NPP, decreased the nucleation induction times to 30 ± 4 (n = 4), 19 ± 3 (n = 4), and 11 ± 3 (n = 4) min, respectively (Figure 5).
Figure 5.
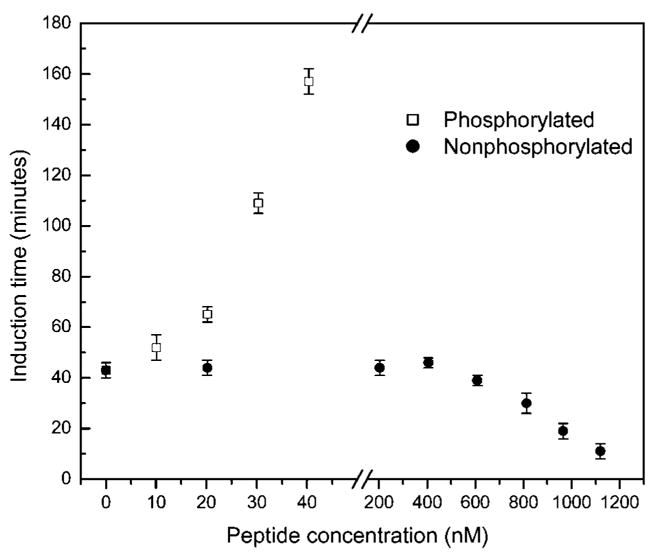
Plots of CC induction time against PP and NPP peptide concentration for COM crystal nucleation (σCOM = 1.53, I = 0.15 M, pH = 7.0, 37 °C).
According to classical nucleation theory (CNT), nucleation is the initial appearance of a new phase during a first-order phase transition in which small nuclei, formed spontaneously in a supersaturated solution, overcome a nucleation barrier. However, unless their size exceeds a critical value—the so-called critical nucleus—they redissolve rather than grow.22,23 Before these small nuclei or embryos (metastable clusters of structural units with a broad size distribution) can reach a critical radius, they remain unstable even when a positive thermodynamic driving force is applied (Δμ/kT, Δμ = μf − μs, μf and μs are the chemical potentials of the solute molecules in the fluid phase and in the solid phase, respectively. k is Boltzmann’s constant, and T is temperature). According to CNT, the total free-energy cost to form a spherical crystallite with radius r is given by eq 6:22,24
| (6) |
where ρs is the number-density of the solid (ρs = 1/Ω, where Ω is the volume per molecule). The first term on the right-hand side of eq 3 is a “bulk” term that expresses the fact that the solid is more stable than the supersaturated fluid. This term is negative and proportional to the volume of the crystallites. The second is a “surface” term that takes into account the free-energy cost of creating a solid–liquid interface. This term is positive and proportional to the surface area of the crystallites. The function ΔG goes through a maximum where dΔG/dr = 0 at rc = 2γ/ρs|Δμ|, and the height of the homogeneous nucleation barrier is22
| (7) |
The technical difficulties involved in directly evaluating crystal nucleation rates (J) have led to other approaches to study initial crystallization events. One of the most common ways to characterize the kinetics of nucleation is to measure the induction period (ts) prior to nucleation based on the equation J = 1/(tsV), where V is the volume of the system. Taking into account the effect of the additive molecule on both the nucleation barrier and the transport process, the nucleation induction time is given by23
| (8) |
where κ[= 16πγcf3Ω2/3(kT)3] remains constant under a given set of conditions. N0 is the density of the additive molecules, B is a kinetic constant, and f(m, x) is the interfacial correlation factor. This factor will depend upon supersaturation, the interfacial interaction parameter m, and the relative size of foreign particles x. m = (γaf − γac)/γcf, where γaf, γac, and γcf are the interfacial free energies between the foreign particle (a) and fluid (f), and crystal nucleus (c) interface; x = Rs/rc (where rc the critical size of nuclei, rc = 2Ωγcf/kT ln(1 + σ), and Rs is the average radius of spherically shaped foreign particles).25,26 Under our nucleation conditions, the only variable relating ts and σ that can be altered by PP is the interfacial energy. Thus, the results indicate that the interfacial energy (γcf) of a COM crystal nucleus is increased by the adsorption of PP. More recently, Grohe et al. used a rat bone OPN peptide fragment (amino acids 220–235) containing no (P0), one (P1), or all three (P3) (pSHEpSTEQSDAIDpSAEK) phosphates and found that these peptides adsorbed preferentially to (−101) faces of COM and inhibited growth of these faces in a phosphorylation-dependent manner.17 The atomic-scale molecular-dynamics simulation of a peptide–COM crystal interaction showed that P3 adsorbs to the (−101) face much more rapidly than P1, which in turn adsorbs more rapidly than P0.17 It has also been proposed, on the basis of 15N and 31P NMR experiments, that phosphorylated serine (pS) in a statherin fragment is capable of binding to HAP surfaces through calcium chelation.27,28 Monte Carlo simulation showed that both phosphoserines in the N-terminal residues of statherin interact strongly with HAP surfaces, even coordinating a surface calcium in a manner similar to that of bulk HAP phosphate groups. The favorable portion of this interaction is dominated almost entirely by electrostatics and to a lesser extent by van der Waals forces.29 Liu et al. concluded that adsorbed proteins/peptides on crystal faces inhibit COM nucleation by increasing the kink kinetics barrier.30 Obviously, during embryo growth, the solvent and foreign species (additive molecules) that have been adsorbed on both the kink sites (active adsorption sites) and the surfaces of the growth units must be desolvated. Thus, the kink energy barrier will be enhanced so as to remove solvent and adsorbed foreign species, particularly at kink sites. Consequently, the integration of growth units into crystal nuclei/embryos at the kink site will be significantly suppressed or even terminated.31,32
In contrast to PP, the homogeneous and heterogeneous nucleation barriers are almost same in the absence and presence of NPP at concentrations below 609 nM (Figure 5), suggesting that these molecules exert almost no influence on the nucleation barrier ( ). According to eq 7, the height of this free-energy barrier is proportional to γ3/|Δμ|2 emphasizing how strongly the nucleation rate depends on both supersaturation and interfacial energy. Thus, NPP does not influence the interfacial energy, indirectly suggesting that its binding ability (adsorption) to COM crystal faces is very weak or even absent. However, the shorter induction times with further increase of NPP concentration from 812 to 1120 nM may be understood in terms of the structure of NPP in solution. Dynamic light scattering results in Figure 6 show that under similar COM nucleation experimental conditions (pH 7.00, 37 °C, and I = 0.15 M), NPP is more likely to aggregate than PP with increase of NPP concentrations, rather than adsorbing on COM crystal faces. A hydrodynamic radius (RH) determined in Tris buffer solution containing 0.147 M NaCl and 5.2 × 10−4 M CaCl2 increased from 1.0–2.4 nm (maximum distribution at 1.8 nm) at NPP concentration of 1118 nM to 97.8–136.1 nm (maximum distribution at 115.4 nm) at an NPP concentration of 30 μM. However, PP at even higher concentrations (up to 30 μM) exhibited size distributions with RH of 1.0–1.3 nm in Tris buffer containing 0.147 M NaCl and 5.2 × 10−4 M CaCl2. Low PP concentration under the same solution conditions maintained constant sizes (data not shown). The theoretical hydrodynamic radius RH of PP (14 residues) is 1.0 nm using the equation RH = (4.75 ± 1.11)N0.29 ± 0.02 (N is the number of residues in the protein/peptide).33 If PP formed a dimer (28 residues), the RH would be 1.3 nm. Therefore, PP probably exists as a monomeric conformer under our experimental conditions. No DLS signals were detected when both peptide concentrations were smaller than 1118 nM. Due to relatively imprecise DLS measurements at very low peptide concentrations, we further used SAXS with high sensitivity at lower concentrations to explore possible peptide aggregation in diluted (900 nM) solutions of both peptide segments at room temperature. Three weak SAXS peaks at q = 0.031, 0.049, and 0.068 Å−1 were observed for NPP (Figure 7b), corresponding to interparticle distances of 202, 128, and 92 Å (d = 2π/q), respectively. No clear peaks were detected for PP at the same concentration as compared with the control (Figure 7a). The Guinier approximation analysis (log(I) vs q2 plot and corresponding slopes of the Guinier fitting) of SAXS intensity for NPP gave estimated hydrodynamic radii (RH) of approximately 16, 11, and 10 Å, respectively. This directly suggests that dimer or trimer species were already formed in the 900 nM NPP solution in addition to monomer. No SAXS signals were detected when both peptide concentrations were less than 900 nM.
Figure 6.

Hydrodynamic radius of NPP and PP in Tris buffer solution containing 0.147 M NaCl and 5.2 × 10−4 M CaCl2 (close to the CC nucleation experimental conditions) by dynamic light scattering (DLS).
Figure 7.

SAXS scattering intensities recorded in capillaries containing (a) 900 nM phosphorylated or (b) 900 nM nonphosphorylated OPN peptide segments (green circles) in buffer solutions containing 0.147 M NaCl and 5.2 × 10−4 M CaCl2 compared with control (buffer solution with NaCl and CaCl2 only) (brown circles).
In addition to crystallite fragments, there are many factors that will influence nucleation such as the presence of a foreign surface which may act as a nucleator. An increase of foreign particle (additive molecule) size may lower f(m, x) (a parameter in eq 8 describing the influence of foreign particles on the nucleation barrier)and promote nucleation at lower supersaturations.32,34 Cacciuto et al. have found that, to be effective crystallization promoters, foreign seed particles need to exceed a well-defined minimum size.35 Just above this size, seed particles act as crystallization “catalysts”, whereas smaller seeds hardly affect the height of the nucleation barrier; the precritical nuclei break away from the foreign seed surface, and the critical nuclei are formed only in the bulk solution.35,36 This may account for the NPP aggregation, reaching the critical size with increase of NPP thus promoting COM nucleation in a CC heterogeneous crystallization system.
Conclusions
The present CC study is an in vitro mimic; it does not address all in vivo aspects of biological control of kidney stone formation. Because of differences in the solution conditions used in our CC crystallization system and the physiological microenvironment in the urinary tract, it is difficult to directly relate our in vitro results to in vivo stone formation. However, this in vitro study provides important clues to further understandings of how individual domains (the 14 amino acid OPN fragment) within an OPN molecule control COM nucleation and growth at the earliest stages of mineralization.
Collectively, our results suggest that, in bulk crystallization, inhibition by a phosphorylated 14-mer OPN peptide segment is manifested by longer induction times for delayed nucleation and by lowering the growth rates for seeded COM growth. The local binding of negatively charged phosphate side chains to crystal faces rather than any secondary peptide structure is responsible for the crystallization inhibition. The phosphorylation-deficient form of this segment fails to inhibit COM crystal growth. Conversely, increasing the concentration of this non-phosphorylated segment promotes COM nucleation. The DLS and SAXS results demonstrate that aggregation of nonphosphorylated peptide segment takes place in solution at sufficiently high concentrations (>900 nM), and the aggregated peptide particles may exceed a well-defined minimum size and thus readily promote COM nucleation. The free-energy barrier associated with the CC nucleation accounts for the observed phenomena in a heterogeneous nucleation system. The paucity of precise information concerning the possible formation of critical-sized NPP aggregates (812 < [NPP] < 900 nM) and COM nucleation seeded by these critical-sized NPP aggregates has been due in part to the limitations of existing experimental approaches.
Acknowledgments
This work was supported by NIH Grant DE03223. The authors thank Dr. Froso for assistance in DLS and SAXS measurements and analyses. The authors also dedicate this publication to the memory of Dr. John R. Hoyer for his lasting contributions in the advancement of understanding of kidney stone formation.
References and Notes
- 1.Asplin JR, Arsenault D, Parks JH, Coe FL, Hoyer JR. Kidney Int. 1998;53:194. doi: 10.1046/j.1523-1755.1998.00739.x. [DOI] [PubMed] [Google Scholar]
- 2.Min W, Shiraga H, Chalko C, Goldfarb S, Krishna G, Hoyer JR. Kidney Int. 1998;53:189. doi: 10.1046/j.1523-1755.1998.00745.x. [DOI] [PubMed] [Google Scholar]
- 3.Wesson JA, Johnson RJ, Mazzali M, Beshensky AM, Stietz S, Giachelli C, Liaw L, Alpers CE, Couser WG, Kleinman JG, Hughes J. J Am Soc Nephrol. 2003;14:139. doi: 10.1097/01.asn.0000040593.93815.9d. [DOI] [PubMed] [Google Scholar]
- 4.Qiu SR, Wierzbicki A, Orme CA, Cody AM, Hoyer JR, Nancollas GH, Zepeda S, De Yoreo JJ. Proc Natl Acad Sci USA. 2004;101:1811. doi: 10.1073/pnas.0307900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang LJ, Zhang W, Qiu SR, Zachowicz WJ, Guan X, Tang R, Hoyer JR, De Yoreo JJ, Nancollas GH. J Cryst Growth. 2006;291:160. [Google Scholar]
- 6.Wang LJ, De Yoreo JJ, Guan X, Qiu SR, Hoyer JR, Nancollas GH. Cryst Growth Des. 2006;6:1769. [Google Scholar]
- 7.Wang LJ, Qiu SR, Zachowicz WJ, Guan X, De Yoreo JJ, Nancollas GH, Hoyer JR. Langmuir. 2006;22:7279. doi: 10.1021/la060897z. [DOI] [PubMed] [Google Scholar]
- 8.Sodek J, Ganss B, McKee MD. Crit Rev Oral Biol Med. 2000;11:279. doi: 10.1177/10454411000110030101. [DOI] [PubMed] [Google Scholar]
- 9.Christensen B, Nielsen MS, Haselmann KF, Petersen TE, Sørensen ES. Biochem J. 2005;390:285. doi: 10.1042/BJ20050341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sørensen ES, Højrup P, Petersen TE. Protein Sci. 1995;4:2040. doi: 10.1002/pro.5560041009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keykhosravani M, Doherty-Kirby A, Zhang C, Brewer D, Goldberg HA, Hunter GK, Lajoie G. Biochemistry. 2005;44:6990. doi: 10.1021/bi050109p. [DOI] [PubMed] [Google Scholar]
- 12.Hunter GK, Kyle CL, Goldberg HA. Biochem J. 1994;300:723. doi: 10.1042/bj3000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gericke A, Qin C, Spevak L, Fujimoto Y, Butler WT, Sørensen ES, Boskey AL. Calcif Tissue Int. 2005;77:45. doi: 10.1007/s00223-004-1288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jono S, Peinado C, Giachelli CM. J Biol Chem. 2000;275:20197. doi: 10.1074/jbc.M909174199. [DOI] [PubMed] [Google Scholar]
- 15.Pampena DA, Robertson KA, Litvinova O, Lajoie G, Goldberg HA, Hunter GK. Biochem J. 2004;378:1083. doi: 10.1042/BJ20031150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoyer JR, Asplin JR, Otvos L. Kidney Int. 2001;60:77. doi: 10.1046/j.1523-1755.2001.00772.x. [DOI] [PubMed] [Google Scholar]
- 17.Grohe B, O’Young J, Ionescu DA, Lajoie G, Rogers KA, Karttunen M, Goldberg HA, Hunter GK. J Am Chem Soc. 2007;129:14946. doi: 10.1021/ja0745613. [DOI] [PubMed] [Google Scholar]
- 18.Wierzbicki A, Sikes CS, Sallis JD, Madura JD, Stevens ED, Martin KL. Calcif Tissue Int. 1995;56:297. doi: 10.1007/BF00318050. [DOI] [PubMed] [Google Scholar]
- 19.Sheng X, Ward MD, Wesson JA. J Am Chem Soc. 2003;125:2854. doi: 10.1021/ja029575h. [DOI] [PubMed] [Google Scholar]
- 20.Jung T, Sheng X, Choi CK, Kim WS, Wesson JA, Ward MD. Langmuir. 2004;20:8587. doi: 10.1021/la0488755. [DOI] [PubMed] [Google Scholar]
- 21.Sheng X, Jung T, Wesson JA, Ward MD. Proc Natl Acad Sci USA. 2005;102:267. doi: 10.1073/pnas.0406835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auer S, Frenkel D. Nature. 2001;409:1020. doi: 10.1038/35059035. [DOI] [PubMed] [Google Scholar]
- 23.Kelton KF. In: Solid State Physics. Ehrenreich H, Turnbull D, editors. Vol. 45. Academic; Boston, MA: 1991. p. 75. [Google Scholar]
- 24.Liu XY, Lim SW. J Am Chem Soc. 2003;125:888. doi: 10.1021/ja020355d. [DOI] [PubMed] [Google Scholar]
- 25.Liu XY. In: Nanoscale Structure and Assembly at Solid-Fluid Interfaces. Liu XY, De Yoreo JJ, editors. II. Kluwer Academic Publishers; Boston, MA: 2004. p. 109. [Google Scholar]
- 26.Liu XY. Langmuir. 2000;16:7337. [Google Scholar]
- 27.Naganagowda GA, Gururaja TL, Levine MJ. J Biomol Struct Dyn. 1998;16:91. doi: 10.1080/07391102.1998.10508230. [DOI] [PubMed] [Google Scholar]
- 28.Gibson JM, Raghunathan V, Popham JM, Stayton PS, Drobny GP. J Am Chem Soc. 2005;127:9350. doi: 10.1021/ja050910m. [DOI] [PubMed] [Google Scholar]
- 29.Makrodimitris K, Masica DL, Kim ET, Gray JJ. J Am Chem Soc. 2007;129:13713. doi: 10.1021/ja074602v. [DOI] [PubMed] [Google Scholar]
- 30.Liu JF, Jiang HD, Liu XY. J Phys Chem B. 2006;110:9085. doi: 10.1021/jp057403b. [DOI] [PubMed] [Google Scholar]
- 31.Jia YW, Liu XY. J Phys Chem B. 2006;110:6949. doi: 10.1021/jp0536089. [DOI] [PubMed] [Google Scholar]
- 32.Liu XY. J Chem Phys. 2000;112:9949. [Google Scholar]
- 33.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Biochemistry. 1999;38:16424. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 34.Liu XY. J Chem Phys. 2000;113:8807. [Google Scholar]
- 35.Cacciuto A, Auer S, Frenkel D. Nature. 2004;428:404. doi: 10.1038/nature02397. [DOI] [PubMed] [Google Scholar]
- 36.Davey RJ. Nature. 2004;428:374. doi: 10.1038/428374a. [DOI] [PubMed] [Google Scholar]