

Abstract
Tumour suppressor genes (TSGs) inhibiting normal cellular growth are frequently silenced epigenetically in cancer1. DNA methylation is commonly associated with TSG silencing1, yet mutations in the DNA methylation initiation and recognition machinery in carcinogenesis are unknown2. An intriguing possible mechanism for gene regulation involves widespread non-coding RNAs such as microRNA, Piwi-interacting RNA and antisense RNAs3-5. Widespread sense-antisense transcripts have been systematically identified in mammalian cells6, and global transcriptome analysis shows that up to 70% of transcripts have antisense partners and that perturbation of antisense RNA can alter the expression of the sense gene7. For example, it has been shown that an antisense transcript not naturally occurring but induced by genetic mutation leads to gene silencing and DNA methylation, causing thalassaemia in a patient8. Here we show that many TSGs have nearby antisense RNAs, and we focus on the role of one RNA in silencing p15, a cyclin-dependent kinase inhibitor implicated in leukaemia. We found an inverse relation between p15 antisense (p15AS) and p15 sense expression in leukaemia. A p15AS expression construct induced p15 silencing in cis and in trans through heterochromatin formation but not DNA methylation; the silencing persisted after p15AS was turned off, although methylation and heterochromatin inhibitors reversed this process. The p15AS-induced silencing was Dicer-independent. Expression of exogenous p15AS in mouse embryonic stem cells caused p15 silencing and increased growth, through heterochromatin formation, as well as DNA methylation after differentiation of the embryonic stem cells. Thus, natural antisense RNA may be a trigger for heterochromatin formation and DNA methylation in TSG silencing in tumorigenesis.
To test the hypothesis that antisense RNA may trigger epigenetic silencing of mammalian genes, we first searched the UCSC Genome Browser for the existence of antisense transcripts of 21 well-known TSGs, identifying antisense transcripts for each TSG (Supplementary Table 1). For example, p15, a well-documented TSG in many tumours9, has several annotated antisense transcripts (Supplementary Table 1). We next established a nuclear sense-antisense transcript library from HeLa cells (Supplementary Fig. 1). After polymerase chain reaction (PCR) sequencing of 192 randomly chosen library clones, we identified 111 genes containing sense-antisense pairs with lengths between 100 and 200 base pairs (bp), including p15 and E-cadherin (Supplementary Table 2). Based on these results, we focused further experiments on p15 because it is frequently deleted or hypermethylated in a wide variety of tumours including leukaemia, melanomas, gliomas, lung cancers and bladder carcinomas9. Interestingly, up to 60% of leukaemias show epigenetic silencing and methylation of p1510, although the initiating events in this process are unknown.
To confirm the existence of a naturally occurring p15AS RNA, we performed PCR with reverse transcription (RT-PCR) with a strandspecific primer and identified p15AS transcripts in two leukaemia cell lines (Fig. 1a). We then performed rapid amplification of 5′/3′ complementary DNA (cDNA) ends (RACE), which identified a 34.8-kilobase (kb) p15 antisense transcript (Supplementary Fig. 2). To uncover the possible mechanistic connection between endogenous p15AS transcripts and human disease, we examined both p15 and its antisense in acute lymphoblastic leukaemia and acute myeloid leukaemia leukocytes, because these two types of leukaemia are frequently accompanied by p15 epigenetic silencing. We found that 11 out of 16 patient samples (69%) showed relatively increased expression of p15AS and downregulated p15 expression (6/11 in acute myeloid leukaemia and 5/5 in acute lymphoblastic leukaemia). In contrast, 16 normal controls showed high expression of p15 but relatively low expression of the p15AS (Fig. 1b). Additionally, the two acute myeloid leukaemia lines, which displayed high p15AS and low p15 expression (Fig. 1a), also exhibited DNA hypermethylation and typical heterochromatic features for silencing in the p15 promoter region (Supplementary Table 3).
Figure 1. p15 antisense expression in leukaemia cells.
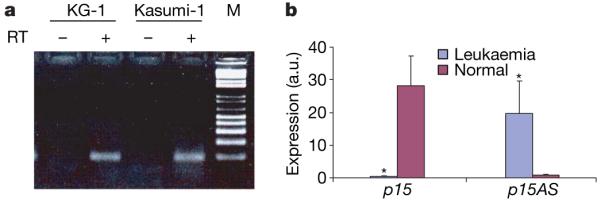
a, Native p15AS transcripts were found in leukaemia cell lines. Lanes 1 and 3 are negative controls without reverse transcriptase. Lanes 2 and 4 show 97 bp bands amplified from the cDNA reverse-transcribed from RNA by a strand-specific primer from leukaemia cell lines KG-1 and Kasumi-1. M is a 100 bp ladder marker. b, p15 and p15AS expression in leukaemic (n = 16) and normal lymphocytes (n = 16), analysed by real-time RT-PCR. Leukaemia samples showed higher expression of p15 antisense and lower expression of p15 than normal lymphocytes. Both p15 and its antisense expression levels were normalized with GAPDH. Error bars, s.e.m.; a.u., arbitrary units; *P < 0.05.
As a result, we hypothesized that p15AS might play a mechanistic role in p15 silencing in tumour cells. To test this, we engineered three reporter constructs (Fig. 2a): (1) pP15 contains the p15 promoter, the p15 first exon and a green fluorescent protein (GFP) reporter gene; (2) pP15-AS, with the same features but containing a portion of p15AS driven by a cytomegalovirus (CMV) promoter located between the p15 promoter and the GFP gene; (3) pP15-ASΔ contains a stop sequence downstream of the CMV promoter that prevents p15AS transcription. These constructs were transfected into HCT116 cells, selected and evaluated by fluorescence-activated cell sorting (FACS), which was also used to sort cells for further culture, after which we evaluated p15 promoter activity by monitoring GFP expression through a second FACS analysis. Both FACS scan results showed that cells stably transfected with pP15-AS have significantly decreased percentages of GFP-positive cells, and this was progressive over time (Fig. 2b, c). Repeat transfections in HeLa cells yielded similar results (Supplementary Figs 3 and 4). Thus, the p15 antisense transcript appears to induce partner sense gene silencing and this effect is relatively stable.
Figure 2. p15 promoter is silenced by the expressed p15 antisense transcript in transfected HCT116 cells.

a, Maps of constructs. Arrows indicate the direction of transcription. b, Downregulation of GFP (under control of p15 promoter) in cells transfected with pP15-AS and selected for 3 weeks. The transfected cells were analysed by FACS. The percentage of GFP-positive cells with p15 antisense expression was lower than that without the antisense expression (0.3% versus 5.8% and 6.6%) (P < 0.001). c, The second FACS showed downregulation of GFP in expressing cells selected from b and cultured for an additional 3 weeks. The percentage of GFPpositive cells with p15 antisense expression remained lower than that without the antisense expression (11.0% versus 51.1% and 59.3%) (P < 0.0001). d, Downregulation of GFP in cells with a tetracycline-inducible pP15-AS-Tre. Expression was reduced 68% on addition of tetracycline, which persisted after tetracycline withdrawal.
Real-time RT-PCR showed that the expressed p15AS transcript downregulated both exogenously (Supplementary Fig. 5) and endogenously (Supplementary Fig. 6), which was confirmed by western blotting (Supplementary Fig. 7). This effect was specific to p15, as the endogenous p16 transcript level was unaffected (Supplementary Fig. 8). We repeated the transfections with new vectors in which four nucleotides were replaced by site-directed mutagenesis (pP15′ and pP15AS′; Supplementary Fig. 9a, b), allowing us to distinguish exogenous from endogenous transcripts. The ratio of antisense to sense expression after transfection was about one-quarter the ratio in leukaemia compared with normal lymphocytes (Supplementary Table 4). Furthermore, the exogenous antisense transcript abrogated expression of the exogenous p15 and reduced the endogenous p15 by about two-thirds (Supplementary Fig. 9c). Thus, the antisense transcript showed a strong cis effect and weaker trans effect on expression, which we then confirmed by a new vector separating the sense and antisense genes on different vectors (Supplementary Fig. 10).
To test the stability of silencing induced by the antisense transcript, we constructed a fourth vector pP15-AS-Tre which placed the p15AS under a Tet-inducible expression system (Fig. 2a). When antisense expression was induced with tetracycline for 10 days in stably transfected T-Rex-HeLa cells, p15-GFP-expressing cells showed a significant decrease in p15 promoter activity. Interestingly, tetracycline withdrawal did not reactivate p15 promoter activity (Fig. 2d), suggesting that antisense RNA may function in initiation of epigenetic silencing rather than maintenance of silencing. A loxP/Cre deletion experiment further confirmed that removal of the CMV promoter for the p15AS did not reactivate p15 promoter activity once silencing had been established (Supplementary Fig. 11), further confirming that stable silencing of p15 by p15AS persisted after the p15AS expression was eliminated.
Next, we tested for a possible alteration in DNA methylation, because p15 silencing often accompanies frequent hypermethylation of its promoter in leukaemia10. However, bisulphite sequencing analysis failed to show any significant changes in DNA methylation in the p15 promoter (Supplementary Fig. 12). To identify an alternative mechanism, we analysed histone modifications in the p15 promoter region by chromatin immunoprecipitation (ChIP), because it has been reported that short interfering RNAs can induce methylation of histone 3 lysine 9 (H3K9) and demethylation of histone 3 lysine 4 (H3K4) experimentally in human cells11,12. Using the pP15′ and pP15AS′ vectors (Supplementary Fig. 9a, b) and sequence-specific probes, we were able to distinguish the exogenous sequences from the endogenous sequences through the analysis of immunoprecipitated DNA. The exogenous p15 promoter showed a marked increase in dimethylation of H3K9 and a decrease in dimethylation of H3K4 in cells with antisense transcripts; this effect was found in both the p15 promoter and exon 1 regions (Fig. 3a, b), suggesting that the antisense RNA may trigger heterochromatin formation that leads to the transcriptional silencing of the sense p15. Interestingly, altered chromatin modifications affected not only the exogenous p15 promoter and exon 1 (Fig. 3a, b), but also the endogenous p15 promoter (Fig. 3c, d), although the magnitude of chromatin modification of the endogenous promoter was less than that of the exogenous promoter (Fig. 3a-d). Thus, the antisense RNA may have both a cis- and trans-acting function in heterochromatin formation. Furthermore, for the endogenous p15 promoter, a much larger region could be examined, which revealed that the decrease in H3K4 methylation was over a much smaller interval (about 1 kb) than the increase in H3K9 methylation (about 6 kb). Chromatin modifications were stable, because cells transfected with a tetracycline-inducible p15AS vector (Fig. 2a) showed reduced H3K4 methylation and increased H3K9 methylation with p15 silencing. After tetracycline withdrawal, H3K4 methylation remained reduced at the same level, and H3K9 was somewhat reduced but still significantly elevated over control (Supplementary Fig. 13).
Figure 3. Heterochromatin formation induced by p15AS.

a, Antisense expression induced an increase in H3K9 dimethylation and a decrease in H3K4 dimethylation in the exogenous p15 promoter region. ChIP enrichment was measured using real-time PCR, normalized by input DNA. The endogenous and exogenous genes were distinguished with probes specific for sense (pP15′) and antisense (pP15-AS′) vectors transfected after modification by site-directed mutagenesis (see Supplementary Fig. 9). b, Antisense expression induced an increase in H3K9 dimethylation and a decrease in H3K4 dimethylation in the exogenous p15 exon 1 region. c, Antisense expression induced a decrease in H3K4 dimethylation in the proximal endogenous p15 promoter, examined by ChIP followed by realtime PCR at six locations, normalized by input DNA. The numbers on the x axis represent PCR sites relative to the transcription start site of p15. The site (+253) near the transcriptional start site was examined by vectors to distinguish endogenous and exogenous genes, as in a. d, Antisense expression induced an increase in H3K9 dimethylation in a large region (about 6 kb) of the endogenous sequence around the p15 transcriptional start site. e, Reactivation of antisense-silenced p15 promoter by 5-aza-2′-deoxycytidine (AZA) and trichostatin A (TSA). p15 promoter activity was measured by FACS for GFP after 5-aza-2′-deoxycytidine and trichostatin A treatment. The top two panels represent negative controls: HCT116 cells only and untreated HCT116 cells with pP15-AS, respectively. The bottom three panels show the result of treatment of the pP15-AS-transfected cells with either 5-aza-2′-deoxycytidine, or trichostatin A or 5-aza-2′-deoxycytidine + trichostatin A. All treatments reactivated the p15 promoter. Significant differences were tested compared with pP15 (n = 3). Error bars, s.d.; *P < 0.05; **P < 0.01; ***P < 0.001.
Recent reports show that both 5-aza-2′-deoxycytidine and trichostatin A can relieve H3K9 methylation-induced repression13,14. The treated cells were subsequently evaluated by FACS and showed that exposure to either 5-aza-2′-deoxycytidine or trichostatin A could restore p15 promoter activity (Fig. 3e, and Supplementary Fig. 14). Interestingly, the epigenetic silencing could not be reactivated by transforming-growth-factor-β, an activator of p1515 (Supplementary Fig. 15).
One mechanism for carcinogenesis is thought to involve the clonal expansion of cancer stem cells, and epigenetic modifications may arise early in such cells within individual tissues16,17. Thus, we investigated the effect of p15AS expression in mouse embryonic stem cells. After selection of embryonic stem cells, FACS demonstrated results similar to those observed in HCT116 and HeLa cells (Fig. 4a), which was confirmed by real-time RT-PCR (Supplementary Fig. 16) and ChIP (Fig. 4b). Furthermore, there was no change in promoter DNA methylation (Fig. 4c), suggesting that heterochromatin rather than DNA methylation is a prerequisite for gene silencing. However, after differentiating embryonic stem cells for one week in vitro to form embryoid bodies, we were surprised to discover hypermethylation at the p15 promoter region (Fig. 4c). Finally, when transfected mouse embryonic stem cells were cultured individually, we found that when stably transfected with pP15-AS they had significantly increased growth rates compared with pP15 control cells (Supplementary Figs 17 and 18). These results provide functional confirmation that the p15AS transcript represses p15 sense expression.
Figure 4. p15 silencing, heterochromatin formation and DNA methylation induced by p15 antisense transcript in mouse embryonic stem cells.

a, Downregulation of GFP in mouse embryonic stem cells transfected with pP15-AS constructs. The transfected cells were analysed by FACS. The top panel is the negative control, mouse embryonic stem cells without transfection; the middle panel shows mouse embryonic stem cells transfected with pP15; the bottom panel is mouse embryonic stem cells with pP15-AS. Green dots in the R2 area represent GFP-positive cells. The numbers in the brackets show the percentages of GFP-positive cells. b, Antisense expression induced a decrease in H3K4 dimethylation and an increase in H3K9 dimethylation in the exogenous p15 promoter region (blue, pP15 mouse embryonic stem cells; red, pP15-AS mouse embryonic stem cells). c, Alterations in DNA methylation of exogenous p15 promoter region in mouse embryoid bodies analysed by bisulphite pyrosequencing. Top two panels represent mouse embryonic stem cells transfected with pP15-AS and pP15. The bottom two panels represent mouse embryoid bodies differentiated from mouse embryonic stem cells transfected with pP15-AS and pP15. The six examined cytosine-guanosine sites (CpGs) are marked and methylation percentages are indicated on the top. Note that hypermethylation is only observed in mouse embryoid bodies differentiated from mouse embryonic stem cells transfected with the pP15-AS construct. Significant differences were tested compared with mES-pP15 cells (n = 3). Error bars, s.d.; *P < 0.05; **P < 0.01.
Dicer is a central component of the short interfering RNA and microRNA silencing pathways18. To determine if it plays a role in epigenetic silencing of p15 induced by antisense RNA, we transfected the pP15-AS vector into murine Dicer knockout (Dicer(-/-)) embryonic stem cells and selected, sorted and assayed these cells as described above. Surprisingly, it was found that the Dicer knockout did not affect antisense-induced silencing of p15 (Supplementary Fig. 19), indicating the mechanism of p15AS RNA-induced silencing was independent of Dicer.
In summary, the results of these experiments suggest that an antisense RNA may trigger transcriptional silencing of a partner sense tumour suppressor gene; this effect occurs both in cis and in trans, and is Dicer-independent. The biochemical mediators of this silencing remain to be determined, but might, for example, involve a Piwi-like protein, as their role in mammalian gene silencing is just beginning to be understood19. The silencing induced by the antisense transcript could occur through a ‘hit and run’ mechanism, because the effect persisted after the antisense transcription was eliminated by either a tetracycline-inducible vector or by loxP/Cre excision. This provides evidence for long-lasting heritable effects on gene expression caused by transient bursts in antisense gene expression, through heterochromatin formation, and that DNA methylation is a secondary effect occurring after differentiation of these cells. The evidence using embryonic stem cells provides a developmental context for this effect, which will need to be ultimately confirmed in tumour stem cells.
These results have practical implications, in that activated antisense transcripts might be potential molecular markers for assessment of cancer risk, as well as serving as novel therapeutic or chemopreventative targets. These data are also consistent with the epigenetic progenitor hypothesis of cancer, which states that tumours arise from epigenetic alterations in progenitor cells that can differentiate into the mass population of cells observed clinically. An example is loss of imprinting in progenitor cells17,20, and in the present case would be antisense silencing and heterochromatinization of a TSG.
METHODS SUMMARY
The vector Pires2-EGFP was used to construct pP15, pP15-AS and pP15-ASΔ. The Tet-inducible vector pcDNA4/TO/myc-His A was used to create pP15-ASTre, which was transfected into the T-REx-HeLa cell line. The transfection was done according to the manufacturer’s instructions. FACS was applied to evaluate p15 promoter activity or sort GFP-positive cells. All transcriptional levels were measured by real-time RT-PCR with internal control. DNA methylation was analysed by bisulphite PCR pyrosequencing. H3K4 and H3K9 methylations were analysed by ChIP and real-time PCR. Mouse embryonic stem-cell differentiation was performed in the absence of leukaemia inhibitory factor (LIF) in bacterial Petri dishes to avoid cell adherence.
Supplementary Material
Acknowledgements
We thank G. Hannon for the Dicer knockout line, R. Ambinder for providing leukaemia specimens from the Johns Hopkins SPORE lymphoma tissue bank, M. Gao for help in detection of DNA methylation, and I. Cui for assistance with the manuscript. This work was supported by a grant from the National Institutes of Health.
Appendix
METHODS
Source of cells
HL-60, KG-1, Kasumi-1, DG-75, Raji and Ramos cell lines were obtained from American Type Culture Collection (Rockville, Maryland), leukaemic cells were obtained from patients in the Hematologic Malignancy Clinic of Johns Hopkins Hospital after informed consent, and mouse embryonic stem cells were obtained from the Johns Hopkins ES Cell Targeting Core Laboratory as described (http://www.hopkinsmedicine.org/core/ES_Targeting/Protocol_Pages/protocolmain.htm).
Plasmid construction
The construction was based on the Pires2-EGFP vector (Clontech). First, the CMV promoter in the Pires2-EGFP vector was replaced by SV40 poly(A) sequence. Three different constructs were made, termed pP15, pP15-AS and pP15-ASΔ. For pP15, the p15 promoter region sequence including exon 1 was amplified from human fetal liver genomic DNA. This 1415 bp fragment was ligated into the BglII and PstI sites of the Pires2-EGFP vector. Because the p15 promoter could transcribe the GFP, GFP expression indicated the activity of the p15 promoter. For pP15-AS, amplified plasmid CMV promoter was ligated into the PstI site of the pP15 construct. The CMV promoter can drive p15AS transcription from p15 exon 1 to the promoter region. To engineer pP15-ASΔ, the CMV promoter and SV40 poly(A) sequences were first amplified separately and then ligated together. The CMV promoter and SV40 poly(A) fragment were then ligated into the PstI site of the pP15 construct. This construct has the CMV promoter but no p15AS transcript because of its downstream transcriptional stop signal. The Tet-inducible vector using a 756 bp fragment of the Tet-inducible CMV promoter from the pcDNA4/TO/myc-His A vector (InVitrogen) was amplified and inserted into the PstI site of the pP15 vector to create pP15-AS-Tre. The pP15 and pP15-AS-Tre vectors were transfected into the T-REx-HeLa cell line (InVitrogen) with Lipofectamine 2000 (InVitrogen). After 3 weeks of G418 selection, GFP-positive cells were sorted and grown in sixwell plates. Tetracycline (1 μgml-1) was added and the medium was changed daily. Ten days later, tetracycline was withdrawn and cells were cultured for an additional 10 days without tetracycline. To distinguish the endogenous and exogenous p15 expression, pP15 and pP15-AS vectors were modified using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer’s instructions.
Transfection and FACS analysis
For HCT116 and HeLa cells, 1.5 × 104 cells were plated in each of six wells with 2 ml medium without antibiotics, on the day before transfection. The cells were 60-70% confluent and the transfection was done with Lipofectamine 2000 according to the manufacturer’s instructions, and changed to complete medium after 3 h. The next day, selective medium containing 400 μgml-1 G418 was added and the cells were cultured for 3 weeks. At that time, cells were analysed and sorted by FACS. GFP-positive cells were either continued in mass culture or picked individually as clones and grown in G418 (200 μgml-1) for three additional weeks. For mouse embryonic stem cells, 2 days before transfection, 1 × 105 mouse embryonic stem cells were plated on mouse embryo fibroblasts (MEFs) in 4 ml growth medium without antibiotics per well in a six-well plate with mouse embryonic stem cells about 70% confluent at the time of transfection. The transfection was done with Lipofectamine 2000 according to the manufacturer’s instructions. The next day, selective embryonic stem medium containing 500 μgml-1 G418 was added to the cells and cultured for 2 weeks. G418-resistant cells were then collected and one part was used to assay GFP expression by real-time RT-PCR, whereas the rest were sorted by FACS for GFP. In addition, a single stable transfected mouse embryonic stem cell in each well of a 96-well plate was plated by automated FACS-based singlecell sorting and cultured for 11 days. To observe the growth rate of the embryonic stem cells, colonies were photographed and the diameters of individual mouse embryonic-stem clones were measured daily.
Real-time quantitative RT-PCR
Real-time quantitative RT-PCR was performed using TaqMan Universal PCR Master Mix (Applied Biosystems) on an ABI Prism 7700 Sequence Detection System (Applied Biosystems), or using SYBR Green. Data were recorded and analysed by Sequence Detector software (Applied Biosystems). All primers are listed in Supplementary Table 5. Endogenous gene expression was normalized with GAPDH in human cells and with mouse β-actin in mouse embryonic stem cells, whereas exogenous GFP and antisense expression levels were normalized with neomycin-resistant gene in transfected cells. Arbitrary units are used to display the normalized relative gene expression levels.
Bisulphite pyrosequencing for DNA methylation analysis
A total of 5-10 μg of DNA was treated with sodium bisulphite according to established methods21. Treated DNA was resuspended in 40 μl of distilled water for PCR. All primers are listed in Supplementary Table 5. PCR products were used directly for pyrosequencing according to the manufacturer’s instruction (Biotage).
Mouse embryonic stem-cell differentiation
Mouse embryonic stem cells were dissociated by trypsinization and cultured with embryonic stem medium in the absence of LIF in bacterial Petri dishes to avoid cell adherence. Embryoid bodies were collected after 1 week of differentiation for the examination of DNA methylation and gene expression.
Chromatin immunoprecipitation assays for histone 3 K4 and K9 methylation
Transfected cells were harvested and crosslinked with 1% formaldehyde when they reached about 80% confluence. ChIP was performed as described previously22 by dimethylated H3H4 and H3K9 antibodies (Abcam). Each experiment was repeated at least twice independently. Immunoprecipitated DNA was analysed by real-time PCR normalized by input DNA. The sequences of primers are listed in Supplementary Table 5.
- 21.Cui H, et al. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62:6442–6446. [PubMed] [Google Scholar]
- 22.Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
References
- 1.Jones PA, Laird PW. Cancer epigenetics comes of age. Nature Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 2.Feinberg AP, Tycko B. The history of cancer epigenetics. Nature Rev. Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 3.Novina CD, Sharp PA. The RNAi revolution. Nature. 2004;430:161–164. doi: 10.1038/430161a. [DOI] [PubMed] [Google Scholar]
- 4.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 5.Willingham AT, Gingeras TR. TUF love for “junk” DNA. Cell. 2006;125:1215–1220. doi: 10.1016/j.cell.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 6.Rosok O, Sioud M. Systematic identification of sense-antisense transcripts in mammalian cells. Nature Biotechnol. 2004;22:104–108. doi: 10.1038/nbt925. [DOI] [PubMed] [Google Scholar]
- 7.Katayama S, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 8.Tufarelli C, et al. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nature Genet. 2003;34:157–165. doi: 10.1038/ng1157. [DOI] [PubMed] [Google Scholar]
- 9.Nobori T, et al. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 10.Lubbert M. Gene silencing of the p15/INK4B cell-cycle inhibitor by hypermethylation: an early or later epigenetic alteration in myelodysplastic syndromes? Leukemia. 2003;17:1762–1764. doi: 10.1038/sj.leu.2403045. [DOI] [PubMed] [Google Scholar]
- 11.Ting AH, Schuebel KE, Herman JG, Baylin SB. Short double-stranded RNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nature Genet. 2005;37:906–910. doi: 10.1038/ng1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberg MS, et al. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA. 2006;12:256–262. doi: 10.1261/rna.2235106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gius D, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell. 2004;6:361–371. doi: 10.1016/j.ccr.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 14.Wozniak RJ, et al. 5-Aza-2′-deoxycytidine-mediated reductions in G9A histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene. 2007;26:77–90. doi: 10.1038/sj.onc.1209763. [DOI] [PubMed] [Google Scholar]
- 15.Sandhu C, et al. Transforming growth factor beta stabilizes p15INK4B protein, increases p15INK4B-cdk4 complexes, and inhibits cyclin D1-cdk4 association in human mammary epithelial cells. Mol. Cell. Biol. 1997;17:2458–2467. doi: 10.1128/mcb.17.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nature Rev. Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 17.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nature Rev. Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 18.Lee YS, et al. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell. 2004;117:69–81. doi: 10.1016/s0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- 19.Peters L, Meister G. Argonaute proteins: mediators of RNA silencing. Mol. Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Sakatani T, et al. Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science. 2005;307:1976–1978. doi: 10.1126/science.1108080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.