

Abstract
Cellular senescence suppresses cancer by stably arresting the proliferation of damaged cells1. Paradoxically, senescent cells also secrete factors that alter tissue microenvironments2. The pathways regulating this secretion are unknown. We show that damaged human cells develop persistent chromatin lesions bearing hallmarks of DNA double-strand breaks (DSBs), which initiate increased secretion of inflammatory cytokines such as interleukin-6 (IL-6). Cytokine secretion occurred only after establishment of persistent DNA damage signaling, usually associated with senescence, not after transient DNA damage responses (DDR). Initiation and maintenance of this cytokine response required the DDR proteins ATM, NBS1 and CHK2, but not the cell cycle arrest enforcers p53 and pRb. ATM was also essential for IL-6 secretion during oncogene-induced senescence and by damaged cells that bypass senescence. Further, DDR activity and IL-6 were elevated in human cancers, and ATM-depletion suppressed the ability of senescent cells to stimulate IL-6-dependent cancer cell invasiveness. Thus, in addition to orchestrating cell cycle checkpoints and DNA repair, a novel and important role of the DDR is to allow damaged cells to communicate their compromised state to the surrounding tissue.
Cellular senescence limits the proliferation of damaged cells that are at risk for neoplastic transformation by imposing an essentially irreversible growth arrest1. Senescent cells also develop a complex senescence-associated secretory phenotype (SASP), in culture and in vivo3-6. The SASP can disrupt normal mammary differentiation4,7, promote endothelial cell invasion8, and stimulate cancer cell growth and invasion in culture and tumor growth in vivo4-6. Additionally, some SASP factors can reinforce the senescence arrest by autocrine or paracrine mechanisms9-12. Among the SASP factors, IL-6 and IL-8 are of particular interest. These cytokines initiate inflammatory responses, such as those associated with normal healing in damaged tissues, as well as many age-related pathologies, including cancer13.
Normal cells undergo senescence in response to severe or irreparable DNA damage, especially DNA DSBs. Among the first proteins to respond to DSBs is ATM (Ataxia Telangiectasia Mutated), a member of the phosphoinositide-3 kinase-like kinase (PIKK) family. ATM substrates include H2AX, a nucleosomal histone variant, and p53 binding protein-1 (53BP1), which facilitates checkpoint activation and repair. Phosphorylated H2AX (γH2AX) and 53BP1 rapidly localize to DSBs, forming characteristic foci. ATM also phosphorylates the DDR kinase CHK2 (checkpoint kinase-2), which promotes growth arrest; NBS1 (Nijmegen breakage syndrome), a member of the MRN (MRE11-RAD51-NBS1) complex that reinforces the DDR and participates in DNA repair; and p53, a tumor suppressor and transcriptional regulator that orchestrates repair and cell cycle arrest (reviewed in14). DSBs that cannot be repaired (e.g., uncapped telomeres) cause constitutive DDR signaling, prolonged p53-dependent growth arrest, and eventually an essentially irreversible senescence arrest1,15.
Despite understanding much about how the senescence arrest is initiated and maintained, little is known about how the SASP is regulated. We recently showed that the SASP is correlated with genotoxic stress, suggesting it is controlled by DDR signaling6. However, p53, a key DDR effector, was not required for the SASP6, raising the possibility that the SASP and DDR are not linked. Here, we asked whether DDR components upstream of p53 regulate the SASP, focusing on two principal SASP factors, the inflammatory cytokines IL-6 and IL-8.
We synchronously damaged DNA in HCA2 human diploid fibroblasts using ionizing radiation (X-ray). A relatively low dose (0.5 Gy) rapidly induced multiple 53BP1 foci in all nuclei and engaged the DDR, as indicated by p53 serine15 phosphorylation and upregulation of p21, a cell cycle inhibitor and p53 target (Fig. 1a,b). Both indicators returned to near-baseline levels within 10 h, as cells resolved the 53BP1 foci (Fig. 1a-c) and resumed growth (Supplementary Information, Fig. S1a,b). A senescence-inducing dose (10 Gy) also triggered a rapid DDR; however, several 53BP1 foci failed to resolve, and increased in size (Fig. 1a-c). These persistent DNA damage foci (PDDF) contained γH2AX, and remained for days (Supplementary Information, Fig. S1d) and months (not shown), while cells remained arrested (Supplementary Information, Fig. S1a-b). PDDF also contained activated (phosphorylated) ATM and ATM/ATR substrates (Fig. 1d). Therefore, low dose radiation induced a transient DDR, from which cells recovered, whereas a high dose generated PDDF, localized but constitutive DDR signaling, and senescence.
Figure 1. Persistent DNA damage signaling correlates with IL-6 secretion.
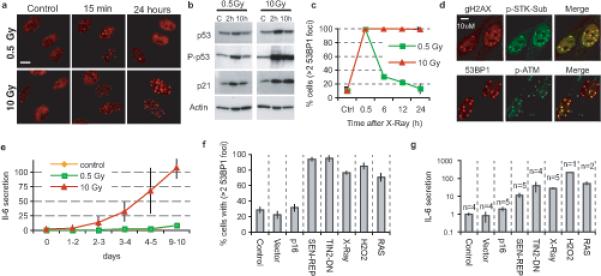
(a) HCA2 fibroblasts were untreated, irradiated with 0.5 Gy, or irradiated with 10 Gy and allowed to recover. Cells were fixed and stained for 53BP1 foci (red) at the indicated intervals after irradiation.
(b) Whole cell lysates were prepared at the indicated intervals after irradiation and analyzed by western blotting for the indicated proteins. P-p53, serine 15-phosphorylated p53.
(c) HCA2 cells were irradiated with 0.5 or 10 Gy and allowed to recover for the indicated intervals before being fixed and stained for 53BP1 foci. Nuclei with 3 or more foci were quantified.
(d) HCA2 cells were irradiated with 10 Gy and analyzed 6 d later for γ-H2AX (red) and the Ser/Thr phosphorylated ATM/ATR substrate motif (p-STK Sub; green) (top panels), or 53BP1 (red) and activated (phosphorylated) ATM (p-ATM; green) (bottom panels). The merged red and green channels display co-localization in yellow.
(e) HCA2 cells were irradiated with 0.5 or 10 Gy, and IL-6 secretion was assessed using ELISA over 10 d for the indicated 24 h windows. The data are reported as the fold increase in secretion relative to untreated control cells.
(f) Cells were infected with lentiviruses or treated as indicated (expression of p16INK4A, p16; replicative senescence, SEN-REP; telomere uncapping using a dominant-negative TIN2 protein,TIN2-DN; X-irradiation, X-Ray; hydrogen peroxide, H2O2; expression of activated RASV12, RAS). Ten days later, cells were analyzed by immunofluorescence to quantify the fraction of cells with 3 or more 53BP1 foci. For representative images of 53BP1 damage foci and for other senescence markers see Supplementary Information, Figs. S1e, S1f-I.
(g) Conditioned media from the cell populations described in Fig. 1F were collected over a 24 h interval, and analyzed for IL-6 by ELISA. The data are reported as fold increase in secretion relative to untreated control cells, and are plotted on a log scale (n= number of cell populations analyzed).
Low dose radiation did not increase IL-6 secretion, which remained at control levels. A high dose, in contrast, increased IL-6 and IL-8 secretion 5- to 6-fold within 2−4 d, and to replicatively senescent levels within 3−5 d (Fig. 1e; Supplementary Information, Fig. S1c). Human WI-38 fibroblasts behaved similarly (not shown). Thus, DNA damage and the DDR alone do not induce inflammatory cytokine secretion; rather, secretion develops, after a delay, when the damage is sufficient to generate PDDF and persistent DDR signaling.
To test this idea further, we used lentiviruses to express p16INK4a, a cyclin-dependent kinase inhibitor that causes a senescence arrest1. The infected cells displayed few PDDF and secreted little IL-6 (Fig. 1f-g; Supplementary Information, Fig. S1e), yet expressed other senescence markers, including increased intracellular reactive oxygen species (Supplementary Information, Fig. S1g-j). Conversely, >2 PDDF and high IL-6 secretion was evident in cells induced to senesce by conditions that caused DNA damage (Fig. 1f-g). IL-8 secretion behaved similarly (Supplementary Information, Fig. S1k).
HCA2 fibroblasts undergo p53-dependent replicative senescence owing to short (dysfunctional) telomeres1,16 (Supplementary Information, Fig. S2a). Accordingly, early passage cells had few PDDF, while senescent cells had three or more (p<10−9, two-tailed student T-test for unpaired samples, Supplementary Information, Fig. S2b-c). As the cells proliferated, PDDF accumulated gradually (Fig. 2a, top), DNA synthesis declined, and IL-6 (Fig. 2a, bottom) and IL-8 (Supplementary Information, Fig. S2d) secretion increased. IMR-90 human fibroblasts behaved similarly (Supplementary Information, Fig. S2e). We used immunostaining to assess cytokine expression, growth arrest (absence of DNA synthesis) and PDDF in single cells within senescing HCA2 populations. Proliferating cells can acquire dysfunctional telomeres prior to arresting growth17. Indeed, PDDF-positive cells did not necessarily fail to synthesize DNA, although they synthesized DNA less frequently than cells without PDDF (Fig. 2b-c). IMR90 fibroblasts behaved similarly (Supplementary Information, Fig. S2f). Further, in late passage populations, many cells that synthesized DNA also showed robust immunostaining for IL-6 (Fig. 2d) and MMP3, another SASP factor (Supplementary Information, Fig. S2g). Together with the p16INK4a results (Fig. 1f-g), these findings suggest that PDDF, rather than the senescence arrest per se, correlate with inflammatory cytokine secretion.
Figure 2. Telomeric PDDF promotes increased IL-6 secretion.
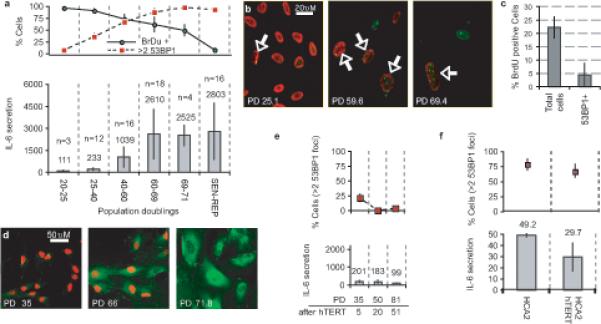
(a) HCA2 populations were cultured until replicative senescence (PD>71). Top panel: Populations were sampled at the indicated PDs (lower panel X axis). Single cells were analyzed for DNA synthesis (BrdU incorporation over 24 h) and 53BP1 foci (cells with 3 or more foci). Lower panel: Conditioned media were collected over a 24 h interval from the populations analyzed in the top panel. IL-6 was measured by ELISA, and is reported as 10−6 pg/cell/day (n= number of cell populations analyzed).
(b) HCA2 cells at the indicated PDs were fixed and single cells analyzed simultaneously for 53BP1 foci (green) and BrdU incorporation over a 24 h interval (red). Arrows mark nuclei that simultaneously harbor 53BP1 foci and BrdU staining.
(c) Cells at PD35 were pulsed with BrdU for 1 h and fixed. Single cells were analyzed simultaneously for 53BP1 foci and BrdU incorporation. BrdU incorporation was scored independent of 53BP1 foci (Total cells) or only in cells with 53BP1 foci (53BP1+).
(d) Cells at the indicated PD levels were pulsed with BrdU for 24 h and fixed. Single cells were analyzed by immunofluorescence for intracellular IL-6 (green) and BrdU incorporation (red).
(e) Cells were infected with a retrovirus expressing hTERT, selected and cultured for the indicated doublings (PD after hTERT). At the indicated PDs, cells were analyzed for 3 or more 53BP1 foci (top panels) or IL-6 secretion (lower panels), as described for Fig. 2a.
(f) Early passage HCA2 or HCA2 cells expressing hTERT were irradiated with 10 Gy, allowed to recover for 8 d, and analyzed for 53BP1 foci (top panel) and IL-6 secretion as described for Fig. 2a. IL-6 secretion is reported as the fold increase compared to unirradiated control cells. There was a non-significant reduction in 53BP1 foci and slight reduction in IL-6 levels (p=0.036, two-tailed student T-test for unpaired samples) in hTERT cells.
To corroborate this idea, we suppressed telomeric PDDF in early passage HCA2 cells by expressing telomerase (catalytic subunit, hTERT) for 20 population doublings (PDs). In contrast to control cells (Fig. 2a; compare PD25−40 to PD40−60), hTERT-expressing cells displayed slightly decreased PDDF but no increase in IL-6 secretion (Fig. 2e). Moreover, when hTERT-expressing cells exceeded the PD level at which unmodified cells completely senesce (PD71−75), PDDF and IL-6 secretion were similar to, or lower than, early passage cells (Fig. 2e). To confirm that rescued telomere dysfunction, rather than hTERT per se, caused the decline in PDDF and IL-6 secretion, we induced senescence in PD50 hTERT-expressing cells by X-irradiation. Radiation generated PDDF and greatly increased IL-6 secretion in these cells (Fig. 2f; Fig. 1f-g). Thus, hTERT reduced telomeric PDDF, but not radiation-induced PPDF (most of which are presumably non-telomeric). These results strengthen the idea that PDDF and persistent DDR signaling are responsible for inflammatory cytokine secretion.
To determine whether the tumor suppressors p53 and pRb were required for IL-6 secretion, we used lentiviruses to express either a short-hairpin RNA against p53 (shp53), GSE22 (a dominant peptide suppressor of p53 activity1) or SV-40 T antigen (SV40LT, which inactivates p53 and pRb1) in replicatively senescent HCA2 cells. As expected1 for HCA2, which express low levels of endogenous p16INK4a, p53 inactivation reversed the senescence growth arrest and drove cells into crisis (Supplementary Information, Fig. S2a; not shown). 3−4 PDs after infection, IL-6 secretion did not decline, but rather increased (Fig. 3a). Thus, neither p53, pRB nor the senescence arrest was required for IL-6 secretion.
Figure 3. Loss of p53 accelerates PPDF formation and IL-6 secretion.
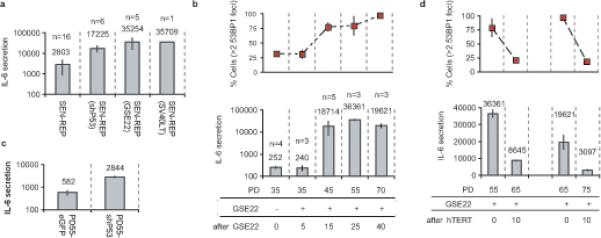
(a) Replicatively senescent HCA2 cells were infected with the indicated lentiviruses and IL-6 secretion was analyzed 3−4 PDs following reversal of the senescence arrest. IL-6 secretion is reported as 10−6 pg/cell/day on a log scale (n= number of cell populations analyzed).
(b) Early passage HCA2 cells were infected with a GSE22-expressing retrovirus, selected for 4 d (∼3 PDs) and IL-6 secretion was measured ∼2 PDs later. Cells were cultured for the indicated PDs and assessed for 53BP1 foci (top panel) and IL-6 secretion (lower panel) (10−6 pg/cell/day on a log scale) (n= number of cell populations analyzed). Because GSE22 prevents p53 tetramerization, which is required for transactivation and rapid degradation, GSE22-expressing cells contained abundant inactive p53 protein (see Supplementary Information, Fig. S3a).
(c) Early passage HCA2 cells (PD35) were infected with lentiviruses expressing either shp53 or eGFP, selected and cultured until PD55, and analyzed for IL-6 secretion (10−6 pg/cell/day on a log scale).
(d) Two HCA2-GSE22 populations were infected with a retrovirus expressing hTERT and analyzed for 53BP1 foci and IL-6 secretion as described above.
Likewise, we inactivated p53 (see Supplementary Information, Fig. S3a) in early passage HCA2 cells using retrovirally-delivered GSE22. There was no change in either PDDF or IL-6 secretion after 5 PDs (Fig. 3b). However, 15 PDs after p53 inactivation, both IL-6 secretion and PDDF increased markedly (Fig. 3b). These increases also occurred following p53 depletion by shp53 (Fig. 3c), although they were more modest (perhaps owing to residual p53 levels; Supplementary Information, Fig. S3b). Thus, p53 was not required to initiate PDDF-driven cytokine secretion, and loss of p53 function in proliferating cells gradually increased the levels of both PDDF and secreted IL-6.
To determine the contribution of telomeric PDDF to the increased IL-6 secretion by p53-deficient cells, we expressed hTERT in GSE-expressing cells that had developed high levels of IL-6 secretion. hTERT reduced both PDDF and IL-6 secretion (Fig. 3d), as it did for cells with wild-type p53 (Supplementary Information, Fig. S3c). Cells expressing hTERT and GSE22 did not grow faster than cells expressing GSE22 only, ruling out the possibility that hTERT enabled the expansion of a few clones with serendipitously low PDDF/IL-6 secretion. hTERT only partially reduced IL-6 and PDDF levels in p53-deficient cells, suggesting that dysfunctional or near-dysfunctional telomeres account for some, but not all, the PDDF in proliferating p53-deficient cells (Supplementary Information, Fig. S3d). Nevertheless, even in actively dividing p53-deficient cells, reduced PDDF coincided with reduced IL-6 secretion.
ATM is a key responder to DNA damage and prominent component of PDDF (Fig. 1d). We tested the idea that PDDF drive cytokine secretion by providing sustained DDR signaling. We infected HCA2 cells with lentiviruses encoding shRNAs against GFP (shGFP, control) or ATM (shATM) (Fig. 4a). ATM depletion (80−90%) prevented the increased IL-6 secretion that normally occurs 9−10 d following 10 Gy X-irradiation (Fig. 4b). We obtained similar results using WI-38 fibroblasts (Supplementary Information, Fig. S3e-f). Additionally, ATM depletion in already (replicatively) senescent cells effectively abolished IL-6 secretion (Fig. 4c). Finally, primary A-T fibroblasts, from patients carrying an inactivating mutation in ATM (ataxia telangiectasia), had low but detectable basal IL-6 secretion levels and completely lacked the 2−3 d and 9−10 d cytokine responses following 10 Gy X-irradiation (Fig. 4d).
Figure 4. DDR signaling is required for the cytokine response to PDDF.
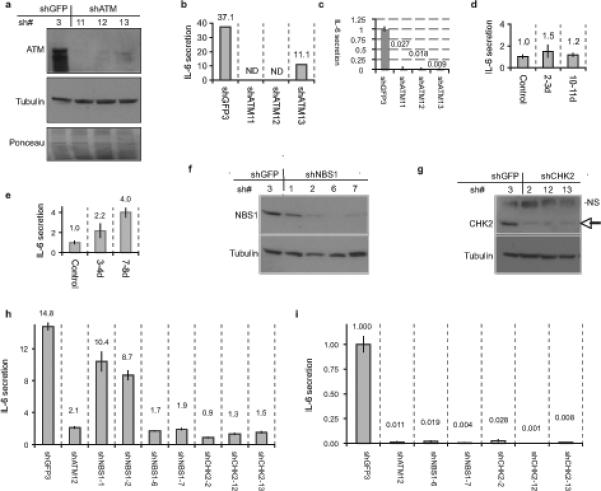
(a) HCA2 cells were infected with lentiviruses expressing shRNAs against GFP (control, shGFP3) or ATM (shATM-11, −12, or −13) and allowed to recover for 5 d. Whole cell lysates were analyzed by western blotting for the indicated proteins. Ponceau staining shows the total proteins.
(b) HCA2 cells were infected as above and irradiated with 10 Gy X-Ray. After 9 d, CM were collected over 24 h and assessed for IL-6 by ELISA. IL-6 secretion is reported as fold change over unirradiated shGFP control. ND=not detected.
(c) Replicatively senescent HCA2 were infected as above and allowed to recover for 6 d. Conditioned media CM were collected over 24 h and assessed for IL-6 secretion (reported as fold change over shGFP control).
(d) A-T cells were irradiated with 10 Gy and allowed to recover for 2 or 10 d. CM were collected over 24 h, and analyzed for IL-6 by ELISA (reported as fold increase compared to unirradiated A-T cells; numerical values are given above the bars).
(e) Seckel syndrome cells were irradiated and analyzed for IL-6 as described above for A-T cells. (f-g) HCA2 cells were infected with lentiviruses expressing shRNAs against GFP (control, shGFP3), NBS1 (shNBS1−1, −2, −6, or −7)) or CHK2 (shCHK2−2, −12, or −13), selected and allowed to recover for 7 d. Whole cell lysates were analyzed by western blotting for the indicated proteins. The arrow indicates CHK2; NS indicates a non-specific band detected by the antibody.
(h) HCA2 cells from (Fig4f-g) with the indicated shRNA-expressing lentiviruses were irradiated with 10 Gy X-Ray. After 2 d, CM were collected over 24 h and assessed for IL-6 using ELISA (reported as fold change over unirradiated shGFP control; numerical values are given above the bars).
(i) Replicatively senescent HCA2 cells were infected with indicated shRNA-expressing lentiviruses and allowed to recover for 6 d. CM were collected over 24 h and assessed for IL-6(reported as fold change over shGFP control; numerical values are given above the bars).
ATM shares many substrates with ATR, another PIKK, which is preferentially activated when cells are damaged during S-phase14. To determine whether ATR was also important for the DNA damage cytokine response, we measured IL-6 secretion by primary fibroblasts from a Seckel syndrome patient. These cells have almost undetectable ATR levels owing to a splicing mutation. They also had relatively high basal levels of IL-6 secretion, but, nonetheless, IL-6 secretion increased after X-irradiation (10 Gy) (Fig. 4e). The magnitude of the increase was smaller than the extent to which IL-6 secretion increased in wild-type cells, possibly because IL-6 secretion is already high in these cells or because ATR partly contributes to the cytokine response. Whatever the case, these findings support the idea that persistent DDR signaling drives IL-6 secretion, and that, while ATR might contribute to this response, ATM is essential.
To determine whether other DDR components were necessary for the DNA damage cytokine response, we depleted cells of either NBS1, an MRN component required for optimal ATM activity, or CHK2, another DDR kinase and downstream target of ATM (Fig. 4f-g). Similar to the effects of ATM depletion, NBS1 or CHK2 depletion essentially prevented the increased IL-6 secretion following 10 Gy X-irradiation and abolished the high IL-6 secretion by already senescent cells (Fig. 4h-i). Thus, three major DDR components (ATM, NBS1 and CHK2) are essential for both establishing and maintaining the cytokine response to DNA damage.
To identify which SASP components respond to DDR signaling, we used antibody arrays to interrogate 120 cytokines and other factors secreted by senescent HCA2 cells. We focused on 16 factors that were significantly modulated by X-irradiation, the majority being upregulated (Fig. 5a). We compared the secretion levels of these 16 factors in control and ATM-depleted cells induced to senesce by X-irradiation (10 Gy). ATM depletion reduced the secretion of 7 of these 16 SASP factors, reducing IL-6 secretion 50-fold and IL-8 secretion 10-fold. Nine factors were unchanged by ATM depletion (≤1.4-fold the secretion level of non-depleted cells) (Fig.5b). Thus, ATM signaling does not regulate the entire SASP, but is required for a subset of SASP components, including the major inflammatory cytokines.
Figure 5. ATM modulates a SASP subset.
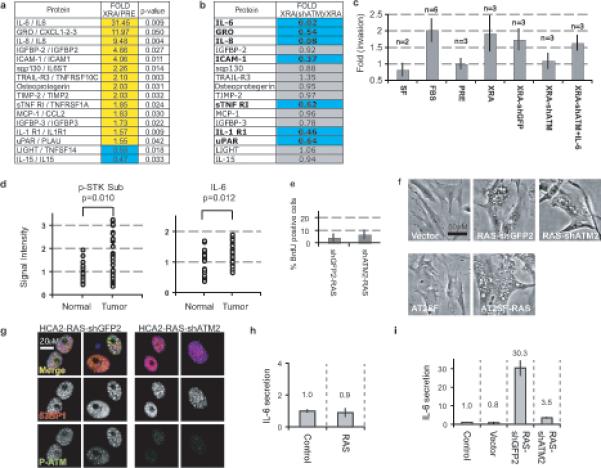
(a) Factors secreted by presenescent (PRE) or senescent HCA2 (10 d following 10 Gy) were analyzed using antibody arrays6. Signals above the PRE baseline are in yellow, signals below baseline in blue. Factors that differed significantly between PRE and XRA cells are displayed (p<0.05, student unpaired T-Test).
(b) SASP factors in (A) were analyzed in X-irradiated ATM-depleted cells (XRA(shATM)) and compared to irradiated-senescent HCA2-shGFP cells (XRA). Changes are reported as XRA(shATM) divided by XRA levels. Signals below the XRA baseline are in blue, factors not substantially modulated by ATM-deficiency in gray.
(c) T47D invasion stimulated by presenescent (PRE) HCA2 CM was given a value of 1. Compared to PRE and serum-free (SF) media, invasion was significantly (p<0.05) stimulated by 10% serum (FBS), and CM from irradiated HCA2 (XRA), HCA2-shGFP (XRA-shGFP), and HCA2-shATM plus IL-6 (XRA-shATM+IL-6), but not by CM from HCA2-shATM (XRA-shATM) (n=number of samples analyzed). Recombinant IL-6 was added to levels detected by ELISA in XRA-shGFP (3ng/ml).
(d) Human breast tissues were analyzed for ATM/ATR serine threonine kinase substrates (p-STK Sub) or IL-6 by immunofluorescence. Data are reported as arbitrary units, with the average normal signal=1 (Signal Intensity). Significance was determined by two-tailed student T-test for unpaired samples using representative fields from 21 normal and 37 cancer tissues.
(e) HCA2 cells were untreated or infected with lentiviruses expressing RAS and shRNAs against GFP (shGFP2) or ATM (shATM2), and pulsed with BrdU for 24 h 9−10 d after infection.
(f) HCA2 or A-T cells were untreated or infected as in (e) and photographed 6 d later.
(g) HCA2 cells infected as in (e) were fixed 10 d later and analyzed for 53BP1 (red), phospho-ATM (green) and DNA (DAPI, blue). Top panel=merge; bottom panels=grayscale for 53BP1 or p-ATM.
(h) A-T fibroblasts were untreated or infected as in (e). CM was collected for 24 h 9−10 d after infection and analyzed for IL-6 (fold increase relative to untreated cells; magnitude is given over the bars).
(i) HCA2 cells were untreated or infected as in (e) and IL-6 secretion analyzed as above.
The SASP can promote cancer cell invasion, largely due to secreted IL-66. To determine the biological significance of the DDR-dependent cytokine response, we used conditioned medium (CM) from control and senescent (X-irradiated) ATM-depleted cells in invasion assays. As expected, human breast cancer cells (T47D) were stimulated to invade a basement membrane when exposed to CM from control senescent cells (Fig. 5c). This stimulatory activity was deficient, however, in CM from ATM-depleted senescent cells, but was largely restored by supplementing this CM with recombinant IL-6. Thus, DDR-dependent SASP factors can have a significant biological consequence.
Persistent DDR signaling has been detected in vivo in premalignant and malignant lesions in human breast, lung, skin, bladder and colon18,19. To model premalignant cells, we used p53-defective HCA2-GSE22 fibroblasts after they spontaneously developed PDDF and increased IL-6 secretion (Fig. 3b). ATM depletion in these cells reduced IL-6 secretion by 70% (Supplementary Information, Fig. S3g), supporting the idea that DDR signaling can drive inflammatory cytokine secretion during neoplastic transformation. To determine whether IL-6 secretion and DDR signaling are linked in vivo, we used immunostaining to assess DDR/ATM activity and IL-6 expression in human breast cancer specimens. Both phosphorylated ATM/ATR substrates and IL-6 levels were significantly elevated in invasive ductal carcinomas compared to normal human breast tissue (Fig. 5d; Supplementary Information, Fig. S4). Thus, DDR signaling and inflammatory cytokine secretion correlated in vivo.
Inflammatory cytokine secretion is also a feature of cells that senesce due to oncogene activation (oncogene-induced senescence; OIS) in culture6,11,12, and of preneoplastic lesions in human colon, which presumably harbor activated oncogenes in vivo11. To determine whether DDR signaling is required for OIS-induced IL-6 secretion, we used lentiviral vectors to simultaneously express oncogenic RAS and shATM in HCA2 cells (Supplementary Information, Fig. S3h). ATM-deficient cells undergo rapid replicative senescence20 but react to OIS depending on the oncogene and context21-23. As previously observed21, oncogenic RAS-expressing fibroblasts underwent OIS regardless of their ATM status (Fig. 5e). Additionally, the RAS-expressing ATM-deficient cells developed a typical OIS morphology (enlargement, vacuolization) and 53BP1 foci that lacked detectable activated ATM (Fig. 5f-g). Oncogenic RAS also caused OIS and 53BP1 foci in primary A-T cells (Fig. 5f; not shown). Therefore, ATM depletion had no discernible effects on OIS phenotypes, including growth arrest and PDDF. However, ATM depletion effectively prevented OIS-driven IL-6 secretion in both A-T (Fig. 5h) and HCA2 cells (Fig. 5i). Thus, ATM controls IL-6 secretion caused by multiple forms of damage-induced senescence, including OIS, which is known to occur in vivo (reviewed in14).
Our findings identify a novel response to persistent DNA damage – the secretion of factors that allow damaged cells to communicate with their microenvironment. This response is associated with cellular senescence, but also occurs in damaged cycling cells that are near, or have bypassed, senescence. Our results suggest a model (Supplementary Information, Fig. S3i) in which mild genotoxic stress (e.g., 0.5 Gy X-ray, which generates ∼17 DSBs/nucleus24) causes a DDR, damage foci, transient cell cycle arrest and repair, but does not induce inflammatory cytokine secretion. More severe genotoxic stress (e.g., dysfunctional telomeres, 10 Gy X-ray) produces PDDF and persistent DDR signaling, which establishes and maintains the p53-dependent senescence growth arrest. After several days, this DDR signaling also initiates the p53-independent cytokine response via ATM, NBS1 and CHK2. p53-deficient cells can initiate the cytokine response in the absence of growth arrest. By contrast, cells induced to senesce by p16INK4a expression, but in the absence of DNA damage, do not initiate a cytokine response. Thus, the DDR can independently control at least two important phenotypes: the p53-dependent growth arrest and senescence-associated extracellular inflammatory signaling. Our results suggest that functions previously attributed to growth arrested senescent cells, specifically their ability to perturb the local microenvironment, can be acquired by damaged cells, whether or not they are senescent or competent to proliferate.
DDR signaling drives only a subset of SASP factors, but those include the potent inflammatory cytokines IL-6 and IL-8. IL-6 was particularly important for the ability of senescent cells to promote cancer cell invasion. Hallmarks of persistent DNA damage are seen in pre-cancerous and malignant lesions18,19, which are presumed to harbor activated oncogenes, and in aging mammalian tissues25,26. Our results suggest DDR signaling drives the inflammation that is also a hallmark of premalignant, malignant and aging tissues. During aging, damaged cells might cause or contribute to tissue dysfunction, including dysfunctional stem cell niches27. In cancer, such cells might promote inflammation, angiogenesis or other phenotypes of cancer progression2.
Why do damaged cells mount a cytokine response? One demonstrated possibility is to reinforce a growth arrest 9-12. Presenescent levels of cytokines receptors are apparently sufficient to contribute to the rapid DDR growth arrest12. However, early experiments showed that senescent cells do not secrete factors that strongly inhibit the growth of nearby presenescent cells28. Thus, cytokines might reinforce a senescence arrest only when cells are moderately damaged or near senescent. The cytokine response might also act in a paracrine manner4-8 to suppress or promote the proliferation of neighboring cells in damaged tissues, allowing damaged cells to communicate their compromised state to surrounding cells, and perhaps mobilize the immune system for their clearance. In support of this idea, tumors induced to senesce by genetic manipulation or chemotherapy regress, with evidence of activated innate immune cells29, and senescent cells in injured liver appear to limit fibrosis before being cleared by the immune system30. In this context, the delayed cytokine response to severe DNA damage could allow cells to attempt DNA repair before generating potent immune-mobilizing signals.
AUTHOR CONTRIBUTIONS
FR designed, performed and analyzed the experiments, JPC and CP performed antibody arrays, JPC and WAMH performed ELISAs, SRR analyzed immunofluorescence, AF analyzed tissue arrays, ARD, DPM and EC generated and tested p53 and ATM RNAi constructs, JC analyzed data, FR and JC wrote the paper.
MATERIAL AND METHODS
Cells
HCA2 foreskin fibroblasts were obtained from J. Smith (University of Texas, San Antonio) and cultured under ambient oxygen levels in Dulbecco's modified Eagle's media (DMEM) supplemented with 10% fetal bovine serum, 2.5 μg/ml fungizone and 100 U/ml streptomycin/penicillin. Early passage is defined as having completed <35 PD and having a 24 h BrdU labeling index of >75%. Cell populations were considered replicatively senescent when they had 24 h labeling indices of <5%. Primary A-T (AT2SF) and Seckel syndrome (GM09812) fibroblasts were obtained from the Coriell Institute and used at early passages (24 h BrdU labeling index >75%). Cumulative PDs of primary cells were determined as follows: current PD = last PD + log2(cell number/cells seeded). 293FT packaging cells (Invitrogen) were used to generate lentiviruses and PT67 cells (Clontech) were used to generate retroviruses1.
Viruses and infections
Retroviruses or lentiviruses encoding dominant negative TIN2 (TIN2−15C), GSE22, SV40LT, p16INK4A, oncogenic RASV12 and hTERT were described1,31. TIN2DN-ires-eGFP, eGFP, p16INK4A and RASV12 were subcloned into a lentiviral vector with puromycin selection (670−1). Custom RNAi short hairpins were subcloned into vectors 749−3 (shp53, zeocin selection) and W17−1 (shGFP2 and shATM2, hygromycin selection) (Campeau et al., submitted; for transgene expression see Supplementary Information, Fig. S5a-b). Lentiviruses encoding shRNAs against GFP, ATM, CHK2 and NBS1 were purchased from Open Biosystems. shRNA target sequences are provided in supplemental material and methods. Virus titers were adjusted to infect 95%−99% of cells1.
Irradiation
Cells were X-irradiated with total doses of either 0.5 or 10 Gy at rates equal to or above 0.75 Gy/min using a Pantak X-ray generator (320 kV/10 mA with 0.5 mm copper filtration).
Immunofluorescence
Cells were cultured in 4 well chamber-slides (Nunc), fixed in Formalin for 10 min at room temperature and permeabilized in PBS-0.2% Triton for 10 min. Slides were blocked for 1 h in PBS containing 1% BSA and 4% normal donkey serum. Primary antibodies were diluted in blocking buffer and incubated with fixed cells overnight at 4° C. The cells were washed, incubated with secondary antibodies for 1 h at room temperature, washed, and mounted with slow-fade gold (Molecular Probes). Images were acquired on an Olympus BX60 fluorescence microscope with the spotfire 3.2.4 software (Diagnostics Instruments) and processed with Photoshop CS2 (Adobe).
Frozen section tissue arrays
Tissue arrays were purchased from Biochain Institute Inc. (arrays #T6235700 and #B112136). Frozen slides were brought to room temperature and processed as described for immunofluorescence, except primary antibodies were diluted in blocking buffer and slides were mounted in vectashield with DAPI (Vector laboratories). Details regarding quantification of the immunofluorescence signals are provided in supplemental material and methods.
Antibodies
Primary antibodies targeted 53BP1 (Bethyl, BL182), γ-H2AX (upstate, JBW301), p53 (Oncogene Research Products, DO-1), Ras (BD Biosciences, 610001), p16 (Neomarkers, JC8), p21 (BD Biosciences, 556430), actin (Chemicon, MAB3128), tubulin (Sigma, T5168), IL-6 (R&D Systems, MAB2061), IL-8 (R&D systems, MAB208), ATM (Abcam Y-170), phospho-ATM (Upstate, #05−740), phospho-p53 (Cell signaling #9284) or phospho-ATM/ATR STK substrates (Cell signaling #2851). Donkey secondary antibodies conjugated to Alexa Fluors were purchased from Molecular Probes (Alexa 350, 488 and 594). Where noted, DAPI was used to label nuclear DNA.
Labeling indexes
Cells were seeded in 4 well chamber-slides, allowed to recover at least 48 h, and labeled with BrdU for 24 h in complete culture media. BrdU incorporation was measured using a kit and manufacturer's protocol (Roche BrdU labeling kit I – immunofluorescence detection).
ELISA
Conditioned media (CM) were prepared by washing cells 3 times in PBS and incubating them in serum-free DMEM containing antibiotics for 24 h. CM were filtered and stored at −80° C. Cell number was determined in every experiment. ELISAs were performed using kits and procedures from R&D (IL-6 #D06050 and IL-8 #D8000C). The data were normalized to cell number and reported as 10−6 pg secreted protein per cell per day.
Invasion assays
Assays were performed as described6 using modified Boyden chambers with 8-micron pore filter inserts for 24-well plates (BD Bioscience). Filters were coated with 12 μl ice-cold 20% basement membrane extract (Matrigel, BD Bioscience). Epithelial cells (5 ×104) were added to the upper chamber in 200 μl serum-free medium, while the lower chamber was filled with 300 μl CM (collected over 24 h at 1×105 cells/ml). After 16−24 h, epithelial cells on the under side of the filter were fixed with 2.5% glutaraldehyde in PBS and stained with 0.5% toluidine blue in 2% Na2CO3. Cells on the underside of the filter were counted using light microscopy.
Antibody arrays
Cultures were washed and incubated in serum-free DMEM for 24 h to generate CM, which was collected, and cells were counted. CM was filtered (0.2 mm pore), stored at −80° C, and analyzed using antibody arrays (Chemicon, Human Arrays VI and VII, cat #AA1001CH-8) essentially as per the manufacturer's instructions, but with modifications that yielded a numerical intensity for each protein detected by the array and allowed comparisons among arrays6.
Western blots
Cells were lysed in 50 mM Tris-HCl pH 6.8, 100 mM DTT, 2% SDS, 10% glycerol. The mixture was briefly sonicated (30 sec) followed by boiling (10 min) and centrifugation. Samples were loaded on 5% or 4−15% gradient tris-glycine SDS-polyacrylamide gels and separated by electrophoresis. Proteins were electroblotted overnight onto PVDF membranes and probed overnight at 4° C in PBS 5% milk. Secondary antibodies conjugated to horseradish peroxidase were used at a dilution of 1:15000 (Jackson Immunoresearch). Blots were developed using the supersignal pico reagent (PIERCE).
Statistics
Error bars on all graphs represent the standard deviation of 3 independent measurements, except where indicated otherwise in the figures.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs. Christian Beausejour (grant CIHR # MPO-79317) for help in the design of a modified lentiviral expression system, Robert Teachenor, Victoria Chu and Genevieve Tang for valuable technical assistance, and Dr. Pierre Desprez for insightful comments on the manuscript. This work was supported by National Institutes of Health grants AG017242 (JC) and AG025708 (Buck Institute), a CABCRP grant 11IB-0153 (ARD), a Larry L. Hillblom Foundation fellowship (CKP), the Netherlands Organization for International Cooperation in Higher Education (Nuffic, HSP-TP 06/78) and Dutch Cancer Society (WAMH), and the Department of Energy under contract DE-AC03−76SF00098 to the University of California.
REFERENCES
- 1.Beausejour CM, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–3126. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- 5.Bavik C, et al. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006;66:794–802. doi: 10.1158/0008-5472.CAN-05-1716. [DOI] [PubMed] [Google Scholar]
- 6.Coppe JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–29574. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 9.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 12.Acosta JC, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 13.Caruso C, Lio D, Cavallone L, Franceschi C. Aging, longevity, inflammation, and cancer. Ann N Y Acad Sci. 2004;1028:1–13. doi: 10.1196/annals.1322.001. [DOI] [PubMed] [Google Scholar]
- 14.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 15.Rodier F, Kim SH, Nijjar T, Yaswen P, Campisi J. Cancer and aging: the importance of telomeres in genome maintenance. Int J Biochem Cell Biol. 2005;37:977–990. doi: 10.1016/j.biocel.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 16.d'Adda di Fagagna F, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 17.Beliveau A, et al. p53-dependent integration of telomere and growth factor deprivation signals. Proc Natl Acad Sci U S A. 2007;104:4431–4436. doi: 10.1073/pnas.0700260104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 19.Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 20.Xu Y, Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev. 1996;10:2401–2410. doi: 10.1101/gad.10.19.2401. [DOI] [PubMed] [Google Scholar]
- 21.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–48. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 23.Bartkova J, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 24.Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A. 2003;100:5057–5062. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sedelnikova OA, et al. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004;6:168–170. doi: 10.1038/ncb1095. [DOI] [PubMed] [Google Scholar]
- 26.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 27.Rodier F, Campisi J, Bhaumik D. Two faces of p53: aging and tumor suppression. Nucleic Acids Res. 2007;35:7475–7484. doi: 10.1093/nar/gkm744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 29.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krizhanovsky V, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SH, et al. TIN2 mediates the functions of TRF2 at telomeres. J. Biol. Chem. 2004;279:43799–43804. doi: 10.1074/jbc.M408650200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.