

Abstract
Mitochondrial dysfunction and altered transmembrane potential initiate a mitochondrial respiratory stress response, also known as mitochondrial retrograde response, in a wide spectrum of cells. The mitochondrial stress response activates calcineurin, which regulates transcription factors, including a new nuclear factor-κB (NF-κB) pathway, different from the canonical and noncanonical pathways. In this study using a combination of small interfering RNA-mediated mRNA knock down, transcriptional analysis, and chromatin immunoprecipitation, we report a common mechanism for the regulation of previously established stress response genes Cathepsin L, RyR1, and Glut4. Stress-regulated transcription involves the cooperative interplay between NF-κB (cRel: p50), C/EBPδ, cAMP response element-binding protein, and nuclear factor of activated T cells. We show that the functional synergy of these factors requires the stress-activated heterogeneous nuclear ribonucleoprotein (hnRNP) A2 as a coactivator. HnRNP A2 associates with the enhanceosome, mostly through protein–protein interactions with DNA-bound factors. Silencing of hnRNP A2 as well as other DNA binding signature factors prevents stress-induced transcriptional activation and reverses the invasiveness of mitochondrial DNA-depleted C2C12 cells. Induction of mitochondrial stress signaling by electron transfer chain inhibitors also involved hnRNPA2 activation. We describe a common mechanism of mitochondrial respiratory stress-induced activation of nuclear target genes that involves hnRNP A2 as a transcription coactivator.
INTRODUCTION
Mitochondrial biogenesis requires a coordinated interplay between proteins encoded by the nuclear and mitochondrial genomes. At least two different mechanisms have been described for the intergenomic cross-talk between these spatially separated genetic systems: anterograde and retrograde signaling (Liao et al., 1991; Liu and Butow, 2006). The anterograde intergenomic regulatory circuit involves nonmitochondrial signals that activate nuclear transcription factors to regulate both nuclear and mitochondrial gene expression (Poyton and McEwen, 1996; Kelly and Scarpulla, 2004; Spiegelman, 2007). The role of peroxisome proliferator-activated receptor γ coactivator-1 family of coactivator proteins in mitochondrial biogenesis and respiration is an example of this type of regulation (Puigserver et al., 1998). Regulation of cellular respiration by reduced mammalian target of rapamycin signaling probably represents another example of anterograde signaling (Bonawitz et al., 2007). In contrast, the retrograde intergenomic regulatory circuit involves the regulation of nuclear gene expression by mitochondrial stress signals that are initiated by metabolic stress, respiratory changes, or mitochondrial DNA damage (Liao and Butow, 1993; Jia et al., 1997; Biswas et al., 1999; Liu et al., 2001; Amuthan et al., 2002; Butow and Avadhani, 2004; Liu and Butow, 2006).
The mammalian retrograde pathway is initiated by disruption of mitochondrial membrane potential (ΔΨm), which can be induced by treatment with mitochondrial uncouplers such as carbonyl cyanide m-chlorophenylhydrazone (CCCP), partial or complete depletion of mitochondrial DNA (mtDNA), mtDNA mutations, hypoxia-induced mitochondrial dysfunction, or mitochondrial protein misfolding (Sciacco et al., 1994; Biswas et al., 1999; Amuthan et al., 2001, 2002; Arnould et al., 2002; Zhao et al., 2002; Biswas et al., 2003, 2005; Petros et al., 2005; Shidara et al., 2005; Srinivasan and Avadhani, 2007). Disruption of ΔΨm causes release of cytosolic free calcium ([Ca2+]c), activation of calcineurin (Cn), and transcriptional activation by nuclear factor of activated T cells (NFAT), nuclear factor-κB (NF-κB) (cRel:p50), C/EBPδ, and cAMP response element-binding protein (CREB) (Biswas et al., 1999; Amuthan et al., 2002; Arnould et al., 2002; Biswas et al., 2003; Butow and Avadhani, 2004). This retrograde response culminates in wide-ranging changes in nuclear gene expression in metazoan cells (Marusich et al., 1997; Amuthan et al., 2001; Delsite et al., 2002; Biswas et al., 2003, 2005; Crimi et al., 2005; Jahangir Tafrechi et al., 2005; Guha et al., 2007; van Waveren and Moraes, 2008). Similar to yeast, the transcriptional targets of the retrograde pathway in mammalian cells are diverse and include proteins that regulate Ca2+ storage and release, glucose uptake and metabolism, mitochondrial energy transduction, cell survival, and cytoskeletal organization (Biswas et al., 1999; Dey and Moraes, 2000; Amuthan et al., 2001, 2002; Arnould et al., 2002; Biswas et al., 2005, 2008b; Guha et al., 2007). Although 120 candidate target genes of the mammalian retrograde mitochondrial stress pathway have been identified in C2Cl2 pluripotent myoblasts by using microarray analysis, the full genetic footprint of mitochondrial stress has not been characterized (Biswas et al., 2005, 2008b). Characterization of the genes regulated by the intergenomic retrograde pathway is central to understanding the putative role of mitochondrial stress in cellular resistance to apoptosis, aging, cancer progression, and neural and bone degenerative diseases (Dey and Moraes, 2000; Delsite et al., 2002; Petros et al., 2005; Shidara et al., 2005; Wallace, 2005; Lin and Beal, 2006; Ohta, 2006; Pelicano et al., 2006).
In this study, we validated that the genes encoding Cathepsin L, RyR1, and Glut4 respond to mitochondrial stress signaling, identified the mitochondrial stress response promoter elements, and investigated the mechanisms of transcriptional activation for these genes. We chose to examine the activation of Cathepsin L, RyR1, and Glut4 genes because their activation is a marker for stress-induced cellular changes, including tumor invasion, regulation of Ca2+ homeostasis, and altered cellular metabolism, respectively. The regulation of mitochondrial respiratory stress-responsive nuclear genes requires physical interactions and functional synergy between the transcriptional activators NF-κB (cRel: p50), C/EBPδ, CREB, and NFAT that are activated under mitochondrial stress conditions. In addition, we have found that the functional synergy of these factors in the mitochondrial stress pathway requires coactivation of the heterogeneous nuclear ribonucleoprotein (hnRNP) A2. hnRNP A2 is a protein with known functions in RNA processing/trafficking, telomere maintenance, and oncogenesis (Dreyfuss et al., 1993; Fielding et al., 1999; He et al., 2005; Kosturko et al., 2005; Moran-Jones et al., 2005). Here, we show a new function for this protein as transcription coactivator, which is mediated mostly through interaction with other transcription factors.
MATERIALS AND METHODS
Plasmid Constructs and Cloning
The mouse Cathepsin L gene promoter DNA (sequence −273 to +47) (Amuthan et al., 2001) and the mouse RyR1 promoter (sequence −205 to +63) (accession FJ480190) were amplified from mouse genomic DNA (Amuthan et al., 2001) and cloned into the pGL3 mammalian expression vector (Promega, Madison WI). The human Glut4 promoter (sequence −1209 to −168) (Liu et al., 1992) was subcloned in the pGL3 vector. Mutations in the transcription factor binding sites (NF-κB, GATGCGAATCC; C/EBPδ, GACTACGAC; and CREB, CGCTAACCCT) were introduced using the site-directed mutagenesis kit from Stratagene (La Jolla, CA). The cRel, C/EBPδ, NFAT, and CREB genes were cloned into the pCMV4 expression vector. hnRNP A2 cDNA was also subcloned from pET28a (+) vector into pCI for transfections of C2C12 cells. The gal4 fusion constructs were generated by cloning the full-length (1-342 aa) and the deletion constructs (1-180 aa, 90-242 aa, 178-342 aa, and 240-342 aa) in frame into the EcoRV and HindIII sites of the pBIND gal4 dbd (Checkmate Mammalian 2-hybrid system, Promega).
Cell Lines and Transient Transfections
Murine C2C12 skeletal myoblasts (CRL1772; American Type Culture Collection, Manassas, VA) were grown in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and 0.1% gentamicin. mtDNA-depleted clones containing ∼80% reduced mtDNA contents were generated as described previously (Biswas et al., 1999) and grown in the presence of 1 mM sodium pyruvate and 50 μg/ml uridine. Reverted cells represent the mtDNA-depleted cells (with 80% DNA depletion) grown for 30 cycles in the absence of ethidium bromide until the mtDNA content reached 20% of the control cell level. In some experiments, an alternate method of mitochondrial stress induction by treatment with mitochondria specific uncoupler, CCCP (25 μM) was used. In these experiments, CCCP treatment was carried out for up to 10 h as specified. Transfections were carried out using the FuGENE 6 reagent (Roche Molecular Biochemicals, Indianapolis, IN) using the manufacturer's suggested protocol. Promoter DNA constructs cloned in pGL3 vector (1 μg) and 0.5 μg of a Renilla luciferase construct (Promega) as an internal control were used in each transfection. The luciferase activity was assayed using the Dual-Luciferase reporter assay system (Promega). Cotransfections with various cDNAs were carried out using 0.2 μg of cDNA constructs.
Small Interfering RNA (siRNA) Design, Cloning, and Transfection
Three siRNAs were directed to the mouse hnRNP A2 mRNA sequence (accession NM_016806) by using the siRNA Design software (Ambion Technologies, Foster City, CA). The siRNA sequences were cloned into the pSilencer2.1neo vector (Ambion Technologies). A target sequence with no known homology to any mouse transcript was also cloned and used as a control. The siRNA sequence which knocked down the hnRNP A2 mRNA level to ∼80% was selected to generate the stable cell lines. Stable cell lines were generated after transfection of mtDNA-depleted C2C12 cells with hnRNP A2si or scrambled sequence cloned into the pSilencer2.1neo vector containing a neomycin resistance gene. Transfected cells were grown in a medium containing Geneticin (G418; 1 mg/ml) for 14 d, and resistant clones were individually picked and expanded. These clones were then screened for hnRNP A2 mRNA levels by real-time polymerase chain reaction (PCR), and the clone with 70% knockdown was used for further studies.
For experiments using CCCP as a stress inducer, control C2C12 cells were transiently transfected for 24 h either with pSilencer 2.1neo empty vector or pSilencer2.1neo-hnRNP A2siRNA vector. After 24 h of transfections, cells were treated with CCCP (25 μM) for 10 h, and mRNA was isolated for real-time PCR analysis.
For mRNA silencing by transient transfections, predesigned siRNAs for mouse cRel (sc-29858), C/EBPδ (sc-37723), and CREB (sc-35111) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and double-stranded scrambled negative siRNA control was purchased from Integrated DNA Technologies (San Diego, CA). Control and mtDNA-depleted cells (1 × 106) were transfected with preannealed double-stranded siRNAs at a final concentration of 25 nM by reverse transfection as described previously (Guha et al., 2007). Transient transfections were carried out in triplicate by using siPORT NeoFX reagent (Ambion Technologies). After 48 h, total cell lysates were analyzed for knockdown of the respective protein levels by immunoblot. A parallel set of transfected cells were used for chromatin immunoprecipitation (ChIP) assays.
Enhanceosome Pull-Down
Nuclear extracts were prepared by the method of Dignam et al. (1983). RNA bound to the proteins was removed by RNAse treatment. DNA sequences for the Cathepsin L promoter (−273 to −53) and RyR1 promoter (−205 to +63) were end labeled using T4 polynucleotide kinase and were coupled to cyanogen bromide-activated Sepharose 4B as described previously (Kadonaga and Tjian, 1986). Approximately 1 mg of protein was loaded per DNA-Sepharose column as described previously (Masternak et al., 2000), and bound proteins were eluted with 0.2–0.4 M KCl buffer containing 25 mM HEPES, pH 7.8, 12.5 mM MgCl2, 1 mM dithiothreitol, 20% (vol/vol) glycerol, and 0.1% (vol/vol) Nonidet P-40. Eluates (200 μl each) were concentrated to 20 μl and run on a 12% SDS-polyacrylamide gel electrophoresis (PAGE) gel and silver stained using a Silver Stain Plus kit (Bio-Rad Laboratories, Hercules, CA). The immunoblots were developed using SuperSignal West Femto maximum sensitivity substrate from Pierce Chemical (Rockford, IL).
Immunoprecipitation
To ensure equal amounts of input hnRNP A2 levels, we used 100 μg of protein from mtDNA-depleted cells and 500 μg of protein from control cells for immunoprecipitation. The nuclear and cytosolic fractions were immunoprecipitated overnight at 4°C, with the respective antibodies as mentioned in figure legends (2 μg/ml). The immune complexes were collected onto protein A-Agarose beads and washed extensively (Sigma-Aldrich, St. Louis, MO). The immunoprecipitates were extracted from the beads with 2× Laemmli buffer devoid of β-mercaptoethanol at 95°C for 5 min and used for further analysis.
ChIP Analysis
ChIP assays were performed following the protocol of Millipore (Billerica, MA). Cells were fixed by adding 1% formaldehyde to the culture medium and incubated at 37°C for 10 min. The cell pellet was suspended in SDS lysis buffer [1% SDS, 10 mM EDTA, and 50 mM tris(hydroxymethyl) aminomethane, pH 8.1], and the cell lysates were sonicated using an ultrasonic processor sonicator (10-s pulses; 20 times, on ice). Diluted aliquots were immunoprecipitated overnight at 4°C. Preimmune immunoglobulin G was used as a negative control, and equal volumes of samples were used as input DNA. The antibody–chromatin complex was immunoprecipitated by protein A-agarose/salmon sperm DNA (50% slurry) (Sigma-Aldrich). The DNA recovered after the reversal of cross-linking was quantified by real-time PCR. Data were analyzed by normalizing with the corresponding input values.
Growth Pattern and In Vitro Invasion Assays
Growth patterns were measured by seeding cells at a density of 1 × 104 cells/well in 24-well culture plates. Cells from three wells at each time point were harvested and counted in a Guava personal cytometer according to the manufacturer's protocols (Guava Technologies, Hayward, CA). The in vitro invasion assays were carried out in Matrigel invasion chambers (BD Biosciences Discovery Labware, Bedford, MA) as described previously (Yagel et al., 1989). Cells (1 × 104) were seeded on top of the Matrigel layer. After incubation for 24 h at 37°C, noninvading cells in the Matrigel layer were removed, and the microporous membranes containing invaded cells were stained and viewed with a BX 61 fluorescence microscope (Olympus America, Center Valley, PA) as described previously (Amuthan et al., 2001). At least six fields were examined for each condition per experiment.
Uptake of 2-Deoxyglucose
Glucose uptake was measured as described previously (Guha et al., 2007). Briefly, 106 cells grown in six-well plates were serum starved for 6 h and incubated in the presence or absence of the indicated levels of insulin for 30 min in glucose-free DMEM medium. One microcurie of 2-[3H]deoxyglucose (1 mCi/0.1 mmol; American Radiolabeled Chemicals, St. Louis, MO) was added to each well containing 2 ml of medium for an additional 15 min. The transport was terminated by washing cells rapidly three times with ice-cold phosphate-buffered saline and lysing in 1 N NaOH. Radioactivity levels of the lysates were assessed in a liquid scintillation counter (Beckman Coulter, Fullerton, CA).
mRNA Quantitation by Real-Time PCR
Total RNA was isolated using TRIzol reagent according to the supplier's protocol (Invitrogen). The cDNA was generated from 5 μg of RNA using the cDNA Archive kit (Applied Biosystems, Foster City, CA), and 50 ng of the cDNA was used as template per reaction. SYBR Green (Applied Biosystems) primers were designed for hnRNP A2, TGFβ, IGF1R, Glut4, RyR1, and Cathepsin L by using the Primer Express 2.0 software (Applied Biosystems). (Sequences can be provided on request.) Data were normalized using β-actin levels as an endogenous control. Results represent three independent experiments.
Statistical Analysis
Data on luciferase activity, mRNA quantitation, and chromatin immunoprecipitation analysis are presented as mean ± SD of three to five independent experiments. Differences between paired variables were determined using two-way analysis of variance. A single asterisk (*) represents p values <0.05 considered statistically significant, and a double asterisk (**) represents p values <0.001 considered highly significant.
RESULTS
Common DNA-binding Proteins Involved in the Transcriptional Regulation of Mitochondrial Stress-responsive Genes
In a previous study, we showed that mouse Cathepsin L promoter exhibited 2.5-fold higher activity in mtDNA-depleted C2C12 cells compared with control cells (Amuthan et al., 2001). In addition, 5′ deletion analyses showed that the proximal promoter region spanning sequence −273 to +47 fully responded to mitochondrial stress as revealed by promoter activity. This minimal promoter region contained sites with full or partial consensus for binding to NFκB (cRel:p50), C/EBPδ, CREB, and NFAT (Amuthan et al., 2001). These same factors are activated under mitochondrial stress as seen by increased nuclear accumulation and also DNA binding (Amuthan et al., 2001; Arnould et al., 2002; Biswas et al., 2003, 2005; Butow and Avadhani, 2004). In the present study, we focused on these same transcription factors and investigated the mechanism of transcriptional activation of three putative mitochondrial stress response target genes for Cathepsin L, RyR1, and Glut4 by using multiple approaches. In initial experiments by 5′-deletion analysis (data not shown), we defined a minimal promoter region for each gene that was capable of responding to mitochondrial respiratory stress. A positive response was defined as a greater than twofold higher transcription rate in mtDNA-depleted C2C12 cells than in control cells (Figure 1, A, C, and D). The minimal promoter regions shown in Figure 1B retained >80% of the promoter activity obtained with the larger 5′-extended promoter DNAs in mtDNA-depleted cells (data not shown). The specificity of the promoter responses to stress was demonstrated by the absence of increased transcription for the pGL3 basic vector containing the simian virus 40 promoter in mtDNA-depleted cells. Furthermore, the activities of all three promoters were markedly reduced to near control cell level in reverted cells with mtDNA content restored to ∼70% of the control. A reversal of activity in reverted cells provides evidence that the altered promoter activity in depleted cells is due to mitochondrial stress induced by mtDNA depletion. Nucleotide sequence analysis using MatInspector indicated that the RyR1 and Glut4 promoters also contained consensus sites for binding to NF-κB (cRel), C/EBPδ, and CREB within their stress response regions (Figure 1B). The RyR1 and Cathepsin L promoters also contained NFAT consensus sites (Figure 1B). The functional significance of these factor binding sites was tested by mutational analysis. The enhancement of Cathepsin L promoter activity was eliminated by mutations in the putative C/EBPδ binding site and greatly reduced by mutations in the NF-κB site (Figure 1C). Mutations in the putative cRel, C/EBPδ, NFAT, and CREB binding sites of the RyR1 promoter likewise reduced the activation of this promoter in mtDNA-depleted cells (Figure 1D). Mutations within the NF-κB, CREB, and C/EBPδ sites of the Glut4 promoter caused similar changes in response to mitochondrial respiratory stress (data not shown). The basal transcription activities of these promoters in control C2C12 cells was not significantly affected by these mutations (Figure 1, C and D), probably because of different set of regulatory controls operating under normal cellular conditions when the steady state levels of stress-activated factors are generally low. These results show that NF-κB, C/EBPδ, CREB, and NFAT are important factors in mediating the effects of mitochondrial respiratory stress.
Figure 1.

Mitochondrial stress-mediated transcriptional activation of RyR1, Cathepsin L, and Glut4 promoters. (A) Relative promoter activity in control cells with normal mtDNA levels (□) and mtDNA-depleted (■) cells. (B) Minimal regions of RyR1, Cathepsin L, and Glut4 promoters required for stress responsive transcriptional activation. (C and D). Effects of mutations at the putative factor binding sites of the promoter on transcription activity in control and mtDNA-depleted cells. Data represent the mean ± SD of three separate experiments. **p < 0.001, represents highly significant difference.
Identification of Cathepsin L and RyR1 Promoter-binding Proteins
We used the enhanceosome pull-down approach (Masternak et al., 2000) to determine the nature of nuclear proteins that bind to the minimal stress response regions of Cathepsin L and RyR1 promoters. The Cathepsin L (−273 to −53) and RyR1 (−205 to +63) promoter DNAs were conjugated to CNBr-activated Sepharose (Kadonaga and Tjian, 1986) to produce affinity columns. Approximately 1 mg of nuclear protein was loaded on each column, and the proteins from mtDNA-depleted and control C2C12 cells were eluted with buffer containing progressively higher salt (0.2–0.4 M KCl), and proteins were resolved by SDS-PAGE (Figure 2, B and D). The gel profile in Figure 2A (bottom) and immunoblot of the gel (Figure 2A, top) with antibody to nuclear p97, represent 1% of the total protein loaded on the Cathepsin L promoter DNA column in Figure 2B. The 0.2 M KCl eluate from mtDNA-depleted cells showed a number of protein bands. Out of these bands, five protein bands with apparent molecular masses of 110, 75, 50, 36, and 28 kDa (marked with arrowheads) were identified as NFAT, cRel, p50, hnRNP A2, and C/EBPδ by using nano-liquid chromatography (LC)/tandem mass spectrometric analysis of tryptic peptides. The LC/mass spectrometric pattern of hnRNP A2 is shown in Supplemental Figure S1. The other minor bands (marked with lines) either represented degradation products or could not be identified because of low abundance. The 0.3 and 0.4 M KCl eluates yielded lower intensity bands. The extract from control C2C12 cells also yielded very low level of DNA-binding proteins, and most of these bands migrated differently from the major bands obtained with nuclear protein fraction from mtDNA-depleted cells. These results are consistent with the higher promoter activity and higher level of activation of these factors in mtDNA-depleted cells. The identities and relative levels of bound proteins were confirmed by immunoblot analysis. High levels of hnRNP A2, C/EBPδ, NFAT, cRel, and p50 were detected in 0.2 M KCl eluate when nuclear extract from mtDNA-depleted cells was used (Figure 2C, right). The 0.3 M KCl elutes also showed some of these proteins, whereas the 0.4M KCl elute showed very low levels. Consistent with the low protein binding observed in Figure 2B, all three column fractions with control cell nuclear extract showed very low to marginal levels of antibody-reactive bands in Figure 2C (left). Very low level of p65 was detected in the immunoblots of bound proteins from control and mtDNA-depleted cell extracts, suggesting a preferential binding of the NFκB proteins cRel:p50 to the promoter DNA. Purification using the RyR1 promoter DNA (−205 to +63) gave essentially similar results (Supplemental Figure S2). Immunoblot analysis of the eluted proteins showed that DNA bound hnRNP A2 was eluted with all three KCl fractions, suggesting a higher affinity interaction with the enhanceosome complex of RyR1 promoter. The reason for this difference in affinity between the Cathepsin L and RyR1 promoter binding remains unclear. NFAT and p50 were eluted from the RyR1 promoter with both 0.2 and 0.3 M KCl (Supplemental Figure S2B).
Figure 2.

Enhanceosome interaction with Cathepsin L promoter DNA. (A) Silver-stained gel pattern of nuclear proteins from control and mtDNA-depleted cells (10 μg each) (bottom panel) and immunoblot of the companion gel with antibody to nuclear p97. (B) Silver-stained gel profile of proteins eluted from the Cathepsin L DNA affinity column by using nuclear proteins from mtDNA-depleted and control C2C12 cells (1 mg of protein each). Each eluate (200 μl) was concentrated to 20 μl and used for gel electrophoresis. (C) Immunoblot analysis of protein fractions bound to Cathepsin L promoter DNA. One hundred microliters of eluate per sample was concentrated to ∼20 μl and used for immunoblotting.
Elevated Nuclear hnRNP A2 in Cells Subjected to Mitochondrial Stress
hnRNP A2, a protein that shuttles between the cytosol and the nucleus, is involved in mRNA processing, RNA transport, and RNA metabolism. The immunoblot in Figure 3A shows that in control, mtDNA-depleted, and reverted C2C12 cells, the levels of hnRNP A2 are significantly higher in the nucleus than in the cytoplasmic compartment. In addition, the hnRNP A2 protein level was almost fivefold higher in the nuclear fraction of mtDNA-depleted cells than in control cells. Reverted cells contained significantly lower hnRNP A2 protein in both the cytosolic and nuclear compartments. Results also show that the nuclear level of another member of hnRNP family proteins, hnRNP D-L, did not increase in response to mitochondrial stress. In both depleted and reverted cell nuclei there was a reduction in the level of hnRNP D-L protein. These results suggest that modulation of hnRNP family protein levels during mitochondrial stress is highly selective and not a general phenomenon. In addition, the increase in nuclear level of hnRNP A2 in mtDNA-depleted cells is indeed in response to stress due to mtDNA depletion, because restoration of mtDNA reduced hnRNP A2 level. Consistent with the increased protein levels, real-time PCR detected fivefold higher hnRNP A2 mRNA in mtDNA-depleted cells than in control and reverted cells (Figure 3B). The induction of mitochondrial stress by CCCP treatment also increased the steady-state levels of hnRNP A2 in 3T3 fibroblasts, RAW 264.7 macrophages, H9C2 cardiomyocytes and A549 lung carcinoma cells (Figure 3C). These results suggest that a high nuclear level of hnRNP A2 is a common consequence of mitochondrial respiratory stress. In compliance with our previous observations (Amuthan et al., 2002; Srinivasan and Avadhani, 2007; Guha et al., 2007), these results also show that mitochondrial respiratory stress induced changes in nuclear activity is a general feature of many cell types.
Figure 3.

Increased nuclear hnRNP A2 in response to mitochondrial stress and its association with other DNA binding transcription factors. (A) Immunoblot analysis of nuclear and cytoplasmic proteins (50 μg each) from control, mtDNA-depleted, and reverted cells with indicated antibodies. The blots were reprobed with antibodies to nuclear protein p97 and cytosolic actin to assess loading levels. (B) hnRNP A2 mRNA levels in control, mtDNA-depleted, and reverted cells. Data represent the average ± SD of three independent experiments. **p < 0.001, represents highly significant difference in mRNA levels between control, mtDNA-depleted, and reverted cells. (C) hnRNP A2 immunoblot analysis of nuclear extracts from control and CCCP (25 μM; 2 h) treated 3T3, RAW264.7 macrophage, C2C12, and H9C2 cells (50 μg of protein). (D) Immunoprecipitates from control and mtDNA-depleted nuclear extracts by using hnRNP A2 antibody were subjected to immunoblot analysis with C/EBPδ, cRel, CREB, or YY1 antibodies (panels from top 1–5). In panels 6–9 from top, nuclear extracts were immunoprecipitated with C/EBPδ, cRel, CREB, and p50 antibodies and probed with hnRNP A2 antibody.
Physical Interaction of hnRNP A2 with the Mitochondrial Respiratory Stress-activated Transcription Factors
Although hnRNP A2 did not directly bind to promoter DNA in EMSA assays (data not shown), it was associated with promoter DNA in the pull-down assay (Figure 2 and Supplemental S2). These results suggest that the association of hnRNP A2 with the stress-responsive promoters is mediated by protein–protein interactions with other DNA-bound transcription factors. Indeed, C/EBPδ, cRel, and CREB were coimmunoprecipitated from nuclear extracts of mtDNA-depleted cells using hnRNP A2 antibody (Figure 3D). The same input levels of hnRNP A2 were used for immunoprecipitation from both cell types. The immunoprecipitate did not contain significant YY-1 transcription factor, which does not respond to mitochondrial respiratory stress or bind to stress-responsive promoters. Coimmunoprecipitation of control extracts with hnRNP A2 antibody immunoprecipitated considerably lower levels of C/EBPδ, cRel, and CREB than stressed cell extracts (Figure 3D). A reciprocal coimmunoprecipitation using antibodies against C/EBPδ, cRel, p50, and CREB also immunoprecipitated hnRNP A2 (Figure 3D), confirming the associations. Lower levels of hnRNP A2 were immunoprecipitated from control cell nuclear extract, confirming a reduced interaction of hnRNP A2 with DNA-binding proteins, and/or reduced nuclear levels of hnRNP A2 in these cells.
Transcriptional Activation of Cathepsin L, RyR1, and Glut4 Promoters by Mitochondrial Stress-responsive Nuclear Factors
We investigated the transcriptional activation of the Cathepsin L (sequence −273 to +47), RyR1 (sequence −205 to +63), and Glut4 (sequence −1209 to −168) promoters in cells transfected with cDNAs for cRel, C/EBPδ, and CREB (Figure 4). Luciferase activity driven by the Cathepsin L promoter was 1.8-, 2.5-, 1.5-, and 1.7-fold higher over basal levels after cRel, C/EBPδ, CREB, and hnRNP A2 transfections, respectively (Figure 4A). Transfection of control cells also modestly increased luciferase activity. Cotransfections of hnRNP A2 cDNA with either cRel or C/EBPδ synergistically enhanced luciferase activity, suggesting a cooperativity between these proteins. The cooperative increase in promoter activity is specific for hnRNP A2, because hnRNP D-L cDNA failed to show such an effect (Figure 4A). In contrast to the marked increase in activity observed with hnRNP A2, a combination of hnRNP D-L and cRel marginally increased the activity in control cells, whereas the activities in mtDNA-depleted cells were marginally repressed. Repeat experiments using luciferase expression driven by the defined RyR1 and Glut4 promoters gave similar results (Figure 4, B and C). Although not shown, hnRNP D-L did not either by itself or in combination with other factors yield an increase in promoter activity.
Figure 4.

Transcriptional activation of Cathepsin L, RyR1, and Glut4 promoters by various factors. Promoter DNA constructs (1.0 μg each) cloned in pGL3 vector were cotransfected with or without 0.2 μg of cRel, CREB, C/EBPδ, NFAT, hnRNP A2, and hnRNP D-L cDNAs into control and mtDNA-depleted cells. A plasmid containing Renilla luciferase (0.5 μg) was used as an internal control for normalization of transfection efficiency between samples. Promoter activity of the Cathepsin L (A), RyR1 promoter (B), and Glut4 promoters (C). Data represent the mean ± SD of three independent experiments. **p < 0.001, represents highly significant difference between control and mtDNA-depleted cells and also in cells cotransfected with hnRNP A2 cDNA. *p < 0.05, represents significant difference.
Reversal of Stress-induced Invasive Behavior, Glucose Uptake, and Gene Expression by hnRNP A2 Silencing
Previous studies have shown that cells subjected to mitochondrial stress exhibit higher growth rates (Amuthan et al., 2001; Guha et al., 2007). To determine whether hnRNP A2 is necessary for mitochondrial stress-induced growth, we first generated mtDNA-depleted cell lines stably expressing siRNA targeted to hnRNP A2 mRNA (Figure 5A). This decreased the protein and mRNA level of hnRNP A2 by 70% (Figure 5A) without any change in levels of other transcription factors (data not shown). Equal numbers of cells (1 × 104) were seeded and their cell viability was assessed at 24-h intervals. At 24 h after seeding, mtDNA-depleted cells had growth rates comparable with control and mtDNA-depleted/hnRNP A2-silenced cells. However, by 48 h the mtDNA-depleted cells had expanded significantly more than the mtDNA-depleted/hnRNP A2-silenced cells, which had viable cell numbers comparable with control C2C12 cells (Figure 5B).
Figure 5.

Reversal of tumorigenic phenotype by hnRNP A2 silencing. (A) Levels of hnRNP A2 mRNA by real time RT-PCR and protein levels by anti-hnRNP A2 immunoblot. (B) Growth rate of control and mtDNA-depleted cells expressing scrambled RNA (mock si) or hnRNP A2 siRNA. (C) Matrigel invasion patterns of control, mtDNA-depleted/mock siRNA, and mtDNA-depleted/hnRNP A2 siRNA cells. (D) Glucose uptake by control, mtDNA-depleted, and mtDNA-depleted/hnRNP A2-silenced cells. Data represent the mean ± SD of three independent experiments. **p < 0.001, represents highly significant difference.
Another important characteristic of mtDNA-depleted cells is increased in vitro invasiveness and increased tumorigenicity (Amuthan et al., 2001, 2002). Because hnRNP A2 plays a role in the activation of the stress-specific marker genes Cathepsin L, RyR1, and Glut4, we evaluated the role of hnRNP A2 in cell invasiveness by using a Matrigel invasion chamber. Figure 5C shows the number of cells that invaded through the Matrigel membrane after 24 h of incubation. Consistent with previous results, mtDNA-depleted cells expressing a mock siRNA showed a higher level of invasion than control cells (Figure 5C). The mtDNA-depleted/hnRNP A2-silenced cells had considerably impaired invasiveness (Figure 5C). The observed decrease in invasiveness of hnRNP A2-silenced cells was not due to a difference in growth rates because the assay was performed after 24 h of growth (Figure 5B). These results suggest that hnRNP A2 plays an important role in the induction of cell growth and invasiveness stimulated by mitochondrial stress. We have shown previously that the increased invasiveness of mtDNA-depleted cells is associated with activation of IGF1R pathway and increased glucose uptake. Figure 5D shows that siRNA-mediated knockdown of hnRNP A2 reduced glucose uptake to near control cell levels, suggesting that hnRNP A2 protein also plays an important role in the cellular metabolic shift triggered by mitochondrial respiratory stress.
To understand the specificity of the effects of hnRNP A2 silencing in mtDNA-depleted cells, we carried out control experiments in which the effects of mRNA silencing in control cells was investigated. Figure 6A shows that siRNA-mediated hnRNP A2 mRNA silencing did not have any significant effects on the growth of control C2C12 cells at 24 and 48 h of growth. However, a marked effect was observed in mtDNA-depleted cells. Although not shown, analysis of cells by annexin staining did not show any measurable increase in cell death both in control and mtDNA-depleted cells. Consistent with the results of promoter pull-down and cotransfection studies (Figures 2 and 4), promoter analysis using Cathepsin L, Glut4, and RyR1 promoter constructs showed only marginal effects of hnRNP A2 silencing in control cells, whereas marked effects were seen in mtDNA-depleted cells (Figure 6B). These results suggest that the effects of hnRNP A2 silencing on invasiveness and other phenotypic parameters are due to selective effects on transcription regulation of stress target genes rather than direct effects on cell growth and survival parameters.
Figure 6.
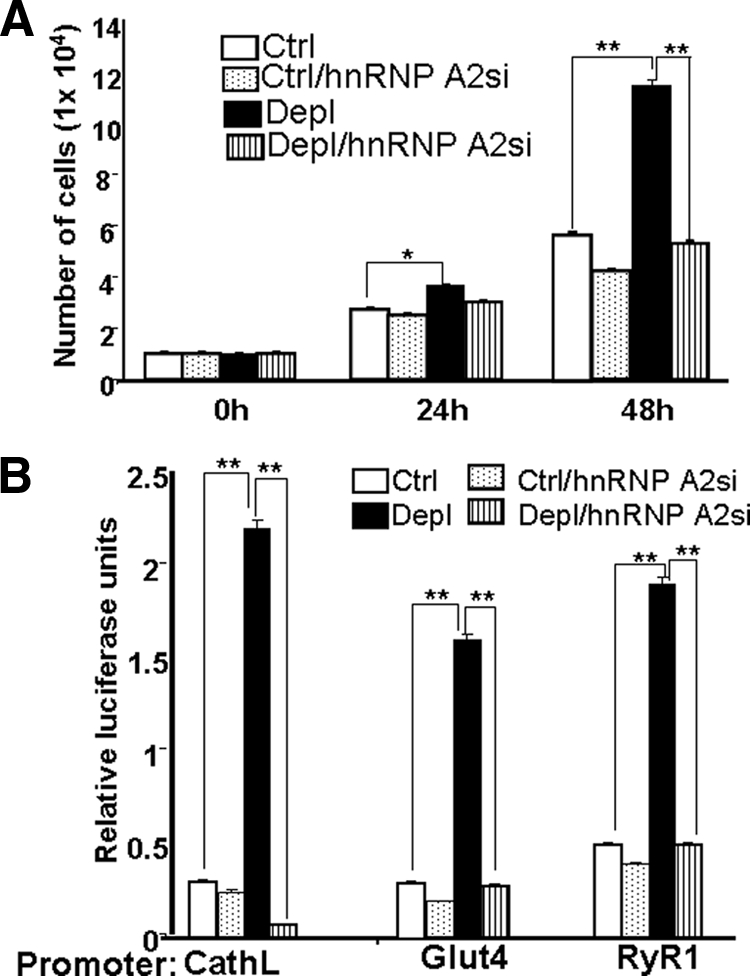
Effects of hnRNPA2 silencing on growth rate and promoter activity in control C2C12 cells. (A) HnRNPA2 mRNA levels were silenced in control and mtDNA-depleted cells by transfection with siRNA expressing plasmid. Cell numbers were measured after 24 and 48 h of growth. (B) The activities for the Cathepsin L, Glut4, and RyR1 promoter constructs in control, mtDNA-depleted and hnRNP A2 mRNA-silenced cells were measured as described in Materials and Methods. Luciferase activities are normalized using Renilla luciferase as an internal control for transfection efficiency. Data represent the mean ± SD of three independent experiments. **p < 0.001, represents highly significant difference. p < 0.05, represents significant difference.
Finally, we measured the production of mRNA encoding several mitochondrial stress responsive genes in hnRNP A2-depleted cells. These marker genes were shown previously to be down-regulated by silencing calcineurin (Cn) A-α or inhibitor of nuclear factor-κB (IκB) β in mtDNA-depleted C2C12 cells (Biswas et al., 2003, 2008b). These two proteins are important upstream factors of this pathway. Using real-time PCR, we detected higher RyR1 (7.3-fold), TGFβ (2.0 fold), Cathepsin L (7.4-fold), IGF1R (9.8-fold), and Glut4 (4.2-fold) mRNA levels in mtDNA-depleted cells than in control cells (Figure 7A). The increase in mRNA and protein levels were effectively reduced by hnRNP A2 mRNA silencing (Figure 7, A and B). The two antibody reactive bands for Glut4 (Figure 7B) probably represent the glycosylated and unglycosylated forms. Figure 7C shows that the alternate approach of stress induction by CCCP treatment also induced the expression of hnRNP A2, Cathepsin L, IGF1R, and Glut4 genes, which was significantly reversed by transient expression of siRNA for hnRNP A2. Both the levels of induction and reversal by hnRNP A2 silencing were marginal for RyR1 and TGFβ mRNAs. The reason for the difference in the levels of mRNA induction by CCCP treatment versus mtDNA depletion remains unclear. Although not shown, transient transfection with siRNA for 10 h caused ∼60% reduction in hnRNP A2 levels. Consistent with the reduced mRNA levels, the transcriptional activities of the Cathepsin L, Glut4, and RyR1 promoters were also reduced to control cell levels in response to hnRNP A2 silencing (Figure 7D). These results confirm that hnRNP A2 plays direct roles in the transcriptional programming as well as the phenotypic changes that occur in cells experiencing mitochondrial respiratory stress.
Figure 7.

Reversal of gene expression and transcriptional activity of target gene promoters by hnRNP A2 silencing. (A) Relative mRNA levels for stress-responsive genes in control, mtDNA-depleted/mock si, and mtDNA-depleted/hnRNP A2 si cells measured by real-time PCR. The mouse β-actin gene was used as an endogenous control. Data shown are the mean ± SD of triplicate experiments. (B) Immunoblot analysis of post-mitochondrial supernatants (30 μg of protein each) of control C2C12, mtDNA-depleted/mock si, and mtDNA-depleted/hnRNP A2si cells. β-Actin is used as loading control. (C) Relative levels of indicated mRNAs in control C2C12 or mtDNA-depleted C2C12 cells transfected with mock si vector or hnRNPA2 siRNA vector with or without treatment with CCCP. mRNA levels were measured by real-time PCR using β-actin mRNA as an internal control. Control C2C12 cells were transiently transfected with either pSilencer2.1 neo vector or with the vector containing hnRNP A2 siRNA for 24 h and treated with CCCP (25 μM; 10 h). (D) Transcriptional activities measured as luciferase units of Cathepsin L, RYR1, and Glut4 promoters in control, mtDNA-depleted/mock si, and mtDNA-depleted/hnRNP A2 si cells. Luciferase activities are normalized using Renilla luciferase as an internal control for transfection efficiency. Data represent the mean ± SD of three independent experiments. **p < 0.001, represents highly significant difference. *p < 0.05, represents significant difference.
Role of hnRNP A2 in Enhanceosome Formation and Integration of Respiratory Stress Signaling
Transcriptional analysis in Figure 8A shows that Cathepsin L promoter activity was two- to threefold higher in mtDNA-depleted cells than in control cells and was reduced to control cell levels by silencing of hnRNP A2 mRNA. Results also show that cotransfection with cRel and C/EBPδ induced Cathepsin L promoter activity by two- to threefold in mtDNA-depleted cells but not in hnRNP A2-silenced cells. A similar transcriptional activation in mtDNA-depleted cells and reversal in hnRNP A2 mRNA-silenced cells was observed for the RyR1 promoter (Supplemental Figure S3A). These results suggest a possible role for hnRNP A2 in coordinating the activities of mitochondrial stress induced DNA binding signature factors.
Figure 8.

Effects of hnRNP A2 silencing on transcriptional activity and occupancy of Cathepsin L promoter sites by DNA binding factors. (A) Transcriptional activities of the Cathepsin L promoter after cotransfection with cRel and C/EBPδ cDNAs in control, mtDNA-depleted/mock si, and mtDNA-depleted/hnRNP A2 si cells. (B). ChIP analysis of the Cathepsin L and β-Actin promoters in control, mtDNA-depleted and reverted cells by using hnRNP A2 antibody. (C) ChIP analysis of the Cathepsin L promoter with hnRNP A2, hnRNP d-L, C/EBPδ, cRel, and CREB antibodies in the indicated cells. Preimmune serum was used as a negative antibody control. Real-time PCR analysis using 10% input DNA for normalization. Values are expressed as -folds relative to the factor binding in control cells. (D) ChIP analysis of the Cathepsin L promoter with hnRNP A2, C/EBPδ, cRel, and CREB antibodies in control, mtDNA-depleted, and mtDNA-depleted cells transfected with cRel, C/EBPδ, and CREB siRNAs. Values are expressed as -folds relative to the factor binding in control cells. Preimmune serum was used as a negative control. Data represent the mean ± SD of three independent experiments. **p < 0.001, represents highly significant difference. *p < 0.05, represents significant difference.
To ascertain the in vivo association of stress-activated transcription factors and hnRNP A2 at the target promoters, we performed ChIP assays in hnRNP A2-silenced cells. Figure 8B shows a control experiment comparing the level of hnRNP A2 antibody pull-down of stress-responsive Cathepsin L promoter and a nonmitochondrial stress responsive Actin promoter in control, mtDNA-depleted, and reverted cells. The reverted cells showed a pattern closer to the control cells, suggesting that observed increase in promoter occupancy of hnRNP A2 is directly in response to mtDNA depletion. ChIP analysis in Figure 8C shows an increased association of cRel, C/EBPδ, CREB, and hnRNP A2 with the Cathepsin L (Figure 8B) promoter in mtDNA-depleted cells. In contrast, hnRNP A2 silencing in mtDNA-depleted cells resulted in marked and uniform reduction in factor binding to the Cathepsin L promoter (Figure 8B). Consistent with the results of promoter analysis in Figure 4, hnRNP D-L was not associated significantly with the promoter, further confirming the specificity of hnRNP A2 association with the enhanceosome. A similar increase in the occupancy of promoter DNA by signature factors in mtDNA-depleted C2C12 cells and reversal by hnRNP A2 mRNA silencing was observed with the RyR1 promoter (Supplemental Figure S3B). These results suggest that hnRNP A2 acts as an adaptor protein that bridges cRel, C/EBPδ, and CREB to the transcription complex. Silencing of hnRNP A2 probably impairs transcription by disrupting this association.
The cooperative nature of factor binding to the enhanceosome and the roles of individual DNA binding signature factors in the recruitment of hnRNP A2 were investigated by ChIP analysis of cells in which individual factors were silenced with specific siRNAs (Figure 8D and Supplemental S3C). The cRel silencing had a marked effect on the association of both C/EBPδ and CREB with the Cathepsin L and RyR1 promoters (Figure 8D and Supplemental S3C). C/EBPδ silencing also had a pronounced effect on association of all three proteins with the Cathepsin L promoter (Figure 8D). CREB silencing also affected the association of cRel in a major way. However, promoter occupancy of hnRNP A2 and C/EBPδ were affected only marginally. (Figure 8D). Thus, the three DNA binding signature factors exhibit varying levels of cooperativity for association with the enhanceosome. Furthermore, the promoter occupancy of C/EBPδ and cRel is critical for hnRNP A2 recruitment to the enhanceosome. A similar pattern of occupancy of RyR1 promoter was observed in mtDNA-depleted and for cRel and C/EBPδ mRNA-silenced cells (Supplemental Figure S3C). However, CREB mRNA silencing had a more profound effect on RyR1 promoter occupancy by other factors (Supplemental Figure S3C) compared with the Cathepsin L promoter. These results suggest significant interindividual variations in their response to different target genes. Although not shown, silencing of the other signature factors p50 and NFAT also cooperated for association of the enhanceosome with the Cathepsin L and RyR1 promoters.
Mapping of The Transcriptional Activation Domain and Protein–Protein Interaction Domains of hnRNP A2
The hnRNP A2 protein has two distinct RNA binding domains (RBD) I and II and a C-terminal glycine-rich domain. We next investigated whether hnRNP A2 contains a transcriptional activation domain and whether this domain, or any other domains, is important for transcriptional synergy. For this purpose, we generated various deletions of hnRNP A2 and fused them to the C-terminal end of the yeast Gal4 DNA binding domain (1-147) in the pBIND mammalian expression vector (Figure 9A) (Yang et al., 2006). Gal4-fusion constructs were cotransfected with a pG5luc synthetic promoter-reporter construct, which reports the transcriptional activation activity of proteins fused to the C terminus of the Gal4 DNA binding domain. The C-terminal end of the glycine-rich domain (240-342 aa) induced a 10-fold greater luciferase activity than the vector control and a twofold higher activity than the full-length hnRNP A2 (1-342aa) fusion (Figure 9A). Fusion of the Gal4-DBD with the glycine-rich region (178-342 aa), RBD I and II (1-180 aa), or RBD II (90-242 aa) produced a lower reporter activity (∼5–20% activity) than the full-length construct. This suggests that hnRNP A2 has its own transcriptional activation domain within the C-terminal Gly-rich region.
Figure 9.

Transcriptional activation and protein-interacting domains of hnRNP A2. (A) Model of hnRNP A2 domain constructs fused to Gal4 protein (left). Luciferase activity of the pG5luc promoter (right). (B) Relative transcriptional activity of the Cathepsin L promoter cotransfected with the Gal4 fusion constructs in mtDNA-depleted cells. (C) ChIP analysis of the Cathepsin L promoter by using cells transfected with Gal4-hnRNP A2 fusion constructs and gal4 antibody. Data shown in A–C are the mean ± SD of at least three independent experiments. **p < 0.001, represents highly significant difference.
To understand the functional role of hnRNP A2 in stress-mediated transcriptional activation of target genes, we tested the same Gal4 fusion constructs for transcriptional activation of the Cathepsin L and RyR1 promoters in which the transcription is driven by the four signature factor binding sites (Figure 9B). In cotransfection studies, the RBD (1-180 aa) construct yielded maximum transcriptional activation of the Cathepsin L promoter (Figure 9B) and the RyR1 promoter (data not shown). The RBD II domain (90-242 aa) yielded marginally lower activity. In contrast, the C-terminal glycine-rich domains (178-342 aa and 240-342 aa) yielded progressively decreasing activities. These results raise the possibility that the RBD I and RBD II, which contain extensive helical structures characteristic of protein–protein interacting domains, are critical for transcription activation of stress response genes.
The association of Gal4-hnRNP A2 fusions with the promoter region of the Cathepsin L gene in vivo was further investigated by ChIP analysis in mtDNA-depleted cells transfected with different fusion constructs. As shown in Figure 9C, the gal4 vector alone showed very low association with the Cathepsin L promoter. The RBD domain fusion (1-180 aa), which interacted very efficiently with cRel, showed the highest level of association, followed by the full-length fusion. These results confirm that the RBD I and II domains play important roles in coordinating the transcriptional activities by regulating protein-protein interactions.
DISCUSSION
Retrograde intergenomic signaling, also known as mitochondrial respiratory stress signaling, regulates glucose uptake/use, fatty acid metabolism, mtDNA maintenance, aging, cancer progression, and neurodegeneration in yeast and metazoan cells. Consistent with the complexity of these functions, genes coding for proteins with diverse functions and cellular destinations have been reported to be regulated by mitochondrial respiratory stress. The nuclear target genes for mitochondrial stress signaling vary depending on cell type and the nature of the mitochondrial mutation/dysfunction causing the stress signaling (Amuthan et al., 2001; Delsite et al., 2002; Crimi et al., 2005; Jahangir et al., 2005; Biswas et al., 2008a; Moro et al., 2009). In C2C12 cells, we and others have observed that mitochondrial stress modulates the expression of ∼120 genes that are associated with wide-ranging cellular functions, including Ca2+ homeostasis, glucose uptake, glycolysis, cytoskeletal organization, insulin/insulin-like growth factor 1 signaling, apoptosis, and cell proliferation (Amuthan et al., 2001, 2002; Guha et al., 2007; Biswas et al., 2008a,b). In this study, we demonstrate that the stress signaling cascade culminates in the transcriptional upregulation of target gene expression using hnRNP A2 as a coactivator.
The use of multiple approaches demonstrated that stress-activated transcriptional up-regulation involves four common sequence specific DNA binding factors NFκB (cRel:p50), C/EBPδ, CREB, and NFAT. The DNA binding consensus sequences for these factors are located in proximity to each other within a region immediately 5′ of the transcription start site of mitochondrial stress response-targeted genes. An important finding of this study was that the RNA binding factor hnRNP A2 is part of this enhanceosome. Our results support a model whereby hnRNP A2 plays a critical role in the functional cooperativity of DNA binding signature factors, and they demonstrate that hnRNP A2 functions as a novel transcriptional coactivator of the mitochondrial respiratory stress signaling pathway.
Although hnRNP A2 was found to be a part of the enhanceosome complex, and induced functional cooperativity with the various DNA binding signature factors, both in vitro-translated and recombinant hnRNP A2 did not bind directly to the RyR1 or Cathepsin L promoter DNA (data not shown). In addition, the C-terminal Gly-rich region of hnRNP A2, which showed maximal transcriptional activation in a synthetic promoter system, failed to support stress-mediated transcriptional activation of the Cathepsin L and RyR1 promoters. Instead, the N-terminal RNA binding domains (RBD I and RBD II) of hnRNP A2 were the critical mediators of hnRNP A2 transcriptional activation. Based on these results, we propose that hnRNP A2 directly recruits signature factors to the enhanceosome, or promotes the assembly of the enhanceosome, by providing sites for binding to critical enhanceosome factors. The role of hnRNP A2 in transcription activation of stress target genes and association with the enhanceosomes of these promoters seems highly selective because hnRNP d-L, another member of this family, failed to induce promoter activity in cotransfection experiments and associate with the enhanceosome of stress-responsive genes as tested by ChIP analysis.
hnRNP A2 belongs to a large family of RNA-binding proteins involved in various aspects of mRNA biogenesis (Dreyfuss et al., 1993; Shyu and Wilkinson, 2000). The ability of hnRNP A2 to shuttle between the nucleus and cytoplasm is required for mRNA processing (Nichols et al., 2000). Increased nuclear translocation of this protein has been reported in cells undergoing transformation in various tumors (Lee et al., 2005). hnRNP A2 is a component of this respiratory stress pathway that induces highly invasive phenotypes (Amuthan et al., 2001, 2002; Guha et al., 2007; Biswas et al., 2008a). Our results show that induction and increased nuclear accumulation of hnRNP A2 are common features of respiratory stress signaling because respiratory stress induced by CCCP also caused marked increases in nuclear hnRNP A2 in 3T3 fibroblasts, A549 lung carcinoma cells, RAW264.7 macrophages, and H9C2 cardiomyocytes (Figure 4A).
In previous studies, we showed that cytosolic Cn and IκBβ are two important mediators of mitochondrial stress signaling (Biswas et al., 2003, 2008b). We and others have also shown that nuclear C/EBPδ, CREB, and NFAT are also activated in response to respiratory stress (Amuthan et al., 2001; Butow and Avadhani, 2004). Selective knockdown of Cn Aα and IκBβ abolished the signaling, and caused a reversal of the metabolic properties and invasive behavior in mtDNA-depleted C2C12 cells (Biswas et al., 2003, 2008b). Here, we show that hnRNP A2 is a critical transcriptional coactivator of stress responsive genes, suggesting that it represents another critical mediator in the respiratory stress response pathway. Activation of hnRNP A2 in response to stress is not restricted to C2C12 cells because CCCP treatment induced the nuclear levels of hnRNP A2 in murine 3T3, RAW264.7, and also H9C2 cells.
There is mounting evidence implicating hnRNP A2 in a variety of cancers, including lung, breast, and pancreatic cancer (Zhou et al., 1996; Fielding et al., 1999; Garayoa et al., 2003; Patry et al., 2003; He et al., 2005; Yamaoka et al., 2006). hnRNP A2 has been found to associate with many oncogenic proteins (TDP-43, TOG2, and SET) in tumor cells (Buratti et al., 2005; Kosturko et al., 2005; Vera et al., 2006), although the functional relevance of these interactions have remained unclear until now. Our results suggest that hnRNP A2 functions as a transcriptional coactivator by associating with enhanceosomes through protein–protein interactions with its N-terminal RNA binding domains. In support of our model, a recent study showed that another member of the hnRNP protein family, hnRNP K, also functions as a transcriptional activator (Moumen et al., 2005; Chan et al., 2009; Yuan et al., 2009). However, hnRNP A2 functions in a distinctly different manner than hnRNP K, which has DNA binding properties (Tomonaga and Levens, 1995).
hnRNP A2 has been suggested to function as a tetrameric nuclear protein (Kosturko et al., 2005; Carson et al., 2006). We find that a fusion protein lacking the C-terminal putative transactivation domain is fully functional as a coactivator, suggesting that the main function of hnRNP A2 is to promote enhanceosome assembly or provide interaction sites for signature transcription factors. Our results also suggest that different domains of hnRNP A2 selectively bind to different signature factors (unpublished data). Based on this, we propose a model (Figure 10) for the respiratory stress-induced transcription activation of nuclear target genes requiring hnRNP A2 as a novel coactivator. We propose that hnRNP A2 plays a critical role in the physical association and functional synergy between NF-κB (cRel:p50), C/EBPδ, and CREB that enhances transcription of mitochondrial respiratory stress nuclear gene targets that regulate cell growth and invasion.
Figure 10.

A proposed model for the mechanism of mitochondrial respiratory stress-induced activation of nuclear target genes involving hnRNP A2 as a coactivator. Mitochondrial stress activates calcium-mediated calcineurin, which results in activation of cRel:p50, C/EBPδ, CREB, and NFAT. HnRNP A2 is activated in response to this stress signaling and is recruited to the transcription complex, which is essential for downstream activation of the stress target genes.
Recently, a family of autosomal recessive disorders have been described that are associated with mtDNA depletion (called mtDNA depletion syndrome [MDS]) either due to mutations in genes involved in DNA replication or nucleotide metabolism/transport (Sarzi et al., 2007; Copeland, 2008; Poulton and Holt, 2009). MDS patients exhibit a variety of severe clinical symptoms including myopathy, neuropathy, CNS disorders, hepatopathy, and progressive external ophthalmoplegia. Fibroblasts from human patients with MDS, showed up to 80% mtDNA depletion, and/or deletions (Taanman et al., 2009). We propose that tissues/cells from MDS patients have activated mitochondrial stress signaling accompanied with the biochemical and physiological changes similar to the mtDNA-depleted cell system used in this study.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Gideon Dreyfuss (University of Pennsylvania, Philadelphia, PA) for providing hnRNP A2 and hnRNP D cDNAs and antibodies, Dr. Graeme I. Bell (University of Chicago, Chicago, IL) for the Glut4 promoter construct, Dr. Michael Greenberg (Children's Hospital, Boston, MA) for the CREB cDNA, Dr. Anjana Rao (Harvard Medical School, Boston, MA) for NFAT cDNA, Dr. Shankar Ghosh (Yale School of Medicine, New Haven, CT) for cRel and p50 cDNAs, and Dr. Michael Atchison (University of Pennsylvania, Philadelphia, PA) for C/EBPδ cDNA. This research was supported by National Institutes of Health grant CA-22762.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-04-0296) on July 29, 2009.
REFERENCES
- Amuthan G., Biswas G., Ananadatheerthavarada H. K., Vijayasarathy C., Shephard H. M., Avadhani N. G. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene. 2002;21:7839–7849. doi: 10.1038/sj.onc.1205983. [DOI] [PubMed] [Google Scholar]
- Amuthan G., Biswas G., Zhang S. Y., Klein-Szanto A., Vijayasarathy C., Avadhani N. G. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 2001;20:1910–1920. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnould T., Vankoningsloo S., Renard P., Houbion A., Ninane N., Demazy C., Remacle J., Raes M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 2002;21:53–63. doi: 10.1093/emboj/21.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G., Adebanjo O. A., Freedman B. D., Anandatheerthavarada H. K., Vijayasarathy C., Zaidi M., Kotlikoff M., Avadhani N. G. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 1999;18:522–533. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G., Anandatheerthavarada H. K., Zaidi M., Avadhani N. G. Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J. Cell Biol. 2003;161:507–519. doi: 10.1083/jcb.200211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G., Guha M., Avadhani N. G. Mitochondria-to-nucleus stress signaling in mammalian cells: nature of nuclear gene targets, transcription regulation, and induced resistance to apoptosis. Gene. 2005;354:132–139. doi: 10.1016/j.gene.2005.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G., Srinivasan S., Anandatheerthavarada H. K., Avadhani N. G. Dioxin-mediated tumor progression through activation of mitochondria-to-nucleus stress signaling. Proc. Natl. Acad. Sci. USA. 2008a;105:186–191. doi: 10.1073/pnas.0706183104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G., Tang W., Sondheimer N., Guha M., Bansal S., Avadhani N. G. A distinctive physiological role for Ikappa Bbeta in the propagation of mitochondrial respiratory stress signaling. J. Biol. Chem. 2008b;283:12586–12594. doi: 10.1074/jbc.M710481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonawitz N. D., Chatenay-Lapointe M., Pan Y., Shadel G. S. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007;5:265–277. doi: 10.1016/j.cmet.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E., Brindisi A., Giombi M., Tisminetzky S., Ayala Y. M., Baralle F. E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005;280:37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- Butow R. A., Avadhani N. G. Mitochondrial signaling: the retrograde response. Mol. Cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- Carson J. H., Blondin N., Korza G. Rules of engagement promote polarity in RNA trafficking. BMC Neurosci. 2006;7(suppl 1):S3. doi: 10.1186/1471-2202-7-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J. Y., Hsieh T. Y., Liu S. T., Chou W. Y., Chung M. H., Huang S. M. Physical and functional interactions between hnRNP K and PRMT family proteins. FEBS Lett. 2009;583:281–286. doi: 10.1016/j.febslet.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Copeland W. C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008;59:131–146. doi: 10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimi M., Bordoni A., Menozzi G., Riva L., Fortunato F., Galbiati S., Del B. R., Pozzoli U., Bresolin N., Comi G. P. Skeletal muscle gene expression profiling in mitochondrial disorders. FASEB J. 2005;19:866–868. doi: 10.1096/fj.04-3045fje. [DOI] [PubMed] [Google Scholar]
- Delsite R., Kachhap S., Anbazhagan R., Gabrielson E., Singh K. K. Nuclear genes involved in mitochondria-to-nucleus communication in breast cancer cells. Mol. Cancer. 2002;1:6. doi: 10.1186/1476-4598-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey R., Moraes C. T. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-x(L) in osteosarcoma cells. J. Biol. Chem. 2000;275:7087–7094. doi: 10.1074/jbc.275.10.7087. [DOI] [PubMed] [Google Scholar]
- Dignam J. D., Lebovitz R. M., Roeder R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G., Matunis M. J., Pinol-Roma S., Burd C. G. hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- Fang J. K., Prabu S. K., Sepuri N. B., Raza H., Anandatheerthavarada H. K., Galati D., Spear J., Avadhani N. G. Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett. 2007;581:1302–1310. doi: 10.1016/j.febslet.2007.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding P., Turnbull L., Prime W., Walshaw M., Field J. K. Heterogeneous nuclear ribonucleoprotein A2/B1 up-regulation in bronchial lavage specimens: a clinical marker of early lung cancer detection. Clin. Cancer Res. 1999;5:4048–4052. [PubMed] [Google Scholar]
- Garayoa M., Man Y. G., Martínez A., Cuttitta F., Mulshine J. L. Downregulation of hnRNP A2/B1 expression in tumor cells under prolonged hypoxia. Am. J. Respir. Cell. Mol. Biol. 2003;28:80–85. doi: 10.1165/rcmb.4880. [DOI] [PubMed] [Google Scholar]
- Guha M., Srinivasan S., Biswas G., Avadhani N. G. Activation of a novel calcineurin-mediated insulin-like growth factor-1 receptor pathway, altered metabolism, and tumor cell invasion in cells subjected to mitochondrial respiratory stress. J. Biol. Chem. 2007;282:14536–14546. doi: 10.1074/jbc.M611693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Brown M. A., Rothnagel J. A., Saunders N. A., Smith R. Roles of heterogeneous nuclear ribonucleoproteins A and B in cell proliferation. J. Cell Sci. 2005;118:3173–3183. doi: 10.1242/jcs.02448. [DOI] [PubMed] [Google Scholar]
- Jahangir Tafrechi R. S., Svensson P. J., Janssen G. M., Szuhai K., Maassen J. A., Raap A. K. Distinct nuclear gene expression profiles in cells with mtDNA depletion and homoplasmic A3243G mutation. Mutat. Res. 2005;578:43–52. doi: 10.1016/j.mrfmmm.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Jia Y., Rothermel B., Thornton J., Butow R. A. A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol. Cell. Biol. 1997;17:1110–1117. doi: 10.1128/mcb.17.3.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadonaga J. T., Tjian R. Affinity purification of sequence-specific DNA binding proteins. Proc. Natl. Acad. Sci. USA. 1986;83:5889–5893. doi: 10.1073/pnas.83.16.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly D. P., Scarpulla R. C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- Kosturko L. D., Maggipinto M. J., D'Sa C., Carson J. H., Barbarese E. The microtubule-associated protein tumor overexpressed gene binds to the RNA trafficking protein heterogeneous nuclear ribonucleoprotein A2. Mol. Biol. Cell. 2005;16:1938–1947. doi: 10.1091/mbc.E04-08-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. H., et al. Identification of the heterogeneous nuclear ribonucleoprotein A2/B1 as the antigen for the gastrointestinal cancer specific monoclonal antibody MG7. Proteomics. 2005;5:1160–1166. doi: 10.1002/pmic.200401159. [DOI] [PubMed] [Google Scholar]
- Liao X., Butow R. A. RTG1 and RTG 2, two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell. 1993;72:61–71. doi: 10.1016/0092-8674(93)90050-z. [DOI] [PubMed] [Google Scholar]
- Liao X. S., Small W. C., Srere P. A., Butow R. A. Intramitochondrial functions regulate nonmitochondrial citrate synthase (CIT2) expression in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991;11:38–46. doi: 10.1128/mcb.11.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. T., Beal M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Liu M. L., Olson A. L., Moye-Rowley W. S., Buse J. B., Bell G. I., Pessin J. E. Expression and regulation of the human GLUT4/muscle-fat facilitative glucose transporter gene in transgenic mice. J. Biol. Chem. 1992;267:11673–11676. [PubMed] [Google Scholar]
- Liu Z., Butow R. A. Mitochondrial retrograde signaling. Annu. Rev. Genet. 2006;40:159–185. doi: 10.1146/annurev.genet.40.110405.090613. [DOI] [PubMed] [Google Scholar]
- Liu Z., Sekito T., Epstein C. B., Butow R. A. RTG-dependent mitochondria to nucleus signaling is negatively regulated by the seven WD-repeat protein Lst8p. EMBO J. 2001;20:7209–7219. doi: 10.1093/emboj/20.24.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusich M. F., Robinson B. H., Taanman J. W., Kim S. J., Schillace R., Smith J. L., Capaldi R. A. Expression of mtDNA and nDNA encoded respiratory chain proteins in chemically and genetically-derived Rho0 human fibroblasts: a comparison of subunit proteins in normal fibroblasts treated with ethidium bromide and fibroblasts from a patient with mtDNA depletion syndrome. Biochim. Biophys. Acta. 1997;1362:145–159. doi: 10.1016/s0925-4439(97)00061-6. [DOI] [PubMed] [Google Scholar]
- Masternak K., Muhlethaler-Mottet A., Villard J., Zufferey M., Steimle V., Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14:1156–1166. [PMC free article] [PubMed] [Google Scholar]
- Moran-Jones K., Wayman L., Kennedy D. D., Reddel R. R., Sara S., Snee M. J., Smith R. hnRNP A2, a potential ssDNA/RNA molecular adapter at the telomere. Nucleic Acids Res. 2005;33:486–496. doi: 10.1093/nar/gki203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro L., Arbini A. A., Yao J. L., di Sant'Agnese P. A., Marra E., Greco M. Mitochondrial DNA depletion in prostate epithelial cells promotes anoikis resistance and invasion through activation of PI3K/Akt2. Cell Death Differ. 2009;16:571–583. doi: 10.1038/cdd.2008.178. [DOI] [PubMed] [Google Scholar]
- Moumen A., Masterson P., O'Connor M. J., Jackson S. P. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005;123:1065–1078. doi: 10.1016/j.cell.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Nichols R. C., Wang X. W., Tang J., Hamilton B. J., High F. A., Herschman H. R., Rigby W. F. The RGG domain in hnRNP A2 affects subcellular localization. Exp. Cell Res. 2000;256:522–532. doi: 10.1006/excr.2000.4827. [DOI] [PubMed] [Google Scholar]
- Ohta S. Contribution of somatic mutations in the mitochondrial genome to the development of cancer and tolerance against anticancer drugs. Oncogene. 2006;25:4768–4776. doi: 10.1038/sj.onc.1209602. [DOI] [PubMed] [Google Scholar]
- Patry C., Bouchard L., Labrecque P., Gendron D., Lemieux B., Toutant J., Lapointe E., Wellinger R., Chabot B. Small interfering RNA-mediated reduction in heterogeneous nuclear ribonucleoparticule A1/A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res. 2003;63:7679–7688. [PubMed] [Google Scholar]
- Pelicano H., et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 2006;175:913–923. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros J. A., et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton J., Holt I. J. 163rd ENMC International Workshop: nucleoid and nucleotide biology in syndromes of mitochondrial DNA depletion myopathy 12–14 December 2008, Naarden, The Netherlands. Neuromuscul. Disord. 2009;19:439–443. doi: 10.1016/j.nmd.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Poyton R. O., McEwen J. E. Crosstalk between nuclear and mitochondrial genomes. Annu. Rev. Biochem. 1996;65:563–607. doi: 10.1146/annurev.bi.65.070196.003023. [DOI] [PubMed] [Google Scholar]
- Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Sarzi E., Goffart S., Serre V., Chrétien D., Slama A., Munnich A., Spelbrink J. N., Rötig A. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann. Neurol. 2007;62:579–587. doi: 10.1002/ana.21207. [DOI] [PubMed] [Google Scholar]
- Sciacco M., Bonilla E., Schon E. A., DiMauro S., Moraes C. T. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum. Mol. Genet. 1994;3:13–19. doi: 10.1093/hmg/3.1.13. [DOI] [PubMed] [Google Scholar]
- Shidara Y., Yamagata K., Kanamori T., Nakano K., Kwong J. Q., Manfredi G., Oda H., Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- Shyu A. B., Wilkinson M. F. The double lives of shuttling mRNA binding proteins. Cell. 2000;102:135–138. doi: 10.1016/s0092-8674(00)00018-0. [DOI] [PubMed] [Google Scholar]
- Spiegelman B. M. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found. Symp. 2007;287:60–63. [PubMed] [Google Scholar]
- Srinivasan S., Avadhani N. G. Hypoxia-mediated mitochondrial stress in RAW264.7 cells induces osteoclast-like TRAP-positive cells. Ann. N. Y. Acad. Sci. 2007;1117:51–61. doi: 10.1196/annals.1402.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taanman J. W., Rahman S., Pagnamenta A. T., Morris A. A., Bitner-Glindzicz M., Wolf N. I., Leonard J. V., Clayton P. T., Schapira A. H. Analysis of mutant DNA polymerase gamma in patients with mitochondrial DNA depletion. Hum. Mutat. 2009;30:248–254. doi: 10.1002/humu.20852. [DOI] [PubMed] [Google Scholar]
- Tomonaga T., Levens D. Heterogeneous nuclear ribonucleoprotein K is a DNA-binding transactivator. J. Biol. Chem. 1995;270:4875–4881. doi: 10.1074/jbc.270.9.4875. [DOI] [PubMed] [Google Scholar]
- van Waveren C., Moraes C. T. Transcriptional co-expression and co-regulation of genes coding for components of the oxidative phosphorylation system. BMC Genomics. 2008;9:18. doi: 10.1186/1471-2164-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera J., Jaumot M., Estanyol J. M., Brun S., Agell N., Bachs O. Heterogeneous nuclear ribonucleoprotein A2 is a SET-binding protein and a PP2A inhibitor. Oncogene. 2006;25:260–270. doi: 10.1038/sj.onc.1209050. [DOI] [PubMed] [Google Scholar]
- Wallace D. C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagel S., Warner A. H., Nellans H. N., Lala P. K., Waghorne C., Denhardt D. T. Suppression by cathepsin L inhibitors of the invasion of amnion membranes by murine cancer cells. Cancer Res. 1989;49:3553–3557. [PubMed] [Google Scholar]
- Yamaoka K., Imajoh-Ohmi S., Fukuda H., Akita Y., Kurosawa K., Yamamoto Y., Sanai Y. Identification of phosphoproteins associated with maintenance of transformed state in temperature-sensitive Rous sarcoma-virus infected cells by proteomic analysis. Biochem. Biophys. Res. Commun. 2006;345:1240–1246. doi: 10.1016/j.bbrc.2006.04.183. [DOI] [PubMed] [Google Scholar]
- Yang C. K., Kim J. H., Stallcup M. R. Role of the N-terminal activation domain of the coiled-coil coactivator in mediating transcriptional activation by beta-catenin. Mol. Endocrinol. 2006;20:3251–3262. doi: 10.1210/me.2006-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W., Xie J., Long C., Erdjument-Bromage H., Ding X., Zheng Y., Tempst P., Chen S., Zhu B., Reinberg D. Heterogeneous nuclear ribonucleoprotein L (HnRNP-L) is a subunit of human KMT3a/Set2 complex required for H3 lys36 trimethylation activity in vivo. J. Biol. Chem. 2009;284:15701–15707. doi: 10.1074/jbc.M808431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Wang J., Levichkin I. V., Stasinopoulos S., Ryan M. T., Hoogenraad N. J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Mulshine J. L., Unsworth E. J., Scott F. M., Avis I. M., Vos M. D., Treston A. M. Purification and characterization of a protein that permits early detection of lung cancer. Identification of heterogeneous nuclear ribonucleoprotein-A2/B1 as the antigen for monoclonal antibody 703D4. J. Biol. Chem. 1996;271:10760–10766. doi: 10.1074/jbc.271.18.10760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.