

Abstract
Biodegradable branched polycationic polymers with varying hydrophilic spacer lengths were synthesized from different triacrylate monomers and the amine monomer 1-(2-aminoethyl)piperazine by Michael addition polymerization. The hydrophilic spacers were varied by the number of ethyleneoxy groups in the triacrylate monomer (E/M) that ranged from 0 to 14. The polymer degradation depended on the spacer length and pH; the amount of ester degraded as determined by 1H-NMR after 14 days was 43.4 ± 2.1% (pH 5.0) and 89.7 ± 1.3% (pH 7.4) for the polymer with 0 E/M compared to 55.7 ± 2.6% (pH 5.0) and 98.5 ± 1.6% (pH 7.4) for the polymer with 14 E/M. Cell viability of rat fibroblasts after exposure to polymer solutions of concentrations up to 1000 μg/mL remained high (above 66.9 ± 12.1% compared to below 7.6 ± 1.1% for polyethylenimine at a concentration of 50 μg/mL or higher) and increased with the spacer length. The polyplexes made with all the synthesized polymers showed higher transfection efficiency (4.5 ± 1.7% to 9.4 ± 2.0%, dependent on the polymer/pDNA weight ratio) with an enhanced green fluorescent protein reporter gene compared to naked pDNA (0.8 ± 0.4%) as quantified by flow cytometry. This study demonstrates that hydrophilic spacers can be incorporated into polycationic polymers to reduce their cytotoxicity and enhance their degradability for non-viral gene delivery.
Keywords: Non-viral gene delivery, Biodegradable polymer, Ester hydrolysis, Hydrophilic spacer, Cytotoxicity, Molecular weight
Introduction
Gene therapy can be used to alter or control the path of cellular action1 by replacing a defective gene or introducing a new gene that encodes for a specific therapeutic protein2. Viral vectors have been shown to be very efficient at delivering DNA. However, due to safety concerns such as immune and toxic response, production complexity and cost of these vectors3,4, non-viral vectors such as polycationic polymers are increasingly being investigated as an alternative method for gene delivery. Such polymers, which frequently contain positively charged protonated amine groups, are able to interact electrostatically with the negative charges of the phosphate groups on the DNA backbone leading to DNA condensation and protection against DNA degradation. Furthermore, positively charged polymer/DNA polyplexes may better adhere to cells by interacting with the negatively charged phospholipids and sulfate groups of the proteoglycans on the cells5,6, which ultimately helps with the internalization of the polyplexes by endocytosis. The pH in such newly formed endosomes gradually decreases and reaches values of 4.5 when the endosome becomes a lysosome6,7. Any amine groups that buffer between pH 7.4 to 5.0 can assist in the escape of the DNA from the endosome and prevent DNA degradation by nucleases in the lysosome through the proton sponge effect6.
Biodegradable polymers with high transfection efficiencies are much needed as more cytocompatible alternatives to current non-biodegradable polycationic polymers, such as polyethylenimine (PEI)8 and poly(dimethylaminoethylmethacrylate) (PDMAEM)9. Although PEI and PDMAEM are known to be effective non-viral vectors, their toxicity and non-degradability presents a problem with repeated dosage and long-term usage due to their accumulation in the endosomal compartment or cell nucleus10,11. Biodegradability of polycationic polymers is important to facilitate the release of encapsulated DNA once in the cell and to excrete the polymers and their degradation products to avoid accumulation in the endosomal compartment or cell nucleus12. An optimal degradation rate of the side chains or part of the main chain of the polymers is a key design criterion of polymers for non-viral gene delivery. If the polymer degrades too quickly, the DNA will disassemble from the polyplex too early during the delivery process. As a result, the degraded polymer will not complex with and condense the DNA, thus making the DNA susceptible to enzymatic degradation. On the other hand, the polymer should degrade fast enough for the DNA to disassemble from the polyplex once the polyplexes have reached the cytoplasm or nucleus to allow for plasmid expression12,13. Degradation of the polymer into lower molecular weight fragments is further desirable as it decreases the cytotoxicity of the polymer by decreasing the density of positive charges, thus, reducing any interaction with cellular compartments14,15.
Biodegradable polymers synthesized by Michael addition polymerization of acrylate and amine monomers have been previously investigated as non-viral gene delivery vectors16-22. These polymers can be synthesized from either diacrylate16-19 or triacrylate20-22 monomers, to produce either linear or branched polymers, depending on the type of amine monomer used in the synthesis. The acrylate monomers contain ester groups, which contribute to the hydrolytic biodegradability of the polymers. The polymers are synthesized in a one-pot reaction that does not require a catalyst, which would have to be isolated during the purification process. In contrast to polycondensation or amidation reactions, addition polymerization preserves basicity of the amine groups, even when they participate in the reaction.
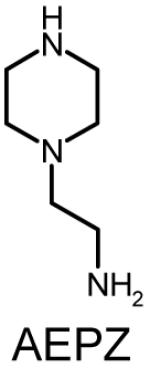
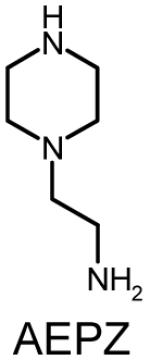
In this work, a series of polymers were synthesized by chemically conjugating different triacrylate monomers with a single amine monomer, 1-(2-aminoethyl)piperazine (AEPZ) to test the hypothesis that hydrophilic spacers can be incorporated into polycationic polymers to produce gene delivery vectors with reduced cytotoxicity and enhanced degradability. The hydrophilic spacers in these polymers consist of ethyleneoxy groups, equivalent to those found in poly(ethylene glycol) (PEG), which is often incorporated into polymers to increase the hydrophilicity and chain flexibility of the polymers23 as well as to decrease the cytotoxicity of polymeric non-viral vectors24,25. To test this hypothesis, we investigated the effects of the hydrophilic spacer lengths (different triacrylate monomers) on the molecular weight, amine equivalents protonating at different pH values, degradability and cytotoxicity of the synthesized polymers as well as the diameter, zeta potential and transfection efficiency of the polyplexes formed by these polymers with plasmid DNA (pDNA), all of which are relevant parameters in designing a gene delivery vector.
Materials and Methods
Materials
Trimethylolpropane triacrylate (TMPTA), 3 types of trimethylolpropane ethoxylate triacrylates (TMPETA) that differ in the average number of ethyleneoxy groups per molecule (TMPETA with 3 ethyleneoxy groups per monomer (3E/M), TMPETA with 7 ethyleneoxy groups per monomer (7E/M) and TMPETA with 14 ethyleneoxy groups per monomer (14E/M)), AEPZ, branched PEI (typical weight average molecular weight, Mw ~ 25 kDa, polydispersity (PDI) of 2.5), chloroform (ACS grade), sterile-filtered dimethyl sulfoxide (DMSO), methyl tetrazolium (MTT) powder, phenol red free Dulbecco’s modified Eagle medium (DMEM), ethidium bromide and Tris-Acetate-EDTA (TAE) were purchased from Sigma-Aldrich (St. Louis, MO). Deuterium oxide (D2O) was obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA). Acetone (ACS grade), isopropanol (ACS grade) and Fisher Certified Buffer pH 5.0 and 7.4 were purchased from Fisher Scientific (Pittsburgh, PA). Fischer rat fibroblast 3T3-like cell line (CRL 1764) was obtained from American Type Culture Collection (Manassas, VA). DMEM, phosphate-buffered saline (PBS), penicillin, streptomycin and amphotericin B were purchased from Gibco Life (Grand Island, NY). Fetal bovine serum (FBS) was obtained from Gemini Bio-Products (Calabasas, CA). Trypsin-ethylenediaminetetraacetic acid (EDTA) (0.25% trypsin/0.02% EDTA) was obtained from Invitrogen (Carlsbad, CA). Poly(ethylene glycol) (PEG) standards with number average molecular weights (Mn) ranging from 102 to 82,500 Da were obtained from Waters (Milford, PA). pDNA with a cytomegalovirus (CMV) promoter and enhanced green fluorescent protein (eGFP) reporter gene (pCMV-eGFP, 4.7 kb, cat no. 6085-1) was obtained from Clontech (Palo Alto, CA). Qiagen Endofree Plasmid Giga Kit was purchased from Qiagen (Valencia, CA).
Polymer Synthesis
Four sets of polymers (P-0E/M, P-3E/M, P-7E/M, P-14E/M) (Table 1) were synthesized by reacting AEPZ with TMPTA (0E/M), TMPETA (3E/M), TMPETA (7E/M), and TMPETA (14E/M) using a synthetic approach published previously22. All polymers were synthesized at a 2:1 molar ratio of amine to triacrylate monomer, which is the same ratio applied previously in the synthesis of polymers with similar architecture and polymerization mechanism20-22. Typically, 10.8 mmole of the amine monomer and 5.4 mmole of the triacrylate monomer were dissolved in 20 mL of chloroform in a round bottom flask. The solution was allowed to react for 8 days at ambient temperature. The polymer was then purified by precipitation in 400 mL of acetone and 5 mL of hydrochloric acid (HCl) (12 M). The product was washed with excess acetone and vacuum dried to remove any remaining solvent.
Table 1.
Polymer composition and structures of the triacrylate and amine monomer building blocks. P-0E/M, P-3E/M, P-7E/M and P-14E/M vary in hydrophilic spacer length. l = m = n = 0 for TMPTA (0E/M), l = m = n = 1 for TMPETA (3E/M), l + m + n = 7 for TMPETA (7E/M) and l + m + n = 14 for TMPETA (14E/M)
| Group | Triacrylate Monomer | Amine Monomer |
|---|---|---|
| P-0E/M | 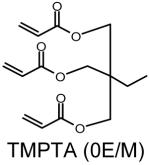 |
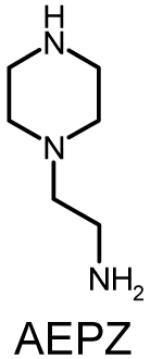 |
| P-3E/M | 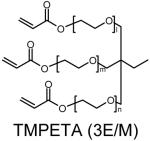 |
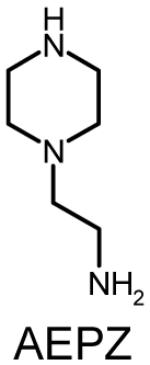 |
| P-7E/M | 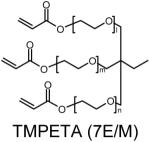 |
 |
| P-14E/M | 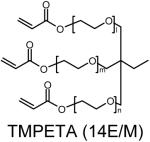 |
 |
Polymer Characterization
Proton Nuclear Magnetic Resonance (1H-NMR)
1H-NMR spectra of the polymers were obtained using a 400 MHz Bruker spectrometer (Bruker Avance 400, Zurich, Switzerland). The samples were dissolved in deuterium oxide (D2O) and analyzed using MesRe-C, an NMR processing software package (Mestrelab Research S. L., Spain). The proton peak of deuterium protium oxide (HDO) was used as the internal shift reference (δ = 4.8 ppm).
Molecular Weight Determination by Size Exclusion Chromatography (SEC)
The weight average molecular weight (Mw), number average molecular weight (Mn) and polydispersity index (PDI) (Mw/Mn) of the polymers were obtained using a Waters Alliance® HPLC system (Waters, Milford, PA) equipped with a differential refractometer. The samples were run through a Shodex OHpak SB-G (6.0 × 50 mm) guard column, a Shodex OHpak SB803 HQ (8.0 × 300 mm) and a Shodex OHpak SB802.5 HQ (8.0 × 300 mm) analytical column placed in series. PEG standards with Mn ranging from 102 to 82,500 Da were used to generate the calibration curve22. A 0.5 M sodium acetate buffer containing 0.1 M sodium nitrate was used as the mobile phase (pH 4.5). The flow rate was set to 0.5 mL/min, with the temperature of the column and sample chamber set at 25°C and 12°C, respectively. The polymer samples were dissolved in the mobile phase and filtered through a 0.2 μm cellulose filter (Alltech, Deerfield, IL). Data analysis was performed using the Empower™ software (Waters, Milford, PA) provided with the instrument. The theoretical average compositions of the polymers were calculated based on the Mn values obtained from SEC by assuming that the reaction occurred at the molar feed ratio of amine to triacrylate monomers of 2:1.
Hydrogen Ion Titration
The number of amine equivalents in a given mass of polymer was evaluated by hydrogen ion titration. In a typical experiment, 50 mg of polymer were dissolved in 10 mL of 150 mM sodium chloride (NaCl) solution. The polymer solution was adjusted to a pH of 2.0 using 0.1 or 1 M HCl solution and/or 0.1 N sodium hydroxide (NaOH) solution. The polymer solution was then titrated by stepwise (200 μL) addition of 0.1 N NaOH under continuous stirring. The pH of the solution was measured after each addition of 0.1 N NaOH solution using an accumet* AP63 Portable pH meter (Fisher Scientific, Pittsburgh, PA). Each titration was done in triplicate, and average pH values were calculated and reported. The amine equivalents (which corresponds to the amount of amines in the polymer) that dissociated in different pH ranges were determined by calculating the amounts of hydroxide ion (OH-) that were required to change between two pH values26.
Polymer Degradation by 1H-NMR Analysis
The rate of hydrolytic ester degradation of the different polymers was determined at pH 5.0 and 7.4 by 1H-NMR analysis22,27,28. In a typical experiment, a polymer sample (7.5 mg) was dissolved in 750 μL of pH 5.0 or 7.4 Fisher Certified Buffer29. Throughout the degradation study, the samples were incubated at 37°C while shaking at 75 rpm. At predefined time points (0 h, 6 h, 12 h, 1 d, 3 d, 7 d and 14 d), the samples were frozen and lyophilized. The dried samples were then dissolved in D2O and analyzed by 1H-NMR. This study was done in triplicate (n=3). At each time point, percent ester degradation was obtained by evaluating the change in the ratio of the integral of the methylene protons in the α-position to the ester to the integral of the methyl protons in the triacrylate monomers in the polymers (I4.2/I1.6). The integral of the protons in the α-position to the ester decreased as the esters degraded. The integral attributed to the methyl protons in the polymers remained unchanged during the hydrolytic ester degradation process. Equation 1 was used to calculate the percentage of ester that was degraded at each time point, and the data was reported as percent ester degraded22,27,28.
| (Equation 1) |
Cytotoxicity Analysis by MTT Assay
The cytotoxicity of the polymers was evaluated using an MTT viability assay30. CRL 1764 rat fibroblasts were expanded in T-75 culture flasks with DMEM supplemented with 10% (v/v) FBS and antibiotics (100 μg/mL penicillin, 100 U/mL streptomycin, and 0.5 μg/mL amphotericin B) and cultured at 37°C in 5% CO2 and 95% relative humidity. Typically, rat fibroblasts were trypsinized with trypsin-EDTA solution (2 mL/flask) and replated on 96-well plates with a density of 8000 cells/well. The plates were incubated (37°C in 5% CO2 and 95% humidity) for 24 hours to allow the cells to reach 80-90% confluency. Polymer solutions with a pH of 7.4 and an osmolarity of 280-320 mosm/kg were prepared in DMEM at six different polymer concentrations (10, 50, 100, 250, 500 and 1000 μg/mL). Branched PEI (25 kDa) at the same concentrations was used as a negative control. PEI 25 kDa has been extensively characterized in the literature and is often used as a control in the development of polymers for non-viral gene delivery22,31. As cytotoxicity and transfection efficiency can vary depending on the cell type and source, PEI was included in this study for experiments that involved cells (i.e., cytotoxicity and transfection efficiency experiments) to allow for direct comparison with the synthesized polymers under the same conditions. To start the experiment, culture media was removed and 100 μL of the polymer solution was added to individual wells of the 96-well plates. The plates were then incubated for 2 and 24 h at 37°C. At each time point, the polymer solution was removed, and the wells were washed twice with PBS to remove any residual polymer solution. 100 μL of MTT solution (1 mg/mL in phenol red free DMEM) was added to each well. After 3 h of incubation under exclusion of light, the MTT solution was removed and 100 μL of DMSO/2-propanol 1:1 (v/v) solution was added to each well to dissolve the formazan crystals formed by the live cells. The plates were agitated to help dissolve the formazan crystals. The absorbance was measured at 570 nm using a microplate reader (Powerwave X340, BIO-TEK Instruments, Winooski, VT). The cell viability was obtained by normalizing the absorbance of the wells containing cells cultured in the presence of polymer solution to the absorbance of the wells cultured with plain media. The average number of live cells was calculated from 5 different samples.
Polymer/pDNA Polyplex Characterization
Amplification and Purification of Plasmid DNA
pCMV-eGFP was amplified in Escherichia coli (E. coli) bacterial cultures and purified using a Qiagen Endofree Plasmid Giga Kit according to the protocols provided by the manufacturer. The yield of the pDNA was determined from the UV absorbance at a wavelength of 260 nm (A260) (NanoDrop™ 1000 Spectrophotometer, Thermo Scientific, Wilmington, DE). To evaluate the plasmid purity, the ratio of the UV absorbance at wavelengths of 260 nm and 280 nm (A260/A280) was determined to be between 1.8 and 2.0. The pCMV-eGFP was used in all the polymer/pDNA polyplex characterization experiments.
Zeta Potential
Polymer/pDNA polyplexes were prepared at different polymer to pDNA weight ratios (5:1, 10:1, 50:1, 100:1, 200:1 and 300:1). The polyplexes were prepared by adding various amounts of polycationic polymer solution in PBS (300 μL) drop-wise to 10 μg of pDNA also dissolved in 300 μL PBS. The mixture was vortexed and then incubated for 1 h at room temperature to allow for polyplex formation. The zeta potential of the resulting particles or larger aggregates was obtained using a Zen 3600 Zetasizer (Malvern Instruments, Worcestershire, UK). Disposable cuvettes were filled with the polyplex solution and run in the zetasizer for a minimum of 10 cycles and a maximum of 100 cycles at 25°C. The zeta potential was calculated using the Smoluchowski equation based on the electrophoretic mobility32. This study was done in triplicate.
Band Retardation with Gel Electrophoresis
Different weight ratios (10:1, 20:1, 30:1, 40:1, 60:1, 80:1 and 100:1) of polycationic polymer to pDNA solutions were prepared. The polyplexes were prepared from 10 μL polymer solution in PBS and 10 μl of a solution of 1 μg pDNA in PBS as described above. A 0.5% (w/w) of agarose gel containing 5 μL of ethidium bromide solution (10 mg/mL) was prepared in TAE buffer (1x). The polyplex sample (20 μL) containing bromophenol blue loading solution was loaded into the wells. The gel was run for 1 h at 80 mV in TAE buffer (1x) and an image of the gel was captured in an UV transillumination box.
Hydrodynamic Polyplex Sizes by Dynamic Light Scattering (DLS)
The hydrodynamic diameter of the polycationic polymers/pDNA polyplexes was evaluated at different polymer to pDNA weight ratios (10:1, 50:1, 100:1, 200:1, 300:1 and 500:1) using a 90PLUS Particle Size Analyzer (Brookhaven Instruments Corporation, Holtsville, NY) with an incident beam of 660 nm wavelength and scattering angle set at 90°. The data were collected using the BIC 32 Bit Software (Brookhaven Instruments Corporation, Holtsville, NY). The polyplexes were prepared from 300 μL polymer solution in PBS and 300 μL of a solution of 10 μg pDNA in PBS as described above. The solution was allowed to equilibrate to 25°C inside the analyzer before the analysis was started. The diameter of the polyplexes or aggregates thereof was obtained using Laplace inverse program Non-Negative Least-Squares (NNLS)31. This study was done in triplicate.
eGFP Transfection of Rat Fibroblasts
CRL 1764 rat fibroblasts were grown in T-75 culture flasks using DMEM supplemented with 10% (v/v) FBS and antibiotics (100 μg/mL penicillin, 100 U/mL streptomycin, and 0.5 μg/mL amphotericin B) at 37°C in an incubator (5% CO2, 95% relative humidity). In a typical experiment, rat fibroblasts were trypsinized (2 mL trypsin-EDTA solution/flask) and replated in 6-well plates at a seeding density of 250,000 cells/well. After 24 h of cell attachment, media was aspirated and 100 μL of the plasmid polyplex solution containing 5 μg of pDNA complexed at different polymer/pDNA weight ratios (10:1, 50:1, 100:1, and 300:1) was added drop-wise to the cells. The following solutions served as controls: PEI/pDNA at a weight ratio of 2:1, naked pDNA and FBS-free media without pDNA. The weight ratio of 2:1 for PEI/pDNA polyplexes corresponds to a ratio of amine groups in the polymer to phosphate groups in the pDNA (N:P) of 11.5:1, which is similar to that used by Wu et al. (N:P of 10:1) as the optimal N:P for transfection with PEI22. The polyplexes were prepared from 50 μL polymer solution in PBS and 50 μl of a solution of 5 μg pDNA in PBS as described before. Immediately after the addition of 100 μL of the polyplex solution, 400 μL of DMEM (FBS free) was added. After 24 h, 2 mL of DMEM/FBS (10% (v/v)) was added to the cells, and the flask was incubated for an additional 48 h. The cells were then washed twice with PBS, trypsinized (500 μL of trypsin-EDTA solution per well), washed with PBS, fixed with chilled formaldehyde solution (1%) and placed on ice for 1 h. After washing with PBS, samples were analyzed using a flow cytometer (Becton Dickenson FACS Scan, BD Biosciences, San Jose, CA) at high flow. CellQuest Pro software (BD Biosciences, San Jose, CA, v 5.1) was used to collect the data. A total of 5000 cells were counted for each sample. The location of the cells on the dot plot was confirmed by running untreated cells in PBS through the cytometer. Markers were placed at 5% of control samples (untreated cells) and the relative rightward shift of the intensity was observed as the percentage of transfected cells. This study was done at n=5. Fluorescence images of selected samples were taken with a confocal microscope (Zeiss LSM 510, Carl Zeiss Jena, Germany) to confirm transfection and expression of the green fluorescent protein. The cells were imaged with a 20x objective after excitation by an argon laser. The transfection study was designed to ensure that a lower mass of polymer per unit area of culture well compared to that tested in the cytotoxicity experiment was used. In the cytotoxicity experiment, cells exposed to the highest mass of polymer per unit area of culture well tested, 300 μg/cm2, were still viable. In the transfection experiment, the highest mass of polymer per unit area of culture well that was tested was 150 μg/cm2 (weight ratio of 300:1).
Statistical Methods
The results were presented as means ± standard deviation. Single-factor analysis of variance (ANOVA) was used to identify if there were any significant differences among groups (p < 0.05). Tukey’s Honestly Significantly Different (HSD) test was then conducted to identify the specific groups that differed statistically significantly.
Results and Discussion
Polymer Synthesis
Polymers were synthesized and precipitated from acetone through the addition of HCl. The precipitates were solid or rubbery and sticky. The protonated polymers were insoluble in organic solvents, thus all characterizations were done in aqueous solutions. A general structure of the synthesized branched polymers is shown in Figure 1. The end groups in the polymer are primary amines from the amine monomer, AEPZ, that did not react with the triacrylate monomer, TMPTA22. Once protonated, these primary amines can participate in the complexation with pDNA. The yields of the polymerization were between 70-85%.
Figure 1.

General structure of a branched polymer obtained by Michael addition reaction of AEPZ with a triacrylate monomer.
Polymer Characterization
Proton Nuclear Magnetic Resonance (1H-NMR)
1H-NMR confirmed that all acrylate groups of the triacrylate monomers converted during the reaction, as the typical signals for olefinic protons (5.8-6.6 ppm) were absent for the polymeric products (not shown). Since the presence of unreacted acrylate groups would contribute to polymer cytotoxicity, it is important to achieve complete reaction of the acrylate groups33. The NMR spectra revealed the following characteristic signal: P-0E/M: 0.9 ppm (t, C-CH2-CH3), 1.4 ppm (m, C-CH2-CH3), 2.5-3.5 ppm (t, -CH2-CH2-NR2- and t, -CH2-CH2-COO-CH2-), 4.0-4.1 ppm (s, C-CH2-OOC-CH2-). P-3E/M, P-7E/M and P-14E/M: 0.9 ppm (t, C-CH2-CH3), 1.4 ppm (m, C-CH2-CH3), 2.5-3.5 ppm (t, -CH2-CH2-NR2- and t, -CH2-CH2-COO-CH2-), 3.6-3.9 ppm (s, C-CH2-O- and t, -O-CH2-CH2-O-), 4.0-4.1 ppm (t, -CH2-CH2-OOC-CH2-).
1H-NMR data was also used to verify that the polymer building blocks, amine and triacrylate monomers, reacted in a molar ratio of 2:1. The integral of the peaks between 2.5-3.5 ppm (derived from six protons: -CH2-CH2-NR2 in the amine monomer, -CH2-CH2-NR2 formed through the reaction, and -CH2-COO-CH2- from the triacrylate monomer) was compared to the integral of the peak at 0.9 ppm (derived from three methyl protons in the triacrylate monomers). These calculations yielded molar ratios for P-0E/M, P-3E/M, P-7E/M and P-14E/M of 1.8, 2.1, 1.9, and 2.3, respectively.
Molecular Weight Determination
Table 2 presents Mn, Mw and PDI of the polymers as obtained by SEC, as well as the theoretical average composition of the polymers calculated based on the experimental Mn values. The SEC traces of the synthesized polymers are shown in Figure 2. It was found that on average the polymers consisted of 6 or 8 amine and 3 or 4 triacrylate monomers. Low molecular weights were obtained for the synthesized polymers. The molecular weights of the polymers were obtained with GPC using linear PEG standards to generate the calibration curve. The calibration with a linear polymer may not account for the effect of branching that may be present in the polymers; thus an underestimation of the molecular weights may have occurred.
Table 2.
Number average molecular weight, weight average molecular weight, polydispersity index (PDI), estimated average number of triacylate and amine monomers per polymer molecule and pKa values of the different polymers examine in this study
| Polymer | Mn (Da)* | Mw (Da)* | PDI* | Number of Triacrylate Monomers† | Number of Amine Monomers† | pKa |
|---|---|---|---|---|---|---|
| P-0E/M | 2,700 | 6,670 | 2.47 | 4 (10) | 8 (20) | 7.85 |
| P-3E/M | 1,980 | 4,800 | 2.42 | 3 (7) | 6 (14) | 7.93 |
| P-7E/M | 2,310 | 8,660 | 3.75 | 3 (10) | 6 (20) | 8.02 |
| P-14E/M | 3,180 | 10,070 | 3.17 | 3 (9) | 6 (18) | 8.08 |
Measured by size exclusion chromatography
Estimated from number average molecular weight, numbers in brackets estimated from weight average molecular weight
Figure 2.

Size exclusion chromatography traces of the synthesized polymers.
As the variable in this study (i.e. hydrophilic spacer length) was altered, the molecular weight and degree of branching in the polymers were potentially affected as well. The molecular weight (Mn and Mw) of the polymers was found to increase with the hydrophilic spacer length of the triacrylate monomer (P-14E/M > P-7E/M > P-3E/M). P-0E/M, which was synthesized from the smallest acrylate precursor (TMPTA), showed comparably high molecular weights and a low PDI and consisted on average of 1 triacrylate and 2 amine monomers more than the other polymers. Although there was an increase in molecular weight as the hydrophilic spacer increased, all the polymers were formed from a similar amount of amine and triacrylate monomer (Table 2). Thus, a large effect of degree of branching on the parameters and polymers investigated in this study is not expected. The difference in Mw likely resulted from the difference in the mass of the triacrylate monomer (i.e. molecular weight of monomer increases with the increase in hydrophilic spacer length) rather than from the amount of potential branching in the polymers. Although this study provided initial insight on the effects of the hydrophilic spacer lengths, further investigation will facilitate a more complete understanding of the individual effects of the various parameters (hydrophilic spacer length, polymer MW and degree of branching).
Determination of Amine Equivalents and Polymer pKa Values by Hydrogen Ion Titration
Hydrogen ion titration of the polymers was conducted from pH 2 to pH 12, instead of from pH 12 to pH 2, to avoid degradation of the polymers at basic pH. The titration curves obtained are shown in Figure 3. Table 2 lists the pKa values determined for the different polymers. For the amine monomer AEPZ, which contains a primary, secondary and tertiary amine, a pKa value of 9.30 was obtained.
Figure 3.

Titration curves (0.1 N NaOH) of the different polymers. The results are expressed as means ± standard deviations for n=3.
The pKa values of the amines change upon conversion in the addition reaction. During the Michael-type reaction the alkyl substitution of the amines is increased, i.e., a primary amine becomes secondary and a secondary amine becomes tertiary. Besides increased alkyl substitution, sterically hindered accessibility also contributes to the observed changes in amine pKa. The synthesized polymers displayed pKa values in a small range between 7.85 and 8.08. pKa values of the polymers increased with the hydrophilic spacer lengths in TMPETA, likely due to improved accessibility of the amines. The titration curves for the four different polymer types were very similar in shape (Fig. 3) which was expected because they all consist of the same amine monomer (Table 1). The polymers in this study were synthesized from an amine monomer, AEPZ, that has a primary, secondary and tertiary amine, where the reaction occurs first at the secondary amine and then at the primary amine19,22. The synthesized polymers have amine groups that have different degrees of substitution and chemical environments and dissociate at different pH values. The buffering by the different amine groups blended into a broad buffering range, where distinct plateau and equivalent points for the different types of amines were not observed, which resulted in one pKa value for the whole polymer. Figure 3 shows that the buffering pH ranges of polymers with shorter hydrophilic spacers were wider as compared to those determined for polymers with longer hydrophilic spacers. This indicates that polymers with shorter hydrophilic spacers have higher buffering capacities.
For a given mass of polymer (typically 50 mg), the amine equivalents dissociating in four different pH ranges were evaluated (Fig. 4). The amine equivalents that dissociate between pH 2.5 and 5.0 were evaluated in order to compare the availability of unprotonated amines at pH 5.0 that may catalyze hydrolytic ester degradation34. Similarly, the amine equivalents that dissociate between pH 2.5 and 7.4 were determined to compare the availability of unprotonated amines which can catalyze the hydrolytic ester degradation at pH 7.4. The amine equivalents that dissociate at pH 5.0 - 7.4 were obtained to estimate the buffering capacity of the polymers at the endosomal/lysosomal pH range and thus, the polymer’s ability to aid in pDNA escape from the endosome prior to lysosomal degradation by nucleases6. The amine equivalents dissociating above pH 7.4 were calculated to determine the amounts of protonated amines that could help with polymer/pDNA complexation when the polyplexes are assembled at pH 7.4.
Figure 4.

Amine equivalents that dissociate at different pH ranges. The results represent means ± standard deviations for n = 3. # indicates a statistically significant difference between one polymer and the other polymers within the same pH interval (p<0.05). * represents a statistically significant difference between two polymers within the same pH interval (p<0.05).
For a given mass of polymer (50 mg), polymers with shorter hydrophilic spacer lengths, which contain a higher density of amines per molecular weight by design, had higher buffering capacities between pH 5.0 and 7.4 as well as between pH 7.4 and 11.0 (Fig. 4). Consequently a higher number of protonated amines is available in the polymers with short spacer lengths to aid in pDNA escape from the endosome and for the assembly of polymer/pDNA polyplexes at pH 7.4. In these polymers, a comparably large number of unprotonated amines is available below pH 7.4 that could assist with polymer hydrolytic ester degradation.
Polymer Degradation (1H-NMR)
In this study, the rate of hydrolytic ester degradation of the polymers, which occurs in the body when the polymers are exposed to the aqueous environment, was investigated. The hydrolytic ester degradation profiles of the different polymers at pH 5.0 and 7.4 are shown in Figures 5A and 5B, respectively. Figure 5C compares the polymers at two different time points (3 and 14 days). Polymers with longer hydrophilic spacers degraded faster than those with shorter spacers at both tested pH values. This is explained by the hydrophilic spacers attracting water molecules to the ester sites. Furthermore, polymers with longer hydrophilic spacers likely form looser structures with more flexible chains and more easily accessible ester sites23. Comparing the polymers synthesized from the different TMPETA monomers, the amount of ester degraded increased as the length of hydrophilic spacer increased. P-3E/M had a significantly higher amount of esters degraded compared to P-7E/M and P-14E/M at pH 5.0 after 3 and 14 days. At pH 7.4, P-3E/M had a significantly higher amount of esters degraded compared to P7E/M and P-14E/M after 3 days, however the amount of ester degraded in the three TMPETA polymers were not significantly different after 14 days. The amount of esters degraded for P-0E/M (no spacer) and P-14E/M (longest spacer) were significantly different from each other at day 14. The percent of ester degraded increased from 43.4 ± 2.1% to 55.7 ± 2.6% at pH 5.0 and 89.7 ± 1.3% to 98.5 ± 1.6% at pH 7.4 with the increase in hydrophilic spacer length. P-0E/M, which was synthesized from TMPTA and contained no ethyleneoxy groups, degraded slower than P-3E/M but faster than P-7E/M for most of the time points and conditions observed. Due to its lower molecular weight compared to P-0E/M, P-3E/M chains may have formed tighter structures and their ester groups might have been more effectively shielded from hydrolytic degradation. In summary, hydrophilic spacers can be incorporated into the polymers to increase their hydrolytic degradation rates. All polymers showed a higher rate of hydrolytic ester degradation at pH 7.4 (cytoplasmal pH) than at pH 5.0 (lysosomal pH). This is explained by a higher number of unprotonated amine groups that are present at the higher pH to catalyze hydrolysis of the ester groups, which is consistent with previous work on poly(ester amine)s10,34. Olefinic protons (5.8-6.6 ppm), which would contribute to polymer cytotoxicity33, were not present in the 1H NMR spectra of polymer degradation samples over the course of the hydrolytic ester degradation study (data not shown), indicating that a reverse Michael addition reaction, which would yield free acrylate groups, did not occur.
Figure 5.

Ester degradation profiles of the different polymers at 37°C and (A) pH 5.0 simulating lysosomal pH and (B) pH 7.4 simulating cytoplasmic pH and (C) degree of degradation after 3 and 14 days at pH 5.0 and 7.4 at 37°C as observed by 1H-NMR. The extent of degradation is express as means ± standard deviation for n = 3. # indicates a statistically significant difference between one polymer and the other polymers degraded under the same conditions (p<0.05). * represents a statistically significant difference between two polymer compositions degraded under the same conditions (p<0.05).
Cytotoxicity Analysis
Cells remained viable after incubation with polymer solutions for 2 and 24 h at all concentrations tested. PEI, which was tested in comparison, however, caused significant cell death (Fig. 6). After 2 h, more than 70% of the cells exposed to the polymers with different hydrophilic spacers were viable (Fig. 6A). After 24 h, more than 70% of the cells were viable except for the cells that were exposed to P-3E/M at 1000 μg/mL concentration (66.9 ± 12.1% viability) (Fig. 6B). In general, polymers with longer hydrophilic spacers (P-7E/M and P-14E/M) were less cytotoxic as compared to those with shorter or no spacer (P-0E/M and P-3E/M) after 24h. This is likely due to the decrease in the charge density in the polymers23. The slow degradation rates observed for P-0E/M and P-3E/M may also contribute to the observed toxicity14. PEI, in contrast, caused significant cell death after 2 h with less than 25% cell viability at a concentration of 250 μg/mL or higher. After 24 h, less than 8% of the cells were viable when exposed to PEI at a concentration of 50 μg/mL or higher. PEI has an 8-13 times higher molecular weight and 2-3 times higher charge density compared to the synthesized polymers. Both parameters certainly contribute to the high toxicity of PEI35,36. The improved cytocompatibility of the developed polycationic polymers will allow for use at high concentrations and high N:P ratios.
Figure 6.

Cytotoxicity of the different polymers (with polyethylenimine (PEI) as negative control) on CRL 1764 rat fibroblasts after (A) 2 h and (B) 24 h as evaluated by a MTT assay. The results are expressed as means ± standard deviation for n = 5.
Polymer/pDNA Polyplex Characterization
Zeta Potential
The surface charge of the polyplexes formed by the different polymers was evaluated to determine optimal polymer to pDNA ratios, which result in a positively charged polyplex that can interact with, instead of repulsing, the negative charges on the cell membrane6,37. The pCMV-eGFP plasmid, which was used in this study, has a zeta potential of -33.1 ± 0.5 mV. All the polyplexes formed at a polymer/pDNA weight ratio of 5:1 had a negative zeta potential ranging from -8.5 ± 0.4 mV to -27.5 ± 2.9 mV (Fig. 7). At a weight ratio of 10:1, P-0E/M, P-3E/M and P-7E/M formed polyplexes that were neutral. P-14E/M, which of all tested polymers provides the least amines per molecule and therefore will form polyplexes at the lowest N:P ratio at a given weight ratio of polymer to DNA, formed a negative polyplex (-4.3 ± 1.9 mV) at a weight ratio of 10:1 and a positive polyplex (7.3 ± 0.5 mV) at a weight ratio of 50:1. The zeta potential of all the polyplexes formed increased as the weight ratio of polymer to pDNA increased, which corresponds with the changing N:P ratios within the polyplexes. Consequently, the polyplexes formed from polymers with shorter hydrophilic spacer lengths had higher zeta potentials compared to polyplexes with polymers with longer hydrophilic spacers at all tested weight ratios.
Figure 7.

Zeta potential of the polyplexes formed with 10 μg pCMV-eGFP DNA at different polymer/pDNA weight ratios. The results are expressed as means ± standard deviation for n = 3. # indicates statistically significant difference between a polyplex and other polyplexes in the same weight ratio (p<0.05). * represents a statistically significant difference between two polyplexes within the same weight ratio (p<0.05).
Band Retardation with Gel Electrophoresis
The electrophoretic patterns of the polymer/pDNA polyplexes were obtained by gel electrophoresis in agarose gel and UV light detection (Fig. 8). Lane 8 in all images represents uncomplexed pDNA (naked pDNA) and shows migration of the plasmid to the positive end of the gels. P-0E/M, P-3E/M and P-7E/M were able to retard the migration of the pDNA at polymer/pDNA weight ratios of 10:1 and higher. In correspondence with the zeta potential results (Fig. 7), the polymers neutralized the charges of the pDNA at a ratio of 10:1. The zeta potential of P-14E/M was negative at a weight ratio of 10:1 and positive at a ratio of 50:1. Neutral polyplexes are consequently expected within this range. Through gel electrophoresis, P-14E/M was found to retard the migration of pDNA at weight ratios of 20:1 or higher. The higher amount of P-14E/M needed to neutralize the charges on the pDNA is explained by the low molar content of amine groups.
Figure 8.

Gel retardation assay of pDNA/polymer polyplexes at different polymer/pDNA weight ratios: (1) 100:1, (2) 80:1, (3) 60:1, (4) 40:1, (5) 30:1, (6) 20:1, (7) 10:1, and (8) naked pDNA.
Hydrodynamic Polyplex Sizes by DLS
The hydrodynamic size (diameter) of the polyplexes formed by the polymers and pDNA at different weight ratios was obtained by DLS (Fig. 9). At low polymer/pDNA weight ratios (10:1 for all the polymers, also 50:1 for P-0E/M), aggregation of the polyplexes was observed as shown by large particle diameters ranging between 923.4 ± 75.6 nm and 1910.5 ± 17.2 nm. At these ratios, zeta potential measurements revealed neutrality of the polyplexes, which favors particle aggregation as electrostatic repulsion is minimal. The zeta potential increased as the polymer/pDNA ratios were increased (Fig. 7) and individual polyplexes were electrostatically stabilized38. Consequently, the diameters of the polyplexes decreased to 147.2 ± 14.1 nm (P-7E/M, polymer/pDNA weight ratio of 300:1). Naked pDNA, in comparison, had a diameter of around 751.6 ± 111.8 nm (data not shown). The results indicate that all the polymers were able to complex with and condense the pDNA at certain weight ratios, which is essential for DNA stabilization and transfection efficiency39. Although the polyplexes formed with the polymers developed in this study may not be as small as those formed by branched PEI (25 kDa) (~20-40 nM)40, the polyplexes formed with the polymers developed in this study are promising, as larger sized particles have been found to sediment onto cells, increasing the interaction with the cells, which can enhance particle uptake41,42. Larger polyplexes may not get internalized through clathrin coated endocytosis43, however they have been shown to get internalized successfully by other mechanisms which have not been elucidated42,44.
Figure 9.

Diameter of the polyplexes formed with 10 μg pCMV-eGFP DNA at different polymer/pDNA weight ratios as evaluated by DLS. The results are expressed as means ± standard deviation for n = 3. # indicates statistically significant difference between a polyplex and other polyplexes in the same weight ratio (p<0.05). * represents a statistically significant difference between polyplexes within the same weight ratio (p<0.05).
Transfection Efficiency with Enhanced Green Fluorescent Protein Reporter Gene
The results from the transfection study are shown in Figure 10. At low polymer/pDNA weight ratios (10:1 and 50:1), only P-3E/M resulted in a significantly higher number of transfected cells than naked pDNA. At a polymer/pDNA weight ratio of 100:1, P-0E/M, P-3E/M and P-14E/M resulted in significantly higher transfection rates (7.2 ± 0.5% to 8.0 ± 1.5%) than naked pDNA (0.8 ± 0.4%). At the highest weight ratio (300:1), P-0E/M, P-3E/M and P-7E/M resulted in significantly higher transfection (6.7 ± 2.0% to 9.4 ± 2.0%) than naked pDNA. The transfection efficiency of polyplexes formed at weight ratios 100:1 and 300:1 were not significantly different from each other. Above a certain weight ratio, no increase in surface charge (Fig. 7) or reduction in the size (Fig. 9) of the polyplexes formed were observed within a polymer group, as also observed in a study by Wu et al.22. This suggests that at a certain polymer/pDNA weight ratio, the amount of polymer that participates in the polyplex formation may reach a critical point where any polymer subsequently added to the polyplex solution will remain uncomplexed. Thus, above that critical point, the addition of polymer will not facilitate additional complexation. Determination of the amount of uncomplexed polymer was not part of the design of this study. However, quantification of the amount of uncomplexed polymer in the future will give more insight on the polymer/pDNA complexation.
Figure 10.

Efficiency of CRL 1764 cell transfection with polyplexes formed from 5 μg pCMV-eGFP DNA at different polymer/pDNA weight ratios. Naked pDNA and polymplexes formed from PEI at a polymer/pDNA weight ratio of 2 served as controls. The results are expressed as means ± standard deviation for n = 4-6. # indicates a statistically significant difference between a polyplex and naked pDNA (p<0.05).
At all the weight ratios tested, the percent of cells transfected by polyplexes made from the different polymers was significantly lower than obtained for polyplexes with PEI at a weight ratio of 2:1. The low molecular weights and charge densities of the synthesized polymers as compared to PEI are factors explaining the lower transfection efficiencies observed35,45. Although low transfection efficiencies were observed in vitro for the synthesized polymers under the conditions explored in this study, they were higher at certain weight ratios than those associated with naked pDNA. We envision that polyplexes formed between pDNA and the polymers explored in this study may be incorporated into and released from tissue engineering scaffolds in vivo. The observed significant differences in the degradation rates of polymers with different spacer lengths in vitro may result in pDNA being released at different times and in different forms (i.e., intact complexes or free pDNA) from the scaffolds in vivo, which could in turn result in significant differences in transfection than were observed upon direct exposure of cells to the polyplexes under the conditions explored in this study in vitro.
Fluorescence micrographs were taken to confirm the transfection results obtained by flow cytometry. As a negative control, a fluorescence image of cells treated with just media was taken (Fig. 11A). There were no transfected cells observed when exposed to naked pDNA (Fig. 11B). Cells transfected with polyplexes of P-0E/M at a polymer/pDNA weight ratio of 300:1 had overall faint eGFP expression as seen in Figure 11C. Arrows point to cells that expressed eGFP. Cells that were transfected with PEI/pDNA (2:1) polyplexes had a more intense expression of eGFP (Fig. 11D).
Figure 11.
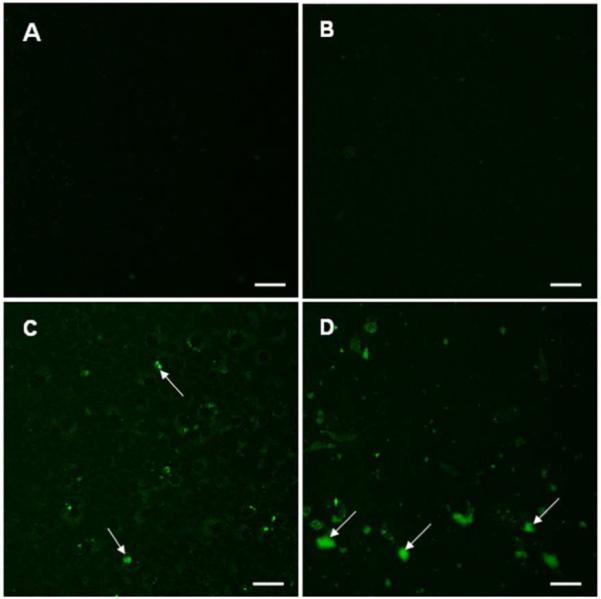
Representative fluorescence images of CRL 1764 cells after exposure to the following media: (A) plain medium, (B) naked pDNA (CMV-eGFP), (C) P-0E/M/pDNA (300:1), and (D) PEI/pDNA (2:1). An amount of 5 μg pCMV-eGFP DNA was used for each media consisting of pDNA (B-D). Green fluorescence in cells represents expression of GFP. Brightly fluorescing cells are indicated by arrows. Scale bars represent 50 μm.
A very low number of live cells, however, were observed after exposure to PEI polyplexes, as seen by light microscopy (data not shown). The cytotoxicity of PEI is well described in the literature and likely attributed to the high MW and charge density of PEI35. Wells treated with the polymers synthesized in this study, even at polymer/pDNA weight ratios as high as 300:1, revealed a high number of live, well spread cells as seen by light micoscopy.
The results indicate that the synthesized polycationic polymers degrade well under physiological and lysosomal conditions and can efficiently complex pDNA and transfect cells. As a result of the low molecular weight of the polymers and the lower charge density than PEI, effective DNA complexation is achieved at high N:P ratios, and the transfection efficiencies are comparably low. At the same time, these parameters contribute to the low cytotoxicity of the developed polymers, which makes them potential vectors for non-viral gene delivery applications. This study suggests that the polymers can be used at higher concentrations compared to PEI and for repeated dosage.
In this study, a limited effect of hydrophilic spacer length on the transfection efficiency of the polymers was observed. However, there were other parameters, such as degradation rate, which were significantly affected by the length of the hydrophilic spacers. In this study, the cells were directly exposed to the polyplexes, but for in vitro or in vivo gene therapy approaches where the polyplexes are first released from a scaffold, the polyplexes may take a longer time to reach the cells. Thus, the significant differences in degradation rates of polymers with different spacer lengths may result in pDNA being released at different times and in different forms (i.e., intact complexes or free pDNA), which could in turn result in significant differences in transfection. Rat fibroblasts were chosen as an initial model cell line in this study, and transfection was performed in the absence of serum; however, transfection efficiency can vary between cell lines and with the absence or presence of serum. Future transfection studies in vitro in different cell lines and as well as in vivo in an appropriate animal model will yield a more comprehensive analysis of the transfection capabilities of these polymers.
Conclusions
This study confirmed our hypothesis that the introduction of hydrophilic spacers to branched biodegradable polycationic polymers yields effective gene delivery vectors with reduced cytotoxicity and increased polymer degradation rates. Polymers with shorter hydrophilic spacers yielded polyplexes with higher N:P ratios, which resulted in polymer/pDNA polyplexes with higher zeta potentials. The percent of cells transfected by polyplexes of the synthesized polymers at certain weight ratios was significantly higher than that associated with naked pDNA, however, not comparable to polyplexes with branched PEI with a weight ratio of 2:1. The lower transfection observed could have resulted from the lower molecular weight and reduced charge density of these polymers compared to PEI. However, a higher density of cells was observed after exposure to the synthesized polymers at high concentration, indicating the capability of the polymer to be used at high concentration as needed for transfection with these polymers. Further work will be done to optimize the Michael addition synthesis to produce polymers with higher molecular weight to study the effect of molecular weight on transfection. The findings of this work can be used for the design of non-viral vectors with tailored structural and degradative characteristics as needed for specific applications.
Acknowledgements
We acknowledge support by a grant from the National Institutes of Health (R21 AR56076). We thank Dr. Joel Moake for the use of the flow cytometer and Dr. Rebecca Richards-Kortum for the use of the DLS instrument. We also thank Dr. Michael R. Diehl, Dr. Ka-Yiu San and Dr. Junghae Suh for the use of their facilities for plasmid DNA amplification.
References
- (1).Kasper FK, Mikos AG. Molecular and Cellular Foundations of Biomaterials. Elsevier; San Diego: 2004. pp. 131–168. M.V., S. [Google Scholar]
- (2).Cartier R, Reszka R. Gene Ther. 2002;9:157–167. doi: 10.1038/sj.gt.3301635. [DOI] [PubMed] [Google Scholar]
- (3).Peel AL, Zolotukhin S, Schrimsher GW, Muzyczka N, Reier PJ. Gene Ther. 1997;4:16–24. doi: 10.1038/sj.gt.3300358. [DOI] [PubMed] [Google Scholar]
- (4).Reschel T, Konak C, Oupicky D, Seymour LW, Ulbrich K. J. Controlled Release. 2002;81:201–217. doi: 10.1016/s0168-3659(02)00045-7. [DOI] [PubMed] [Google Scholar]
- (5).Tanahashi K, Mikos AG. J. Biomed. Mater. Res. A. 2003;67:1148–1154. doi: 10.1002/jbm.a.10147. [DOI] [PubMed] [Google Scholar]
- (6).Brokx R, Gariepy J. Methods Mol. Med. 2004;90:139–160. doi: 10.1385/1-59259-429-8:139. [DOI] [PubMed] [Google Scholar]
- (7).Azzam T, Domb AJ. Curr. Drug Delivery. 2004;1:165–193. doi: 10.2174/1567201043479902. [DOI] [PubMed] [Google Scholar]
- (8).Godbey WT, Wu KK, Mikos AG. J. Controlled Release. 1999;60:149–160. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- (9).Cherng JY, van de Wetering P, Talsma H, Crommelin DJ, Hennink WE. Pharm. Res. 1996;13:1038–1042. doi: 10.1023/a:1016054623543. [DOI] [PubMed] [Google Scholar]
- (10).Kloeckner J, Bruzzano S, Ogris M, Wagner E. Bioconjug. Chem. 2006;17:1339–1345. doi: 10.1021/bc060133v. [DOI] [PubMed] [Google Scholar]
- (11).Godbey WT, Wu KK, Mikos AG. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5177–5181. doi: 10.1073/pnas.96.9.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Luten J, van Nostrum CF, De Smedt SC, Hennink WE. J. Controlled Release. 2008;126:97–110. doi: 10.1016/j.jconrel.2007.10.028. [DOI] [PubMed] [Google Scholar]
- (13).Eldred SE, Pancost MR, Otte KM, Rozema D, Stahl SS, Gellman SH. Bioconjug. Chem. 2005;16:694–699. doi: 10.1021/bc050017c. [DOI] [PubMed] [Google Scholar]
- (14).Breunig M, Lungwitz U, Liebl R, Goepferich A. Proc. Natl. Acad. Sci. U.S.A. 2007;104:14454–14459. doi: 10.1073/pnas.0703882104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Choksakulnimitr S, Masuda S, Tokuda H, Takakura Y, Hashida M. J. Controlled Release. 1995;34:233–241. [Google Scholar]
- (16).Lynn DM, Langer R. J. Am. Chem. Soc. 2000;122:10761–10768. [Google Scholar]
- (17).Lynn DM, Anderson DG, Putnam D, Langer R. J. Am. Chem. Soc. 2001;123:8155–8156. doi: 10.1021/ja016288p. [DOI] [PubMed] [Google Scholar]
- (18).Wu DC, Liu Y, Chen L, He CB, Chung TS, Goh SH. Macromolecules. 2005;38:5519–5525. [Google Scholar]
- (19).Wu DC, Liu Y, He CB, Chung TS, Goh ST. Macromolecules. 2004;37:6763–6770. [Google Scholar]
- (20).Kim T, Seo HJ, Baek J, Park JH, Park JS. Bull. Korean Chem. Soc. 2005;26:175–177. [Google Scholar]
- (21).Kim TI, Seo HJ, Choi JS, Yoon JK, Baek JU, Kim K, Park JS. Bioconjug. Chem. 2005;16:1140–1148. doi: 10.1021/bc0497012. [DOI] [PubMed] [Google Scholar]
- (22).Wu D, Liu Y, Jiang X, Chen L, He C, Goh SH, Leong KW. Biomacromolecules. 2005;6:3166–3173. doi: 10.1021/bm0504983. [DOI] [PubMed] [Google Scholar]
- (23).Yang TF, Chin WK, Cherng JY, Shau MD. Biomacromolecules. 2004;5:1926–1932. doi: 10.1021/bm049763v. [DOI] [PubMed] [Google Scholar]
- (24).Jevprasesphant R, Penny J, Jalal R, Attwood D, McKeown NB, D’Emanuele A. Int. J. Pharm. 2003;252:263–266. doi: 10.1016/s0378-5173(02)00623-3. [DOI] [PubMed] [Google Scholar]
- (25).Mao S, Shuai X, Unger F, Wittmar M, Xie X, Kissel T. Biomaterials. 2005;26:6343–6356. doi: 10.1016/j.biomaterials.2005.03.036. [DOI] [PubMed] [Google Scholar]
- (26).Tseng WC, Fang TY, Su LY, Tang CH. Mol. Pharm. 2005;2:224–232. doi: 10.1021/mp050007t. [DOI] [PubMed] [Google Scholar]
- (27).Forrest ML, Koerber JT, Pack DW. Bioconjug. Chem. 2003;14:934–940. doi: 10.1021/bc034014g. [DOI] [PubMed] [Google Scholar]
- (28).Liu Y, Wu D, Ma Y, Tang G, Wang S, He C, Chung T, Goh S. Chem. Commun. 2003:2630–2631. doi: 10.1039/b309487a. [DOI] [PubMed] [Google Scholar]
- (29).Lu L, Stamatas GN, Mikos AG. J. Biomed. Mater. Res. 2000;50:440–451. doi: 10.1002/(sici)1097-4636(20000605)50:3<440::aid-jbm19>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- (30).Timmer MD, Shin H, Horch RA, Ambrose CG, Mikos AG. Biomacromolecules. 2003;4:1026–1033. doi: 10.1021/bm0300150. [DOI] [PubMed] [Google Scholar]
- (31).Saraf A, Hacker MC, Sitharaman B, Grande-Allen KJ, Barry MA, Mikos AG. Biomacromolecules. 2008;9:818–827. doi: 10.1021/bm701146f. [DOI] [PubMed] [Google Scholar]
- (32).Hosseinkhani H, Aoyama T, Yamamoto S, Ogawa O, Tabata Y. Pharm. Res. 2002;19:1471–1479. doi: 10.1023/a:1020400514990. [DOI] [PubMed] [Google Scholar]
- (33).Yoshii E. J. Biomed. Mater. Res. 1997;37:517–524. doi: 10.1002/(sici)1097-4636(19971215)37:4<517::aid-jbm10>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- (34).Zhong Z, Song Y, Engbersen JF, Lok MC, Hennink WE, Feijen J. J. Controlled Release. 2005;109:317–329. doi: 10.1016/j.jconrel.2005.06.022. [DOI] [PubMed] [Google Scholar]
- (35).Breunig M, Lungwitz U, Liebl R, Fontanari C, Klar J, Kurtz A, Blunk T, Goepferich A. J. Gene Med. 2005;7:1287–1298. doi: 10.1002/jgm.775. [DOI] [PubMed] [Google Scholar]
- (36).Midoux P, Monsigny M. Bioconjug. Chem. 1999;10:406–411. doi: 10.1021/bc9801070. [DOI] [PubMed] [Google Scholar]
- (37).Tanahashi K, Jo S, Mikos AG. Biomacromolecules. 2002;3:1030–1037. doi: 10.1021/bm0200433. [DOI] [PubMed] [Google Scholar]
- (38).Anwer K, Rhee BG, Mendiratta SK. Crit. Rev. Ther. Drug Carrier Syst. 2003;20:249–293. doi: 10.1615/critrevtherdrugcarriersyst.v20.i4.10. [DOI] [PubMed] [Google Scholar]
- (39).Holtorf HL, Mikos AG. Pharmaceutical Perspectives of Nucleic Acid-Based Therapeutics. Taylor & Francis; New York: 2002. pp. 367–387. S.W., K. [Google Scholar]
- (40).Dunlap DD, Maggi A, Soria MR, Monaco L. Nucleic Acids Res. 1997;25:3095–3101. doi: 10.1093/nar/25.15.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Boussif O, Zanta MA, Behr JP. Gene Ther. 1996;3:1074–1080. [PubMed] [Google Scholar]
- (42).Ogris M, Steinlein P, Kursa M, Mechtler K, Kircheis R, Wagner E. Gene Ther. 1998;5:1425–1433. doi: 10.1038/sj.gt.3300745. [DOI] [PubMed] [Google Scholar]
- (43).Anwer K, Rhee BG, Mendiratta SK. Crit. Rev. Ther. Drug Carrier Syst. 2003;20:249–293. doi: 10.1615/critrevtherdrugcarriersyst.v20.i4.10. [DOI] [PubMed] [Google Scholar]
- (44).Pouton CW, Lucas P, Thomas BJ, Uduehi AN, Milroy DA, Moss SH. J. Controlled Release. 1998;53:289–299. doi: 10.1016/s0168-3659(98)00015-7. [DOI] [PubMed] [Google Scholar]
- (45).Godbey WT, Wu KK, Mikos AG. J. Biomed. Mater. Res. 1999;45:268–275. doi: 10.1002/(sici)1097-4636(19990605)45:3<268::aid-jbm15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]