

Abstract
For the past couple of decades, aging science has been rapidly evolving, and powerful genetic tools have identified a variety of evolutionarily conserved regulators and signaling pathways for the control of aging and longevity in model organisms. Nonetheless, a big challenge still remains to construct a comprehensive concept that could integrate many distinct layers of biological events into a systemic, hierarchical view of aging. The “heterochromatin island” hypothesis was originally proposed 10 years ago to explain deterministic and stochastic aspects of cellular and organismal aging, which drove the author to the study of evolutionarily conserved Sir2 proteins. Since a surprising discovery of their NAD-dependent deacetylase activity, Sir2 proteins, now called “sirtuins,” have been emerging as a critical epigenetic regulator for aging. In this review, I will follow the process of conceptual development from the heterochromatin island hypothesis to a novel, comprehensive concept of a systemic regulatory network for mammalian aging, named “NAD World,” summarizing recent studies on the mammalian NAD-dependent deacetylase Sirt1 and nicotinamide phosphoribosyltransferase (Nampt)-mediated systemic NAD biosynthesis. This new concept of the NAD World provides critical insights into a systemic regulatory mechanism that fundamentally connects metabolism and aging and also conveys the ideas of functional hierarchy and frailty for the regulation of aging in mammals.
Introduction
“That the System of Living contrived by me was unreasonable and unjust, because it supposed a Perpetuity of Youth, Health, and Vigour, which no Man could be so foolish to hope, however extravagant he may be in his Wishes. That the Question therefore was not whether a Man would choose to be always in the Prime of Youth, attended with Prosperity and Health, but how he would pass a perpetual Life under all the usual Disadvantages which old Age brings along with it.”
Jonathan Swift, “Gulliver’s Travels,” Part III, Chapter X, 1726
Even 283 years after Swift cast this philosophical question in his famous novel, we are still wondering the same question in our modern society. Although our average lifespan has been almost doubled compared to that in the 18th century, no one can avoid a gradual, global decline in physiological functions through the body, which causes a number of age-associated diseases, such as type 2 diabetes, Alzheimer disease, atherosclerosis, osteoporosis, and cancer. Therefore, we still carry a hope, deep in our minds, for “a Perpetuity of Youth, Health, and Vigour” in our daily life. This hope, however, has recently been empowered by rapidly evolving aging science that employs a variety of powerful genetic tools to analyze the molecular mechanisms of aging and longevity. For the past couple of decades, a number of evolutionarily conserved regulators and signaling pathways have been identified for the control of aging and longevity, using experimental model organisms, such as yeast, worms, flies, and mice [1-9]. Nonetheless, it is still a difficult challenge to reasonably connect many different kinds of molecular, cellular, and physiological events mediated by those regulators and signaling pathways and build up a comprehensive, hierarchical view of aging.
To dissect such a complex aging-regulatory network and ultimately construct a systemic view of aging in mammals, three fundamental components need to be identified and characterized in the presumed hierarchical structure of the aging-regulatory network. First, key molecular regulators that generate genetic and epigenetic alterations in cells and coordinate responses to a variety of nutritional and environmental stimuli through multiple tissues and organs need to be identified. The Sir2 (silent information regulator 2) protein family of NAD-dependent protein deacetylases/ADP-ribosyltransferases, called “sirtuins,” has drawn significant attention as one such regulator in the field of aging research as well as in many other research areas [2, 4, 10]. The role of mammalian sirtuins in the regulation of mammalian aging is currently a major focus. Second, critical signaling molecules and pathways that maintain global communication through tissues/organs and govern the pace of those genetic/epigenetic alterations at a systemic level also need to be identified. The importance of insulin/insulin-like growth factor-I (IGF-I) signaling (IIS) has been well established in worms and flies [1, 3, 5, 9]. In mammals, several genetic models, including IGF-I receptor heterozygous knockout mice [11] and fat-specific insulin receptor knockout (FIRKO) mice [12], provide support for the importance of IIS for the regulation of mammalian aging and longevity. In most cases, however, the results need to be further confirmed to clearly establish the importance of IIS in mammals [13]. Lastly, to elucidate the dynamics of the presumed aging-regulatory network, dominant organs and tissues that play critical roles at the top of the functional hierarchy in maintaining and/or affecting the robustness of the entire system over time need to be identified. No obvious clues have so far been obtained in mammals. In worms and flies, however, it has been suggested that some particular sensory neurons might play a critical role in the regulation of aging and longevity in these organisms [14, 15]. Despite these substantial progresses in the field of aging research, there is currently no comprehensive concept that could provide a hierarchical, systemic view of aging in mammals.
In this review, I will attempt to develop a novel, comprehensive concept of a systemic regulatory network for mammalian aging, named “NAD World,” summarizing recent studies on the functions of the mammalian NAD-dependent deacetylase Sirt1 and nicotinamide phosphoribosyltransferase (Nampt)-mediated systemic NAD biosynthesis in the regulation of metabolism and aging. To achieve this goal, I will follow the chronology of conceptual development towards the NAD World, starting with its prequel.
A conceptual starting point – the heterochromatin island hypothesis
In 1998, Hiroaki Kitano and I proposed a hypothesis named “heterochromatin island hypothesis of aging,” based on our molecular and computational studies on the cellular aging-associated transcriptional regulation of the interstitial collagenase [matrix metalloproteinase 1 (MMP-1)] gene [16]. Cellular aging, also referred to cellular senescence, is an in vitro model of limited cell proliferative capacity that has been suggested to have a link to organismal aging [17, 18]. Whereas the attrition of telomeric structures is one attractive link that has been investigated extensively, another interesting link is the epigenetic alteration of transcriptional regulation [19-21]. One such example is the regulation of MMP-1 gene expression, which is tightly coupled to both processes of cellular and organismal aging in humans [16, 22]. For example, MMP-1 expression and activity significantly increase in aged human fibroblasts in vitro and in vivo [23-25]. The transcriptional changes in MMP-1 expression during cellular aging are mediated through a cis-acting element located 1.7-kb upstream of the MMP-1 promoter, and Oct-1, a member of the POU domain family, mediates the repression of the MMP-1 gene through the interaction with an AT-rich sequence of this cis-element [26, 27]. Interestingly, during the process of cellular aging, the loss of Oct-1-mediated MMP-1 repression is accompanied by the release of Oct-1 from the nuclear periphery, where Oct-1 is colocalized with lamin B, an important component of the nuclear lamina [27]. The cells with no nuclear peripheral localization of Oct-1 show strong signals of the MMP-1 expression, and the number of these cells increases towards the final stage of cellular aging. These findings suggest that Oct-1-mediated transcriptional repressive structures are reorganized in the process of cellular aging, resulting in the induction of specific subsets of genes, including the MMP-1 gene.
On the other hand, we analyzed growth kinetics and gene expression patterns in cellular aging by conducting a series of extensive computer simulations using an object-oriented software system named the Virtual Cell Laboratory [28]. In this system, we were able to simulate growth kinetics and gene expression dynamics in a virtual cell population, not by using equation-oriented calculation, but by modeling over 50,000 virtual cells independently. After a systematic search of more than 30 mathematical models, we found a unique set of possible molecular mechanisms with appropriate parameter ranges. This computational analysis deduced three important features of a possible molecular device that could mediate cellular aging-associated changes in cell growth and gene expression profiles: (a) transcriptional repressive chromatin structures; (b) formed by many repeated functional units; and (c) inheritable through cell division cycles, but changeable at a certain probability. By implementing these features into virtual cells, we were able to reproduce growth kinetics and gene expression profiles that were highly consistent with our experimental data [28].
Importantly, both molecular and computational analyses brought the same emphasis on the importance of global transcriptional repressive structures and their systematic reorganization as a driving force of cellular aging-associated changes (Fig. 1) [16]. Furthermore, these presumed transcriptionally repressive functional units, defined as “heterochromatin islands,” provided fundamental characteristics to explain deterministic (programmed) and stochastic (error-driven) aspects of cellular and organismal aging. The idea that the reorganization in the different hierarchical orders of DNA structures, particularly heterochromatin structures, causes cellular aging has been discussed extensively [29-31]. In the heterochromatin island hypothesis, we speculated that while the polymorphic genomic regions for heterochromatin islands may determine the final potential consequences of aging and their intra- and interspecies varieties, the probabilistic dissociation or redirection of protein components among heterochromatin islands, which might be caused by DNA replication or DNA damage, can generate the heterogeneous onsets of various aging-related phenotypes even in the same genetic background [16].
Figure 1.
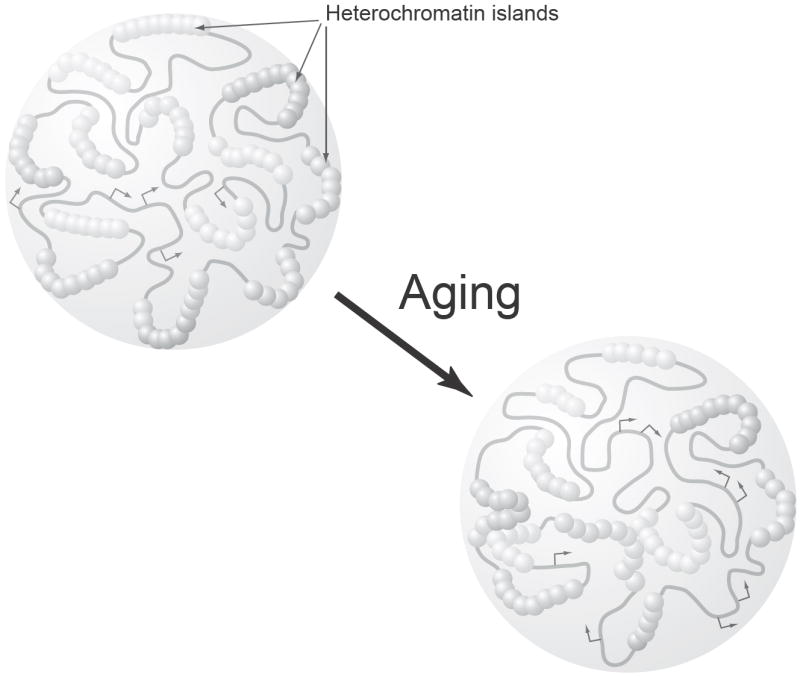
A schematic diagram of the heterochromatin island hypothesis. Heterochromatin islands are defined as global transcriptional repressive chromatin structures that are formed by many repeated functional units and inherited through cell division cycles, but displaced in a stochastic manner. Heterochromatin islands are formed at various loci across the mammalian genome, as depicted as complexes of particles. In the process of aging, protein components of heterochromatin islands are dissociated or redirected to other loci due to DNA damage and/or other systemic alterations, resulting in both silencing and desilencing of gene expression at specific loci (Arrows represent transcribed regions).
To address the heterochromatin island hypothesis, the most important was the identification of evolutionarily conserved regulatory components for heterochromatin islands. When the hypothesis was proposed, there were only a few candidates for such regulatory components, and among them, one strong candidate was the Sir2 family [16]. At that time, Leonard Guarente and his colleagues had reported that the Sir complex is redirected from telomeres to ribosomal DNA regions in cellular aging of the budding yeast Saccharomyces serevisiae [32]. Interestingly, among three components, namely Sir2, 3, and 4, of the yeast Sir complex, only Sir2 was known to have homologs in other species including mammals [33]. However, since its first identification as a critical regulator for yeast mating types in 1979 [34], the biochemical function of Sir2 had been totally unknown despite many researchers’ efforts.
Sirt1, a key regulator for the epigenetic regulation of cellular responses to nutritional and environmental stimuli
In 2000, Leonard Guarente and I made a surprising discovery that yeast and mammalian Sir2 proteins have NAD-dependent deacetylase activity and that this activity is essential for the longevity control in yeast [35]. Later, two other groups also confirmed this novel enzymatic activity [36, 37]. The absolute requirement of NAD for the deacetylase activity of Sir2 proteins immediately suggested that they might function as sensors of the cellular energy status represented by NAD and directly connect energy metabolism to the epigenetic transcriptional regulation [35, 38]. Since this discovery, Sir2 proteins have been emerging as evolutionarily conserved, critical regulators for aging and longevity in a wide variety of experimental organisms. For example, increasing the dosage or activity of Sir2 proteins extends the life spans of yeast, worms, and flies, while mutations of the Sir2 genes shorten their life spans [39-44]. In certain genetic backgrounds, Sir2 proteins are also required for the life span extension mediated by caloric restriction in yeast, worms, and flies [42, 45-48].
The biology of Sir2 proteins, now called “sirtuins,” has been rapidly evolving in many different research areas from bacteria to humans [2, 4, 10, 49]. Sirtuins couple NAD breakdown to deacetylation and/or ADP-ribosylation on lysine residues of target proteins. Their NAD-dependent deacetylation reaction produces nicotinamide, acetyl-ADP-ribose, and deacetylated proteins [50]. Numerous target proteins, including histones and a variety of transcription factors, have already been reported for sirtuins in different species. Mammals have seven sirtuin family members, named Sirt1 through Sirt7 [4, 10, 49]. Sirt1 is the mammalian Sir2 ortholog, and the majority of mammalian sirtuin research has so far focused on the function of Sirt1. Although it is still unclear whether Sirt1 regulates aging and longevity in mammals, increasing lines of evidence have firmly established that Sirt1 plays a critical role in the epigenetic regulation of cellular responses to nutritional and environmental stimuli in a variety of cell types.
The role of Sirt1 as a key metabolic regulator has been well established in multiple tissues, and many review articles have already addressed this aspect of Sirt1 function [49, 51-54]. Most importantly, Sirt1 mediates a variety of metabolic effects in response to nutritional cues, particularly to low nutritional input, and coordinates the production and the secretion of key metabolic hormones, such as insulin [55, 56] and adiponectin [57-59], and essential metabolites, such as glucose [60], cholesterol [61], and fatty acids [62]. These findings place Sirt1 at a central position as one of the fundamental components in the presumed systemic aging-regulatory network, as described above. The importance of Sirt1 as a key epigenetic regulator of metabolism has been further strengthened by recent findings that Sirt1 also regulates the circadian clock oscillatory mechanism and affects the expression of circadian clock genes [63, 64]. The core molecular clock machinery has been demonstrated to be one of the most powerful modifiers of metabolism [65, 66]. In this clock machinery, Clock and Bmal1 are critical regulators that heterodimerize and activate transcription of target genes, including the Period (Per1, 2, and 3) and Cryptochrome (Cry1 and 2) genes, through E-box cis-regulatory elements [67]. Sirt1 forms a complex with Clock and Bmal1, deacetylates Bmal1 or Per2 proteins, and regulates the amplitude and the duration of circadian gene expression. Therefore, through this epigenetic regulation of the core circadian clock oscillatory mechanism, it is likely that Sirt1 affects a number of rhythmic metabolic responses during the course of aging.
Most recently, David Sinclair and his colleagues have provided strong support for the candidacy of Sirt1 as a critical regulator in the heterochromatin island hypothesis [68]. They have demonstrated that in mouse embryonic stem (ES) cells, oxidative stress causes redistribution of Sirt1 across the genome, resulting in the desilencing of Sirt1-bound target genes, including major satellite repeats and seven genes related to metabolism, apoptosis, ion transport, cell motility, and G-protein signaling. Consistent with this finding, modest overexpression of Sirt1 represses six out of seven target genes, while inactivation of Sirt1 conveys opposite changes in their expression. This damage-induced Sirt1 redistribution requires DNA damage signaling through a mammalian PI3-kinase ATM and histone H2AX, and redistributed Sirt1 appears to be involved in DNA repair and genomic stability. These findings are consistent with the relocalization of yeast Sir proteins from the HM silent mating type loci to the DNA breakage sites in a DNA damage checkpoint-dependent manner [69-71]. Interestingly, six out of seven Sirt1-bound target genes that are derepressed by oxidative stress in mouse ES cells are also derepressed in aged mouse brain, and Sirt1 overexpression is able to suppress these age-associated changes. Based on these findings, the authors have proposed that the DNA damage-induced redistribution of chromatin modifiers, such as yeast Sir proteins and mammalian Sirt1, might be a conserved mechanism of aging [21, 68]. Consistent with their findings, we have also found that Sirt1 is able to suppress the MMP-1 gene expression in human fibroblasts (Fig. 2A). Given that the MMP-1 gene is well known to be induced by oxidative stress in human skin fibroblasts in vitro and in vivo [22, 23], it will be of great interest to examine whether Sirt1 can also be redistributed by oxidative stress in differentiated adult cells, such as human skin fibroblasts. This Sirt1-dependent repression of the MMP-1 gene appears to be mediated through the same cis-acting element that was demonstrated to be involved in the cellular aging-associated, Oct-1-mediated repression of the gene (Fig. 2B) [26, 27]. Therefore, it is conceivable that Sirt1 interacts with some specific partners to form heterochromatin islands and regulates the dynamic reorganization of heterochromatin islands in both undifferentiated and differentiated cells (Fig. 2C). Further investigation will be required to address these possibilities.
Figure 2.

Sirt1 as a critical epigenetic regulator in the heterochromatin island hypothesis. (A) Sirt1 suppresses the MMP-1 gene expression in human BJT fibroblasts, a polyclonal derivative of human BJ fibroblasts that are immortalized by ectopic expression of the telomerase enzyme. Real-time quantitative RT-PCR was performed to measure MMP-1 mRNA levels in parental and human Sirt1-overexpressing BJT fibroblasts. MMP-1 expression levels, which were normalized to GAPDH expression levels, were calculated relative to those in BJT cells. Results are expressed as mean ± standard error (n=3). (B) Sirt1 suppresses the transcriptional activity of the MMP-1 promoter in human BJT fibroblasts. Luciferase assays were conducted by transfecting BJT and BJT-hSirt1 fibroblasts with luciferase reporter plasmids driven by indicated MMP-1 promoter fragments. The cellular aging-associated, Oct-1-mediated repression of the MMP-1 gene is regulated by a cis-acting element located 1.7-kb upstream of the MMP-1 promoter [26, 27]. Results are expressed as mean ± standard error (-4400/+63-Luc, n=6; -2300/+64-Luc, n=5; -1600/+63-Luc, n=2). (C) A scheme for Sirt1-mediated heterochromatin islands and their reorganization during the process of aging. Oxidative stress and/or other systemic alterations, such as the reduction in systemic NAD biosynthesis, might trigger the reorganization of those structures.
Nampt-mediated systemic NAD biosynthesis, a global regulatory system for the Sirt1-mediated epigenetic regulation of physiological responses
The heterochromatin island hypothesis and recent findings on Sirt1 have provided important insights into a possible operational system that might comprise the molecular basis for deterministic and stochastic aspects of aging at a cellular level [16, 21]. However, this hypothesis itself is still not enough to build up a hierarchical, systemic view of aging. First, it is still unclear whether DNA damages responsible for the redistribution of Sirt1 are frequent and strong enough to cause irreversible desilencing of the Sirt1 target genes over time and is collectively able to induce a global deterioration in physiological functions at a systemic level. In other words, there might be other systemic events that could cause a significant change in Sirt1 activity and/or localization over time and thereby induce the desilencing of the same Sirt1 target genes through the body. Second, whereas this hypothesis conveys an important concept regarding an aging-inducible epigenetic mechanism at a cellular level, it does not provide any indication on what might control the pace of such epigenetic alterations through tissues and organs at a systemic level. Finally, this hypothesis is not able to predict any hierarchical structure of the presumed systemic aging-regulatory network. It is critical to find which tissues and organs play dominant roles at the top of the functional hierarchy in the regulation of mammalian aging. Therefore, a novel concept that could be complementary to the heterochromatin island hypothesis and provide clues to system dynamics in aging is necessary to construct a comprehensive, hierarchical view of aging.
A key to address those problems is the absolute requirement of NAD for sirtuin enzymatic activity. Due to this unique nature, NAD biosynthesis comes to play a critical role in the regulation of sirtuin functions [72-74]. NAD is synthesized from three major precursors – tryptophan, nicotinic acid, and nicotinamide. While lower eukaryotes and invertebrates, such as yeast, worms, and flies, use nicotinic acid (a form of vitamin B3) as a major NAD precursor, mammals predominantly use nicotinamide (another form of vitamin B3) rather than nicotinic acid for NAD biosynthesis (Fig. 3) [73, 75, 76]. In lower eukaryotes and invertebrates, nicotinamide undergoes deamidation to nicotinic acid, a reaction catalyzed by a nicotinamidase encoded by the Pnc1 gene [45, 77], and nicotinic acid is converted to nicotinic acid mononucleotide (NaMN) by nicotinic acid phosphoribosyltransferase (Npt) (Fig. 3A). There are two more steps to convert NaMN to NAD, and therefore, there are four steps as a total from nicotinamide to NAD in those organisms. It has been demonstrated that enzymes involved in this NAD biosynthetic pathway play a critical role in the regulation of Sir2 activity and aging in yeast [45, 78, 79]. On the other hand, mammals directly use nicotinamide to synthesize NAD, and there are only two steps in this NAD biosynthetic pathway (Fig. 3B). Nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in this pathway [80], initiates NAD biosynthesis by catalyzing the synthesis of nicotinamide mononucleotide (NMN) from nicotinamide and 5-phosphoribosyl-pyrophosphate (PRPP) (Fig. 3B). Nicotinamide/nicotinic acid mononucleotide adenylyltransferase (Nmnat) then completes NAD biosynthesis by converting NMN to NAD. Whereas the pathways starting from tryptophan and nicotinic acid are highly conserved through evolution, the Nampt-mediated pathway from nicotinamide shows a peculiar phylogenetic trait. For example, Nampt has ancient origins as an NAD biosynthetic enzyme, and the entire pyridine nucleotide salvage cycle containing Nampt, Nmnat, and Sir2 homologs exists even in the vibriophage [81]. Despite its ancient origins, no organisms between bacteria and vertebrates have obvious homologs of Nampt, except for a few species [80, 82]. Nevertheless, the homology of Nampt proteins between bacteria and vertebrates is unusually high [80]. Interestingly, the organisms that do not have Nampt homologs, such as yeast, worms, and flies, unanimously have Pnc1 homologs, while vertebrates have no obvious Pnc1 homologs [76, 77]. Therefore, the presence of Nampt clearly distinguishes the dynamics of NAD biosynthesis in mammals from that in lower eukaryotes and invertebrates.
Figure 3.

NAD biosynthetic pathways from nicotinamde and nicotinic acid in yeast, invertebrates, and mammals. (A) The NAD biosynthetic pathways in the budding yeast Saccharomyces cerevisiae and invertebrates, such as C. elegans and Drosophila. Steps from nicotinamide to NAD are shown with grey arrows. Pnc1, Npt1, Nma1 and Nma2, and Qns1 are nicotinamidase, nicotinic acid phosphoribosyltransferase, nicotinic acid mononucleotide adenylyltransferase 1 and 2, and NAD synthetase, respectively. Only Sir2 is shown as a representative NAD-dependent protein deacetylase. NaMN, nicotinic acid mononucleotide. (B) The NAD biosynthetic pathways in mammals. The de novo pathway from tryptophan and the NAD biosynthetic pathway from nicotinic acid are evolutionarily conserved, while the NAD biosynthetic pathway from nicotinamide (grey arrows) is vertebrate-specific and mediated by Nampt and Nmnat. Nicotinamide is the main precursor for NAD biosynthesis in mammals. While multiple enzymes break NAD into nicotinamide and ADP-ribose, only Sirt1 is shown here. NMN, nicotinamide mononucleotide.
In mammals, Nampt has both intra- and extracellular forms (iNampt and eNampt, respectively) [72, 73]. The enzymological, structural, and genetic analyses have firmly established the function of iNampt as an NAD biosynthetic enzyme [80, 82-85]. Whereas eNampt also exhibits robust, even higher NAD biosynthetic activity compared to iNampt [86], eNampt appears to have a cytokine-like function that does not require its enzymatic activity [87]. Indeed, eNampt has been documented as a presumptive cytokine named PBEF (pre-B cell colony-enhancing factor) [88] or a controversial insulin-mimetic hormone named visfatin [89] (the original visfatin paper has recently been retracted because of the irreproducibility of the results [90]). There are several excellent reviews that discuss the function of eNampt as an adipocytokine extensively [72, 73, 91, 92], and therefore, I will focus on the NAD biosynthetic function of both iNampt and eNampt in this review.
Nampt-mediated NAD biosynthesis has been demonstrated to play a critical role in the regulation of Sirt1 activity [72-74]. Increased dosage of iNampt enhances total cellular NAD levels and thereby augments the transcriptional regulatory activity of Sirt1 in mammalian cells [80]. Indeed, increasing the dosage of iNampt or Sirt1 induces common gene expression changes in mouse fibroblasts. It has also been shown that in human vascular smooth muscle cells (SMCs), iNampt regulates NAD biosynthesis and thereby Sirt1 activity and promotes SMC maturation [93]. iNampt also promotes cellular life span of SMCs through the Sirt1-mediated deacetylation and degradation of p53 [94]. In cardiac myocytes, increased dosage of iNampt protects them from cell death through increased NAD production and enhanced Sirt1 activity [95]. Most recently, it has been demonstrated that glucose restriction inhibits differentiation of skeletal myoblasts through the AMP-activated protein kinase (AMPK)-dependent induction of Nampt expression and the resultant activation of Sirt1, indicating that the AMPK-Nampt-Sirt1 pathway might function as a key signaling pathway in response to reduced nutrient availability [96]. iNampt also plays an important role in NAD biosynthesis and sirtuin activation in mitochondria [97]. iNampt expression is induced by a variety of cellular stress and nutrient restriction. While increasing iNampt levels do not prevent depletion of total cellular NAD under genotoxic stress, it maintains mitochondrial NAD levels and protects cells against cell death through mitochondrial sirtuins, Sirt3 and Sirt4. Therefore, Nampt-mediated NAD biosynthesis regulates a wide variety of important biological processes, including cellular differentiation, stress response, and metabolism, through the activation of Sirt1 and other sirtuins in diverse cell types.
Interestingly, both Sirt1 and Nampt-mediated NAD biosynthesis play important roles in the regulation of glucose-stimulated insulin secretion (GSIS) in pancreatic β cells [55, 56, 86]. We and others have demonstrated that Sirt1 regulates GSIS in part by repressing the expression of uncoupling protein 2 (Ucp2), a mitochondrial proton transporter that uncouples respiration from ATP production [55, 56]. Indeed, pancreatic β cell-specific Sirt1-overexpressing (BESTO) transgenic mice show enhanced insulin secretion and improved glucose tolerance in response to glucose challenge [56]. We have also demonstrated that NAD biosynthesis mediated by iNampt and eNampt plays a critical role in the regulation of GSIS in pancreatic β cells [86]. Nampt heterozygous (Nampt+/-) female mice show impaired glucose tolerance due to a defect in GSIS. Primary Nampt+/- islets also show defects in NAD biosynthesis and GSIS. These phenotypes in Nampt+/- mice and islets can be completely ameliorated by administration of NMN, a product of the Nampt reaction (Fig. 3B), strongly indicating that the observed defects are due to a lack of Nampt-mediated NAD biosynthesis. Furthermore, FK866, a potent chemical inhibitor of Nampt, significantly inhibits NAD biosynthesis and GSIS in isolated wild-type primary islets, and administration of NMN restores normal NAD biosynthesis and GSIS in FK866-treated wild-type primary islets. Remarkably, only Nampt+/- females, but not males, have reduced plasma levels of eNampt and NMN as well as defects in glucose metabolism [86]. Although it is currently unclear why and how Nampt+/- males maintain their plasma eNampt and NMN levels, these results suggest that the systemic maintenance of high NMN levels by eNampt in blood circulation is critical for normal β cell function, probably because pancreatic islets have very low levels of iNampt compared to other tissues so that they rely on NMN in blood circulation to maintain sufficient NAD biosynthesis for their function. This particular notion has been further supported by our recent study on aged BESTO mice [98]. We have found that aged BESTO mice completely lose all phenotypes of enhanced β cell function and that Sirt1 activity is significantly reduced in aged β cells. This seems to be due to a significant reduction in plasma NMN levels over age, and consistent with this notion, administration of NMN restores enhanced GSIS and improved glucose tolerance at least in aged BESTO females [98]. Therefore, these findings clearly suggest that Nampt-mediated NAD biosynthesis declines over age at a systemic level, resulting in reduced Sirt1 activity and GSIS in aged β cells.
Importantly, these studies have brought a novel physiological framework that integrates both Nampt-mediated NAD biosynthesis and Sirt1 into a unified systemic regulatory network (Fig. 4A) [52, 73, 99]. While NMN is synthesized from nicotinamide by iNampt in a variety of tissues and organs, a significant amount of NMN may also be synthesized by eNampt in blood circulation and distributed to organs and tissues through circulation. We are now able to detect NMN as well as other NAD intermediates in both mouse and human plasma (unpublished observations). Once NMN is incorporated from blood circulation into each tissue/organ, this exogenous NMN is rapidly converted to NAD by Nmnat, promoting total NAD biosynthesis and thereby contributing to the regulation of Sirt1 activity. Therefore, NAD biosynthesis mediated by iNampt and eNampt functions as a global regulatory system for the Sirt1-mediated epigenetic regulation of physiological responses through the intra- and extracellular biosynthesis of NMN and the distribution of NMN through the entire body (Fig. 4A). In the next section, I will expand these discussions and introduce the concept of the NAD World that provides a systemic, hierarchical view of mammalian aging.
Figure 4.

Conceptual schemes of the NAD World and mammalian aging. (A) The NAD World is a novel systemic regulatory network that fundamentally connects metabolism and aging in mammals. The NAD World comprise two critical components: the NAD-dependent deacetylase Sirt1 as a universal epigenetic regulator that executes various metabolic effects in a tissue/organ dependent manner in response to changes in systemic NAD biosynthesis, and Nampt-mediated systemic NAD biosynthesis as a global regulatory system that fine-tunes Sirt1 activity through intra- and extracellular biosynthesis of NMN and controls the pace of epigenetic alterations in Sirt1-mediated physiological effects. See texts for details. (B) A model of mammalian aging as the process of organismal robustness breakdown triggered by a decline in systemic NAD biosynthesis. This cascade of robustness breakdown might cause a variety of age-associated complications, including type 2 diabetes and dementia. See texts for details.
The NAD World: a novel systemic, hierarchical view of mammalian aging
As discussed in the previous sections, Sirt1 serves as a universal epigenetic regulator that mediates physiological responses to a variety of nutritional and environmental stimuli in a tissue/organ-dependent manner and controls those regulatory effects in response to systemic NAD biosynthesis. On the other hand, Nampt-mediated systemic NAD biosynthesis functions as a global regulatory system that fine-tunes Sirt1 activity in each tissue/organ through intra- and extracellular biosynthesis of NMN and governs the pace of epigenetic alterations in Sirt1-mediated physiological effects at a systemic level. These two critical components comprise a novel systemic regulatory network, named “NAD World,” that fundamentally connects metabolism and aging and also conveys the ideas of functional hierarchy and frailty for the regulation of aging in mammals (Fig. 4A) [99]. Most importantly, the concept of the NAD World enables us to address the following key questions in mammalian aging: 1) which organs and tissues might play dominant roles in the systemic regulation of mammalian aging? 2) what might control the pace of Sirt1-mediated, aging-inducible epigenetic changes at a systemic level? 3) how can we manipulate the process of aging in mammals?
To address the first question, the key is to find frailty points in the NAD World. In the NAD World, frailty points are tissues/organs that do not have adequate amounts of iNampt and therefore rely on circulating NMN to maintain sufficient NAD biosynthesis for their functions. If systemic NAD biosynthesis starts declining, these frailty points would respond first to this change and start having functional problems due to inadequate NAD biosynthesis and thereby reduced Sirt1 activity. Pancreatic β cells and brain (neurons), both of which have very low levels of iNampt [86], are likely the most critical frailty points in the NAD World because of their systemic impacts on many other tissues/organs (Fig. 4B). As discussed above, a decrease in systemic NAD biosynthesis causes reduced Sirt1 activity and GSIS in pancreatic β cells [86, 98], resulting in impaired glucose tolerance and possibly type 2 diabetes when the problem persists. Indeed, a progressive age-associated decline in β cell function has been suggested to be one of the major contributing factors to the pathogenesis of type 2 diabetes, one of the major age-associated complications in our modern society [100-102]. Therefore, pancreatic β cells are clearly an important frailty point in the NAD World that is susceptible to changes in Nampt-mediated systemic NAD biosynthesis. Similarly, brain is likely another critical frailty point in the NAD World. Interestingly, it has long been known that one of the triad symptoms in pellagra, the vitamin B3 deficiency, is dementia [103]. Although it has been unknown why the vitamin B3 deficiency causes dementia, it is very likely that reduced systemic NAD biosynthesis might cause functional deficits in neurons and result in neurological problems, including dementia, in brain. It is also conceivable that Sirt1 might regulate critical molecular processes in brain that contribute to the pathogenesis of neurological problems caused by a decline in systemic NAD biosynthesis. Once pancreatic β cells and brain start having functional problems due to inadequate NAD biosynthesis, other peripheral tissues/organs would also be affected through insulin secretion and central metabolic regulation so that the physiological robustness would gradually deteriorate over age at a systemic level (Fig. 4B). This cascade of robustness breakdown triggered by a decrease in systemic NAD biosynthesis might be the central process of aging. In other words, aging might be the process in which organismal robustness gradually shifts and eventually breaks down according to a functional hierarchy determined by the susceptibility to systemic NAD biosynthesis (Fig. 4B). Therefore, due to their frailty in the NAD World, pancreatic β cells and neurons, two major cell types that are well known to cause serious age-associated complications, might play dominant roles at the top of this functional hierarchy in the systemic regulation of mammalian aging.
In these frailty points, an age-associated decline in systemic NAD biosynthesis must make a much more profound impact on Sirt1 activity compared to other tissues and organs. In this regard, it will be of great interest to examine whether such a global decline in systemic NAD biosynthesis might be able to cause the desilencing of Sirt1-bound target genes in pancreatic β cells and brain in vivo. This presumed desilencing of Sirt1-bound target genes caused by reduced systemic NAD biosynthesis might or might not require the redistribution of Sirt1 across the genome in these cell types. Nevertheless, it will be very important to identify Sirt1-mediated “heterochromatin islands” in those frailty points and examine whether those heterochromatin islands are altered in response to an age-associated decline in systemic NAD biosynthesis. In pancreatic β cells, those presumed Sirt1-mediated heterochromatin islands might be present on the genes that are involved in the regulation of insulin production and secretion. In brain, Sirt1-mediated heterochromatin islands might exist and play an important role in neurons in hippocampus and hypothalamus based on its nuclear localization in these regions (unpublished observations). Although more vigorous studies will be necessary to address these possibilities, one can speculate that in this scenario, circulating NMN may serve as a critical plasma metabolite that controls the pace of Sirt1-mediated, aging-inducible epigenetic changes at a systemic level. Given that circulating NMN is produced by eNampt mainly secreted by adipose tissue (Fig. 4A), there might be a feedback regulatory mechanism by which the production and the secretion of Nampt are regulated among pancreatic β cells, brain, and adipose tissue. It will also be of great importance to elucidate a molecular mechanism by which Nampt-mediated systemic NAD biosynthesis declines over age.
No matter what the mechanism would be for the age-associated decline in Nampt-mediated systemic NAD biosynthesis, NMN administration is likely able to overcome complications caused by reduced systemic NAD biosynthesis, as demonstrated in Nampt+/- females and aged BESTO females [86, 98]. Because the pharmacokinetics of NMN is totally unknown in mammals, it is hard to estimate how much NMN might be necessary to prevent or treat age-associated complications at this moment. Nonetheless, because NMN functions as an important plasma metabolite that regulates NAD biosynthesis and thereby Sirt1 activity in the NAD World (Fig. 4A), this compound and its related physiology will open a new avenue towards the development of effective interventions to manipulate the process of aging in mammals. Investigation is currently underway to examine the efficacy of this attractive compound.
In this review, I followed the chronology of the conceptual development from the heterochromatin island hypothesis to the concept of the NAD World to build up a systemic, hierarchical view of mammalian aging. In coming years, more details will be investigated for the system structure and its dynamics of the NAD World, and the knowledge might enable us to effectively intervene within such a complex systemic regulatory network for mammalian aging and possibly to achieve “a Perpetuity of Youth, Health, and Vigour”, which Swift had once considered foolish to hope. At that point, it might be interesting for us to reconsider how we could answer the challenging question that he cast almost 300 years ago.
Acknowledgments
I thank all members of the Imai lab for their insightful discussions and comments for the concept of the NAD World. I also thank Graziella Mendonsa for her work on the regulation of the MMP-1 gene by Sirt1. I apologize to those whose work is not cited due to the focus of this review and space limitations. This work was supported by grant from the National Institute on Aging (AG024150), Ellison Medical Foundation, and Longer Life Foundation to S. I.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berryman DE, Christiansen JS, Johannsson G, Thorner MO, Kopchick JJ. Role of the GH/IGF-1 axis in lifespan and healthspan: Lessons from animal models, Growth Horm. IGF Res. 2008;18:455–471. doi: 10.1016/j.ghir.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 3.Brown-Borg HM. Hormonal control of aging in rodents: The somatotropic axis. Mol Cell Endocrinol. 2008 doi: 10.1016/j.mce.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imai S, Guarente L. Sirtuins: A universal link between NAD, metabolism, and aging. In: Guarente L, Partridge L, Wallace D, editors. The Molecular Biology of Aging. Cold Spring Habor Laboratory Press; New York: 2007. pp. 39–72. [Google Scholar]
- 5.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Murphy MP, Partridge L. Toward a control theory analysis of aging. Annu Rev Biochem. 2008;77:777–798. doi: 10.1146/annurev.biochem.77.070606.101605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005;126:987–1002. doi: 10.1016/j.mad.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 8.Steinkraus KA, Kaeberlein M, Kennedy BK. Replicative aging in yeast: the means to the end. Annu Rev Cell Dev Biol. 2008;24:29–54. doi: 10.1146/annurev.cellbio.23.090506.123509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- 10.Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 11.Holzenberger M, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 12.Blüher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 13.Liang H, Masoro EJ, Nelson JF, Strong R, McMahan CA, Richardson A. Genetic mouse models of extended lifespan. Exp Gerontol. 2003;38:1353–1364. doi: 10.1016/j.exger.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 14.Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007;8:835–844. doi: 10.1038/nrg2188. [DOI] [PubMed] [Google Scholar]
- 15.Libert S, Pletcher SD. Modulation of longevity by environmental sensing. Cell. 2007;131:1231–1234. doi: 10.1016/j.cell.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Imai S, Kitano H. Heterochromatin islands and their dynamic reorganization: a hypothesis for three distinctive features of cellular aging. Exp Gerontol. 1998;33:555–570. doi: 10.1016/s0531-5565(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 17.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech Ageing Dev. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narita M. Cellular senescence and chromatin organisation. Br J Cancer. 2007;96:686–691. doi: 10.1038/sj.bjc.6603636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23:413–418. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Oberdoerffer P, Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol. 2007;8:692–702. doi: 10.1038/nrm2238. [DOI] [PubMed] [Google Scholar]
- 22.Callaghan TM, Wilhelm KP. A review of ageing and an examination of clinical methods in the assessment of ageing skin. Part I: Cellular and molecular perspectives of skin ageing. Int J Cosmet Sci. 2008;30:313–322. doi: 10.1111/j.1468-2494.2008.00454.x. [DOI] [PubMed] [Google Scholar]
- 23.Fisher GJ, et al. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am J Pathol. 2009;174:101–114. doi: 10.2353/ajpath.2009.080599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burke EM, Horton WE, Pearson JD, Crow MT, Martin GR. Altered transcriptional regulation of human interstitial collagenase in cultured skin fibroblasts from older donors. Exp Gerontol. 1994;29:37–53. doi: 10.1016/0531-5565(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 25.West MD, Pereira-Smith OM, Smith JR. Replicative senescence of human skin fibroblasts correlates with a loss of regulation and overexpression of collagenase activity. Exp Cell Res. 1989;184:138–147. doi: 10.1016/0014-4827(89)90372-8. [DOI] [PubMed] [Google Scholar]
- 26.Imai S, Fujino T, Nishibayashi S, Manabe T, Takano T. Immortalization-susceptible elements and their binding factors mediate rejuvenation of regulation of the type I collagenase gene in simian virus 40 large T antigen-transformed immortal human fibroblasts. Mol Cell Biol. 1994;14:7182–7194. doi: 10.1128/mcb.14.11.7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imai S, et al. Dissociation of Oct-1 from the nuclear peripheral structure induces the cellular aging-associated collagenase gene expression. Mol Biol Cell. 1997;8:2407–2419. doi: 10.1091/mbc.8.12.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitano H, Imai S. The two process model of cellular aging. Exp Gerontol. 1998;33:393–419. doi: 10.1016/s0531-5565(98)00008-4. [DOI] [PubMed] [Google Scholar]
- 29.Macieira-Coelho A. Chromatin reorganization during senescence of proliferating cells. Mutat Res. 1991;256:81–104. doi: 10.1016/0921-8734(91)90003-t. [DOI] [PubMed] [Google Scholar]
- 30.Howard BH. Replicative senescence: considerations relating to the stability of heterochromatin domains. Exp Gerontol. 1996;31:281–293. doi: 10.1016/0531-5565(95)00022-4. [DOI] [PubMed] [Google Scholar]
- 31.Villeponteau B. The heterochromatin loss model of aging. Exp Gerontol. 1997;32:383–394. doi: 10.1016/s0531-5565(96)00155-6. [DOI] [PubMed] [Google Scholar]
- 32.Kennedy BK, et al. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell. 1997;89:381–391. doi: 10.1016/s0092-8674(00)80219-6. [DOI] [PubMed] [Google Scholar]
- 33.Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995;9:2888–2902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 34.Klar AJS, Fogel S, MacLeod K. MAR1-A regulator of the HMa and HMα loci in Saccharomyces cerevisiae. Genetics. 1979;93:37–50. doi: 10.1093/genetics/93.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 36.Landry J, et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USA. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith JS, et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97:6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imai S, Johnson FB, Marciniak RA, McVey M, Park PU, Guarente L. Sir2: An NAD-dependent histone deacetylase that connects chromatin silencing, metabolism, and aging. Cold Spring Harbor Symp Quant Biol. 2000;65:297–302. doi: 10.1101/sqb.2000.65.297. [DOI] [PubMed] [Google Scholar]
- 39.Astrom SU, Cline TW, Rine J. The Drosophila melanogaster sir2+ gene Is nonessential and has only minor effects on position-effect variegation. Genetics. 2003;163:931–937. doi: 10.1093/genetics/163.3.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howitz KT, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 41.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 44.Wood JG, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 45.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin S-J, Defossez P-A, Guarente L. Life span extension by calorie restriction in S. cerevisiae requires NAD and SIR2. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 47.Lin S-J, et al. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Tissenbaum HA. Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech Ageing Dev. 2006;127:48–56. doi: 10.1016/j.mad.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 49.Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med. 2007;39:335–345. doi: 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- 50.Moazed D. Enzymatic activities of Sir2 and chromatin silencing. Curr Opin Cell Biol. 2001;13:232–238. doi: 10.1016/s0955-0674(00)00202-7. [DOI] [PubMed] [Google Scholar]
- 51.Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- 52.Imai S, Kiess W. Therapeutic potential of SIRT1 and NAMPT-mediated NAD biosynthesis in type 2 diabetes. Front Biosci. 2008 doi: 10.2741/3428. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milne JC, Denu JM. The Sirtuin family: therapeutic targets to treat diseases of aging. Curr Opin Chem Biol. 2008;12:11–17. doi: 10.1016/j.cbpa.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 54.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bordone L, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moynihan KA, et al. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 57.Banks AS, et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/EBPalpha transcriptional complex. J Biol Chem. 2006;281:39915–39924. doi: 10.1074/jbc.M607215200. [DOI] [PubMed] [Google Scholar]
- 59.Wang H, Qiang L, Farmer SR. Identification of a domain within peroxisome proliferator-activated receptor gamma regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Mol Cell Biol. 2008;28:188–200. doi: 10.1128/MCB.00992-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 61.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 62.Picard F, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asher G, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 64.Nakahata Y, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134:728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramsey KM, Marcheva B, Kohsaka A, Bass J. The clockwork of metabolism. Annu Rev Nutr. 2007;27:219–240. doi: 10.1146/annurev.nutr.27.061406.093546. [DOI] [PubMed] [Google Scholar]
- 67.Lowrey PL, Takahashi JS. Genetics of the mammalian circadian system: Photic entrainment, circadian pacemaker mechanisms, and posttranslational regulation. Annu Rev Genet. 2000;34:533–562. doi: 10.1146/annurev.genet.34.1.533. [DOI] [PubMed] [Google Scholar]
- 68.Oberdoerffer P, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 70.McAinsh AD, Scott-Drew S, Murray JA, Jackson SP. DNA damage triggers disruption of telomeric silencing and Mec1p-dependent relocation of Sir3p. Curr Biol. 1999;9:963–966. doi: 10.1016/s0960-9822(99)80424-2. [DOI] [PubMed] [Google Scholar]
- 71.Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- 72.Garten A, Petzold S, Körner A, Imai S, Kiess W. Nampt: Linking NAD biology, metabolism, and cancer. Trends Endocrinol Metab. 2009 doi: 10.1016/j.tem.2008.10.004. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Imai S. Nicotinamide phosphoribosyltransferase (Nampt): A link between NAD biology, metabolism, and diseases. Curr Pharm Des. 2009;15:20–28. doi: 10.2174/138161209787185814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang H, Lavu S, Sinclair DA. Nampt/PBEF/Visfatin: a regulator of mammalian health and longevity? Exp Gerontol. 2006;41:718–726. doi: 10.1016/j.exger.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol. 2007;23:164–170. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- 76.Rongvaux A, Andris F, Van Gool F, Leo O. Reconstructing eukaryotic NAD metabolism. Bioessays. 2003;25:683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- 77.Ghislain M, Talla E, Francois JM. Identification and functional analysis of the Saccharomyces cerevisiae nicotinamidase gene, PNC1. Yeast. 2002;19:215–324. doi: 10.1002/yea.810. [DOI] [PubMed] [Google Scholar]
- 78.Anderson R, et al. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J Biol Chem. 2002;277:18881–18890. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- 79.Gallo CM, Smith DL, Jr, Smith JS. Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol Cell Biol. 2004;24:1301–1312. doi: 10.1128/MCB.24.3.1301-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 81.Miller ES, et al. Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. J Bacteriol. 2003;185:5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rongvaux A, et al. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–3234. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 83.Khan JA, Tao X, Tong L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat Struct Mol Biol. 2006;13:582–588. doi: 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- 84.Kim MK, et al. Crystal Structure of Visfatin/Pre-B Cell Colony-enhancing Factor 1/Nicotinamide Phosphoribosyltransferase, Free and in Complex with the Anti-cancer Agent FK-866. J Mol Biol. 2006;362:66–77. doi: 10.1016/j.jmb.2006.06.082. [DOI] [PubMed] [Google Scholar]
- 85.Wang T, Zhang X, Bheda P, Revollo JR, Imai S, Wolberger C. Structure of Nampt/PBEF/visfatin, a mammalian NAD(+) biosynthetic enzyme. Nat Struct Mol Biol. 2006;13:661–662. doi: 10.1038/nsmb1114. [DOI] [PubMed] [Google Scholar]
- 86.Revollo JR, et al. Nampt/PBEF/visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Y, et al. Extracellular Nampt promotes macrophages survival via a non-enzymatic interleukin-6/STAT3 signaling mechanism. J Biol Chem. 2008 doi: 10.1074/jbc.M805866200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–1437. doi: 10.1128/mcb.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fukuhara A, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 90.Fukuhara A, et al. Retraction. Science. 2007;318:565b. doi: 10.1126/science.318.5850.565b. [DOI] [PubMed] [Google Scholar]
- 91.Luk T, Malam Z, Marshall JC. Pre-B cell colony-enhancing factor (PBEF)/visfatin: a novel mediator of innate immunity. J Leukoc Biol. 2008;83:804–816. doi: 10.1189/jlb.0807581. [DOI] [PubMed] [Google Scholar]
- 92.Sethi JK. Is PBEF/visfatin/Nampt an authentic adipokine relevant to the metabolic syndrome? Curr Hypertens Rep. 2007;9:33–38. doi: 10.1007/s11906-007-0007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van der Veer E, Nong Z, O’Neil C, Urquhart B, Freeman D, Pickering JG. Pre-B-cell colony-enhancing factor regulates NAD+-dependent protein deacetylase activity and promotes vascular smooth muscle cell maturation. Circ Res. 2005;97:25–34. doi: 10.1161/01.RES.0000173298.38808.27. [DOI] [PubMed] [Google Scholar]
- 94.van der Veer E, et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem. 2007;282:10841–10845. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 95.Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J Biol Chem. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- 96.Fulco M, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang H, et al. Nutrient-sensitive mitochondrial NAD(+) levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in β cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Imai SI. The NAD World: A New Systemic Regulatory Network for Metabolism and Aging-Sirt1, Systemic NAD Biosynthesis, and Their Importance. Cell Biochem Biophys. 2009 doi: 10.1007/s12013-008-9041-4. Epub on Jan 7, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Basu R, et al. Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes. 2003;52:1738–1748. doi: 10.2337/diabetes.52.7.1738. [DOI] [PubMed] [Google Scholar]
- 101.Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Jarvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab. 1999;84:863–868. doi: 10.1210/jcem.84.3.5542. [DOI] [PubMed] [Google Scholar]
- 102.Muzumdar R, Ma X, Atzmon G, Vuguin P, Yang X, Barzilai N. Decrease in glucose-stimulated insulin secretion with aging is independent of insulin action. Diabetes. 2004;53:441–446. doi: 10.2337/diabetes.53.2.441. [DOI] [PubMed] [Google Scholar]
- 103.Williams AC, Ramsden DB. Pellagra: A clue as to why energy failure causes diseases? Med Hypotheses. 2007;69:618–628. doi: 10.1016/j.mehy.2007.01.029. [DOI] [PubMed] [Google Scholar]