

Abstract
Emerging evidence implicates air pollution as a chronic source of neuroinflammation, reactive oxygen species (ROS), and neuropathology instigating central nervous system (CNS) disease. Stroke incidence, and Alzheimer’s and Parkinson’s disease pathology are linked to air pollution. Recent reports reveal that air pollution components reach the brain. Further, systemic effects known to impact lung and cardiovascular disease also impinge upon CNS health. While mechanisms driving air pollution-induced CNS pathology are poorly understood, new evidence suggests that activation of microglia and changes in the blood brain barrier may be key to this process. Here, we summarize recent findings detailing the mechanisms through which air pollution reaches the brain and activates the resident innate immune response to become a chronic source of pro-inflammatory factors and ROS culpable in CNS disease.
Inflammation is increasingly recognized as a causal factor in the pathology and chronic nature of central nervous system (CNS) diseases 1. While diverse environmental factors have been implicated in neuroinflammation leading to CNS pathology, air pollution may rank as the most prevalent source of environmentally induced inflammation and oxidative stress 2. Traditionally associated with increased risk for pulmonary 3 and cardiovascular disease 4, air pollution is now also associated with diverse CNS diseases, including Alzheimer’s disease, Parkinson’s Disease, and stroke.
Air pollution is a multifaceted environmental toxin capable of assaulting the CNS through diverse pathways. Until recently, the mechanisms responsible for air pollution-induced pathology in the brain were unknown. However, despite the variable chemical and physical characteristics of air pollution and the consequent activation of multiple pathways, inflammation and oxidative stress are identified as common and basic mechanisms through which air pollution causes damage 4, including CNS effects. Furthermore, while multiple cell types in the brain respond to exposure to air pollution, new reports indicate that microglia and brain capillaries may be critical actors responsible for cellular damage. In the following review, we describe the complex composition of air pollution, explain current views on the multifaceted mechanisms through which air pollution impacts the CNS, and discuss the new mechanistic findings implicating innate immunity and chronic neuroinflammation in CNS damage induced by air pollution.
Air Pollution Defined
Air pollution is comprised of a diverse mixture of particulate matter (PM), gases (e.g. ground-level ozone, carbon monoxide, sulfur oxides, nitrogen oxides), organic compounds (e.g. polycyclic aromatic hydrocarbons and endotoxins) and metals (e.g. vanadium, nickel, and manganese) present in outdoor and indoor air 5. Of these components, particle pollution and ground-level ozone are the most widespread health threats and have been heavily implicated in disease 2, 4. In fact, millions of people in the USA and around the world are chronically exposed to concentrations of air pollutants above promulgated standards of safety 5.
PM is especially relevant for CNS effects and is present in urban air pollution as a mixture of solid particles and liquid droplets suspended in the air. The size of PM contributes to their biological effects, where sizes vary from coarse wind blown dust particles to ultrafine particles. Ambient particles are characterized by size and aerodynamic properties: coarse particles with aerodynamic diameter of 2.5 to 10 μm (PM10), fine particles less than 2.5 μm (PM2.5), and ultrafine particles (UFPM) less than 0.1 μm. The PM10 particles are the respirable fraction originating from sources such as road and agricultural dust, tire wear emissions, wood combustion, construction and demolition works, and mining operations 2. PM2.5 are formed from gas and condensation of high temperature vapors during combustion and industrial activities. Thus, PM2.5 are composed of both organic and inorganic compounds, including sulfates, nitrates, carbon, ammonium, hydrogen ions, lipopolysaccharides (LPS), metals, and water 2, characteristics that contribute to their toxicity. Major sources of PM2.5 include oil refineries, metal processing facilities, tailpipe and brake emissions from mobile sources, residential fuel combustion, power plants, and wildfires 2. However, UFPM is widely implicated in PM-associated pathology, as their nanometer size make these particles the most effective size for lung deposition, penetration, and effects extending beyond the respiratory tract 6–8. The primary contributors to UFPM are tailpipe emissions from mobile sources (motor vehicles, aircrafts, and marine vessels) 6. Thus, PM is physically and chemically complex, making analyses of the biological effects of air pollution challenging.
Notably, the two fractions of PM predominantly implicated in CNS effects are PM2.5 and UFPM. Both are inhaled on a regular basis due to air pollution, are acutely toxic to lung and cardiovascular tissue 9, and cross blood-air barrier of the lungs, gaining access to peripheral circulation and the brain 6. Indeed, increasing reports indicate that PM can enter the brain and that PM may be associated with neurodegenerative pathology in vivo 10–14. However, the mechanism responsible for PM entry to the brain is a source of debate, where active transport, a leaky blood-brain barrier, and translocation along the olfactory nerve into the olfactory bulb15 have been proposed.
Despite the complex constitution of air pollution, classic studies in the lung and cardiovascular system have revealed inflammation and oxidative stress as common mechanisms of air pollution-induced damage 3, 4, 6, 9. As discussed in more detail below, not only do recent studies indicate that inflammation and oxidative stress are common denominators in neuropathology and CNS disease 1, but current reports also point to a growing chain of evidence directly associating air pollution with CNS damage.
Air Pollution and CNS Disease
Ischemic Stroke
While it is well known that air pollution affects human health through cardiovascular and respiratory morbidity and mortality, it has only recently been shown that these deleterious effects extend to the brain. The impact of air pollution upon the brain was first noted as an increase in ischemic stroke frequency found in individuals exposed to indoor coal fumes 16. In the United States, stroke is the number one cause of adult disability and the third cause of death, behind only cancer and heart disease 17. While the data on the association between cerebrovascular disease and ambient air pollution is limited, exposure to diverse air pollutants (e.g., particulate matter, ozone, carbon monoxide, and nitrogen dioxide) in the ambient air is epidemiologically associated with enhanced risk for ischemic cerebrovascular events 18–20. In fact, current reports demonstrate that enhanced risk for ischemic stroke correlates with air pollution, even in communities with relatively low pollutant concentrations (below current EPA safety standards) 19, 21. While the mechanisms driving the pathology are unclear, ozone and particulate matter have been shown to rapidly modulate the expression of genes involved in key vasoregulatory pathways in the brain, substantiating the notion that inhaled pollutants induce cerebrovascular effects 10. However, in addition to a neurovascular impact, current reports also indicate that the effects of air pollution invade the brain parenchyma, causing pathology indicative of neurodegenerative disease.
Air Pollution & Neurodegeneration
Air pollution is a prevalent pro-inflammatory stimulus to the CNS that has been largely over- looked as a risk factor for neurodegenerative disease. In the United States alone, an estimated 29 million people are exposed to PM10 and 88 million are exposed to PM2.5 22. Alarmingly, UFPM levels are unmonitored and unregulated in the USA, but exposure is estimated to be high. In addition, millions more are exposed to PM occupationally and in the setting of disasters, including war, fires, and the aftermath of terrorist attacks, such as the attack on the World Trade Center 22. The diseases potentially affected by air pollution, such as Alzheimer disease (AD) and Parkinson’s disease (PD), are also widespread. As the most prevalent neurodegenerative disease 23, AD affects more than 4 million people in the United States and an estimated 27 million are affected worldwide 24. PD is a devastating movement disorder and is the second most prevalent neurodegenerative disease 23, affecting 1–2% of the population over the age of 50 25. Given these statistics, it is of significant concern that recent reports have linked air pollution to neuroinflammation and neuropathology associated with AD and PD.
The first studies exploring whether air pollution is culpable in neurodegenerative disease were investigated in animal (feral dog) populations naturally exposed to polluted urban environments 11. Feral dogs living in regions of high pollution showed enhanced oxidative damage, premature presence of diffuse amyloid plaques, and a significant increase in DNA damage (apurinic/apyrimidinic sites) in olfactory bulbs, frontal, cortex, and hippocampus 11, 12. Further, dogs exposed to high concentrations of urban pollution show tissue damage and accumulated metals (nickel and vanadium) at target brain regions in a gradient fashion (olfactory mucosa > olfactory bulb > frontal cortex), implicating the nasal pathway as a key portal of entry 11. In a striking similarity, both AD and PD share early pathology in the olfactory bulb, nuclei, and pathways, with olfactory deficits being one of the earliest findings in both diseases 26. This work provided the first association between exposure to pollution and acceleration of neurodegenerative disease pathology.
Recently, these findings have now been confirmed and extended in humans and additional animal models. Analysis of brain tissue from individuals residing in highly polluted areas show an increase in CD-68, CD-163, and HLA-DR positive cells (indicating infiltrating monocytes or resident microglia activation), elevated pro-inflammatory markers (Interleukin-1β, IL1-β; cycloxygenase 2, COX2), an increase in Aβ42 deposition (hallmark disease protein of Alzheimer’s disease), blood-brain-barrier (BBB) damage, endothelial cell activation 27, and brain lesions in the prefrontal lobe 28. Interestingly, upregulation of pro-inflammatory markers such as COX2 and IL1-β, as well as the CD-14 marker for innate immune cells, were localized in frontal cortex, substantia nigra and vagus nerves 27. Further, animal studies have also shown that air pollution causes cytokine production 29, 30, increases in MAP kinase signaling through JNK 30, neurochemical changes 31, lipid peroxidation 32, behavior changes 32, and enhanced NFκβ expression 29. Together, these studies clearly indicate that air pollution has CNS effects.
Abnormal filamentous protein aggregates and neuroinflammation are common denominators of both AD and PD 1. While studies have yet to find a direct effect of air pollution on defined Lewy bodies (pathological hallmark of PD) or beta amyloid (Aβ) plaques (pathological hallmark of AD), exposure to urban air pollution has been shown to cause both neuroinflammation and accumulation of Aβ42 (component of Aβ plaques) and α-synuclein (component of Lewy Bodies) in target areas for AD and PD involvement 27. For example, dogs exposed to high levels of air pollution show increased deposits of diffuse amyloid plaques, a decade earlier than their clean air counterpart residents 11, 12. Further, the accumulation of Aβ42 and α-synuclein is reported to commence early in human childhood 27 with exposure to high concentrations of air pollution, supporting that air pollution may cause premature aging in the brain and/or instigate disease processes early in development. One plausible mechanism is that nanoparticles 33–35 and oxidative stress 36, 37 modify aggregation and rate of protein fibrillation, potentially affecting soluble Aβ and α-synuclein. It is possible that these changes in protein aggregation associated with air pollution may mark early pathology in neurodegenerative disease processes.
It has also been proposed that environmental toxicants exert their effects at multiple points across human development to culminate in CNS disease, a theory labeled “the multiple hit hypothesis” 38. Consistent with this premise, studies show that PM begins to impact the CNS early in childhood 28. For example, MRI analyses have revealed structural damage (hyperintense white matter lesions) localized in the prefrontal cortex in children exposed to high concentrations of air pollution, which may be associated with cognitive dysfunction 28. Notably, dogs exposed to the same air pollution also show frontal lesions with vascular/endothelial pathology and neuroinflammation 28. Thus, young humans and animals may be particularly vulnerable to the inflammatory effects of air pollution and these effects may accumulate across an individual’s lifespan.
While ischemic stroke 18–20, Multiple Sclerosis (exposure to second hand smoke promotes risk) 39, and PD 40(manganese content in the air is linked to enhanced risk) 40 are currently the only CNS diseases with established increased epidemiological risk with air pollution exposure, it is likely that many other uninvestigated diseases have an even greater associated risk. These risks may be distributed across individual differences in population susceptibility, as genetic predisposition may confer vulnerability to the CNS effects of air pollution, such as is the case with inherited APOE4 allele carriers 27 in humans and APOE knockout mice 41. However, given the high prevalence of AD and PD, the link between neuroinflammation and AD/PD pathogenesis, the established CNS pathology caused by air pollution, and the common high rate of human exposure to air pollution, extending both mechanistic and epidemiological studies to pursue the risks for other CNS diseases is of pressing concern to human health.
Route of CNS Effects
Recent advances have provided key insight into how air pollution exerts deleterious effects in the brain. Specifically, cerebral vascular damage, neuroinflammation, and neurodegeneration in response to air pollution are believed to occur through the four major pathways (Figure 1) described below.
Figure 1. Air pollution impacts the brain through multiple pathways.
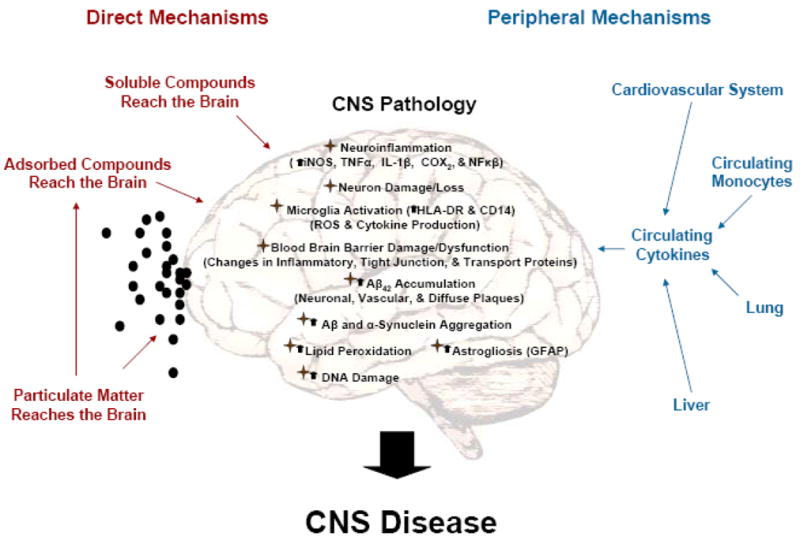
Air pollution is a complex toxin causing diverse CNS pathology through several interrelated mechanisms that may lead to CNS disease. These effects can be categorized into four major groups: 1) Systemic inflammation; 2) Particulate matter; 3) Adsorbed compounds; 4) Ozone. While some CNS effects have been attributed to specific components of air pollution, a single clear pathway responsible for CNS damage has yet to be identified. In fact, due to the complex nature of this environmental toxin it is very likely that CNS pathology is due to the synergistic interaction of the multiple pathways listed here, making air pollution a potent, biologically relevant environmental exposure and a significant challenge for mechanistic inquiry. Black dots depict particulate matter.
1. Systemic Inflammation: Peripheral Impact on the Brain
Systemic inflammation is implicated in stroke42, 43, neurodegenerative diseases44, and sickness behavior45, 46. The peripheral immune system communicates with the CNS through cytokines, where circulating cytokines impact peripheral innate immune cells, activate peripheral neuronal afferents47, and physically enter the brain through diffusion and active transport to impact the CNS46. In addition to cellular damage and modification of the ROS/cytokine milieu in the brain, systemic inflammation has recently been shown to alter the cellular make-up of innate immune cells in the brain. Specifically, in response to peripheral tumor necrosis factor α (TNFα) injection, mice were shown to recruit larger amounts of circulating monocytes to the brain45.
It is becoming increasingly accepted that air pollution causes pro-inflammatory signals originating in peripheral tissues/organs such as the lung 48, liver 49, and cardiovascular system 50, giving rise to a systemic-induced cytokine response 51 that transfers inflammation to the brain 28, 48, 52. Exposure to particulate matter has been shown to elevate plasma cytokine concentrations (IL-1β; Interleukin-6 (IL-6; granulocyte–macrophage colony-stimulating factor, GMCF) which are believed to be released into circulation as a consequence of interactions between particles, alveolar macrophages, and airway epithelial cells4. Further, PM has been shown to mobilize bone-marrow-derived neutrophils and monocytes into the circulation in both human and animal studies4. Given these findings, it is not suprising that air pollution is associated with neuroinflammation.
Circulating cytokines produced in systemic inflammation, such as TNF α and IL-1β, are well known to cause neuroinflammation 53–56, neurotoxicity 53, 55, 56, and cerebral vascular damage 57. For example, chronic, low grade inflammation associated with multiple systemic injections of low concentrations of lipopolysacharide (LPS), a cell wall component of gram negative bacteria that is a potent pro-inflammatory stimulus, in adult mice results in mild neuroinflammation, rendering animals more susceptible to further pro-inflammatory insult 55. However, a single large pro-inflammatory insult in adult animals administered with one IP injection of a high concentration of LPS (and TNFα injection) results in chronic neuroinflammation that persists months after peripheral inflammation abates, resulting in delayed and progressive neuron death, beginning only after 7–10 months post-LPS treatment in mice 53. Animal studies have also shown that exposure to systemic inflammation early in development can both cause and potentiate neuron damage seen later, in adult animals 56. In addition to neuron damage, it is also proposed that systemic inflammation caused by air pollution may contribute to deteriorating olfactory, respiratory, and blood-brain barriers to enhance access to the CNS and further increase neuropathology 11. Thus, systemic inflammation caused by air pollution is very likely to give rise to both neuroinflammation and neuropathology 13 where neurotoxic effects may be cumulative.
2. Particle Effects: Size matters
Ultrafine (nano-size particles) and fine particles are the most notorious of air pollution components, penetrating lung tissue compartments to reach the capillaries and circulating cells, or constituents (e.g. erythrocytes) 7. Experimentally, inhalation or nasal instillation of ultrafine particles in rodents results in the translocation of the particles to the systemic circulation 58 and to the brain 15. The nasal olfactory pathway is believed to be a key portal of entry, where inhaled nanoparticles have been shown to reach trigeminal nerves, brainstem, and hippocampus 59, 60. Very recently, nano-sized particulate matter was identified in the human brain 27. Specifically, particulate matter has been observed in human olfactory bulb periglomerular neurons and particles smaller than 100 nm were observed in intraluminal erythrocytes from frontal lobe and trigeminal ganglia capillaries 27. These observations in highly exposed subjects confirm that air pollution components reach the brain 13, even penetrating deep into the parenchyma.
However, once the particles reach the CNS, there is considerable debate on what the mechanisms of toxicity are. Most hypotheses are derived from traits conferred by the physical and chemical constitution of the particulate matter. For example, ultrafine particles have a large surface-to-volume ratio 8 and easily penetrate cellular membranes 61. This provides insight into why UFPM is able to traverse traditional barriers in the lung and the BBB, including why PM is found in neurons and carried in erythrocytes.
Another hypothesis builds on the premise that the particles themselves may stimulate innate immunity in the brain. Pattern recognition receptors are present on the brain’s resident innate immune cells, microglia, and identify large pathogen associated molecular patterns, such as charge and protein aggregates 1. Studies examining the toxic effects of nanometer-sized carbon (carbon black, a model of PM missing adsorbed compounds) confirm that inhalation of carbon black alone is known to cause inflammation 62, suggesting that something inherent in the particle may be culpable. Indeed, UFPM exposure in mice induces the production of pro-inflammatory cytokines (IL1-β, TNFα, and INFγ) in the olfactory bulbs of exposed animals 63. Work by Veronesi et al. reports that the inflammatory response to PM in both respiratory epithelial cells 64, 65 and microglia 66 (brain macrophages) relates to physiochemical features of the particles, such as surface charge. Thus, particulate matter itself may indeed be a pro-inflammatory stimulus once it reaches the brain.
3. Adsorbed Compounds: The Trojan Horse Effect
As mentioned previously, the particle components of air pollution have several toxic compounds present on their surface (e.g. polyaromatic hydrocarbons) that vary according to the source of the PM, geographic location of sample collection, and season. Interestingly, nanoparticles are proposed as an ideal vehicle to enhance drug entry to the CNS 67. Thus, it has been suggested that the particle components of air pollution may also represent an effective delivery system for diverse environmental toxicants to reach the brain. Additionally, some adsorbed compounds are soluble and may become a toxic stimulus independent of the particle itself 13. Indeed, the toxicity and immune-stimulating characteristics of particulate matter, such as diesel exhaust particles (DEP) in the lung, have been linked to both the adsorbed chemicals on the outside of the carbon particle (e.g. transition metals and lipopolysaccharides) 6, 9 and the physical characteristics of the particle itself 68.
Many of the adsorbed compounds present on PM are neurotoxic. For example, manganese is a component of urban air pollution, where concentrations in the air vary based on location, season, and source 40. Acute manganese exposure typically occurs as an occupational exposure in humans and is liked to dopaminergic neurotoxicity and PD symptoms69. One source of manganese content in the air is industrial –derived, arising due to emissions from ferroalloy production, iron and steel foundries, and coke ovens. In addition, manganese is also dispersed as air pollution due to gasoline engine combustion, when the gasoline contains methycly-clopentadienyl manganese tricarbonyl as an anti-knock agent40. Recently, both traffic and environmentally-derived manganese in air pollution was linked to increased risk for PD diagnosis40, 70. At present, we are just beginning to understand which of these cocktail of factors present in air pollution play a prevalent role in CNS pathology.
4. Ozone: Inhalation of Reactive Oxygen Species
Ozone is a major component of photochemical smog and is derived from multiple sources, including automobile exhaust. While ozone is not a radical, it is a reactive oxygen species and powerful inhaled oxidizing agent. Once in the lung, ozone interacts with proteins and lipids to create modified proteins/lipids, carbon/oxygen centered radicals, and toxic compounds71. For example, breakdown products from the interaction of ozone with lipids produce ozonides and cytotoxic aldehyde byproducts, which have been implicated in the extrapulmonary effects of ozone71, 72. As a consequence, ozone is well known to activate pulmonary macrophages, recruit neutrophils to the lung, and is linked to oxidative stress, airway inflammation, and dysfunction of innate immunity in the lung 73.
However, ozone is also associated with CNS effects. Recent studies with animal models have shown that oxidative stress induced by acute or chronic ozone exposure can lead to brain lipid peroxidation74, 75, dopaminergic neuron death in the substantia nigra76, neuronal morphological damage76, motor deficits75, 77, and memory deficits78. Further, prenatal exposure to ozone has been shown to alter neurotransmitter expression in adult rats79, suggesting there may be a developmental impact on CNS development. In addition, some ozone effects are associated with the cerebral vasculature. For example, ozone exposure in adult rats was shown to cause cytokine production in the brain, where enhanced IL-6 and TNFα expression was localized to astrocytes close to capillary walls80. In addition, ozone exposure upregulated the expression of vascular endothelial growth factor in rat brains, which was believed to be a compensatory and beneficial response 80. Thus, there is increasing experimental evidence that ozone causes neuroinflammation, lipid peroxidation in the brain, neuron damage, memory deficits, and motor deficits.
Because ozone is reactive with a short half-life, it is unlikely to physically reach the brain and molecules derived from ozone and lung tissue interactions have been proposed to mediate non-pulmonary ozone effects40,65. However, the specific signals from the lung to the brain responsible CNS pathology are unknown. While one hypothesis is that radical species generated in the lung enter the blood and transfer to the brain40, this seems unlikely due to the reactivity and again, the consequent short half-life of the radicals. Alternatively, aldehyde ozone-lipid byproducts40, ozone-modified soluble proteins40, activated circulating monocytes, or cytokines from the pro-inflammatory lung response (systemic inflammation) could exert harmful CNS effects. Interestingly, systemic TNFα administration81 causes lipid peroxidation in the brain and TNFα is elevated in brains of animals exposed to ozone80, supporting that a cytokine could link a peripheral response to brain lipid peroxidation, a noted effect of ozone administration in animals. However, animal studies have shown that low levels of ozone exposure have failed to result in a systemic inflammatory response82. Further, ozone administration has been used as a treatment to attenuate pain in humans83 and animals84 with varying results, suggesting that the concentration and duration of ozone exposure may determine the nature of the effects. Thus, while there is clear neuropathology, the mechanisms through which ozone is exerting toxic CNS effects remain poorly understood.
Cellular Mechanisms of Neuroinflammation
In addition to understanding how the effects of air pollution reach the brain, recent studies have also begun to address what cell types mediate air pollution-induced CNS pathology.
Astroglia
In the normal brain, astroglia play essential roles in the integrity of the BBB, providing glia-neuron contact, maintaining ionic homeostasis, buffering excess neurotransmitters, and secreting neurotrophic factors 85. Astroglial activation occurs in response to all types of injuries of the CNS86. Consistent with this, astroglia are reported to be activated in humans chronically exposed to high levels of air pollution, as evidenced by enhanced glial fibrillary acidic protein (GFAP) expression 14, 27. Animal studies investigating ozone exposure have shown that astrocytes localized near brain capillaries have enhanced expression of IL-6 and TNFα 80. In addition, astrocyte exposure to ozone in vitro results in astrocyte death87. However, at this time it is unclear how the astroglia in the brain are activated. Specifically, it is unknown whether the astroglia are responding to the components of air pollution, the inflammation and oxidative stress produced from other cell types, or the cellular damage.
Microglia
Microglia, the resident innate immune cells in the brain, actively survey the brain environment88 and are activated in neurodegenerative diseases, such as AD and PD 89. In fact, human autopsy studies show evidence of increased CD14 expression27 in response to chronic exposure to high concentrations of air pollution, indicating upregulation or activation of either infiltrating monocytes or the resident microglia cells. Microglia are activated in response to endogenous disease proteins (e.g. Aβ and α synuclein), cytokines, neuronal death, and environmental toxicants (e.g. rotenone and paraquat)1, including components of air pollution90–92. Microglia were first shown to recognize and respond to PM in an in vitro study using diesel exhaust particles (DEP)90. Cultures treated with DEP showed microglial activation, determined by changes in morphology and increase in superoxide production, with no TNFα, nitric oxide (NO), or PGE2 detected90. Mixed neuron-glia cultures treated with DEP showed selective dopaminergic neurotoxicity that only occurred in the presence of microglia, indicating that microglia mediated the neuron damage90. Microglia cultures derived from mice missing functional NADPH oxidase, the enzyme responsible for microglial extracellular superoxide production, were insensitive to DEP-induced neurotoxicity, indicating that microglia-derived ROS are key for DEP-induced dopaminergic neurotoxicity90. Microglia are also reported to respond to titanium nanoparticles with ROS 93, which is neurotoxic 66.
Interestingly, microglia exposed in vitro to concentrated ambient air pollution upregulate mRNA of pro-inflammatory cytokines, such as IL-1β and TNFα 91, suggesting that some forms of PM may be able to cause cytokine production. Further, there is evidence that metals associated with air pollution activate microglia, as microglia are activated in vitro by manganese94, a component of industrial-derived air pollution. Microglial activation in response to manganese was also shown to amplify dopaminergic neurotoxicity in vitro95. In addition to neuronal death, disease proteins, and environmental triggers such as the components of air pollution, microglia are also activated in response to systemic inflammation through cytokines53, 56 with disastrous neurotoxic consequences 53, 56 (Figure 2), and cerebral vascular damage 96.
Figure 2. Chronic activation of microglia by air pollution.
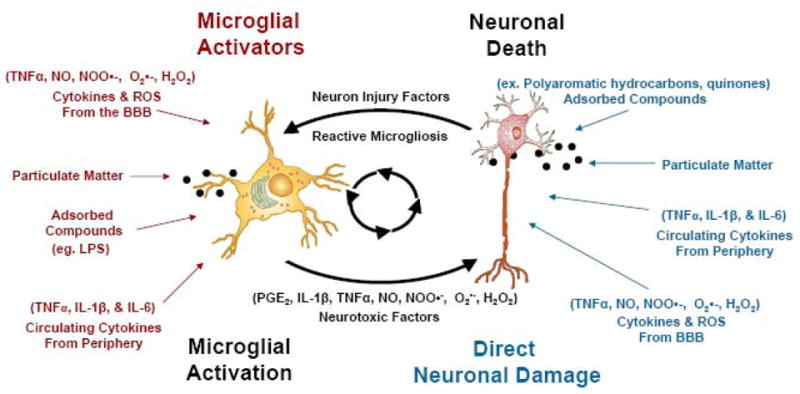
Microglia can become toxically activated by either pro-inflammatory stimuli or in response to neuronal damage. Regardless of how the neuron is damaged, microglia respond to form a chronic cycle of toxic microglial activation called reactive microgliosis. Indeed, air pollution can contribute to toxic microglial activation by triggering the cycle of reactive microgliosis through three mechanisms: 1) components of air pollution may directly activate microglia; 2) cytokines from the peripheral systemic inflammatory response may activate microglia; 3) particles, adsorbed compounds, or cytokines derived from the periphery may directly damage neurons to activate reactive microgliosis. Thus, air pollution components trigger reactive microgliosis at multiple points in the cycle to result in neuronal damage. Black dots depict particulate matter.
While the majority of microglial activation is beneficial, activated microglia can become a chronic source of pro-inflammatory factors (TNFα, PGE2, and INFγ) and oxidative stress (·NO, H2O2, O·2−, ONOO·−/ONOOH) in the brain, driving neurodegenerative diseases 1. The chronic cycle of microglial activation in response to neuron damage is termed reactive microgliosis (Figure 2). Indeed, air pollution can contribute to toxic microglial activation by triggering the cycle of reactive microgliosis through three mechanisms: 1) components of air pollution may directly activate microglia; 2) cytokines from the peripheral systemic inflammatory response may activate microglia; 3) particles, adsorbed compounds, or cytokines derived from the periphery may directly damage neurons to activate reactive microgliosis (Figure 2). Thus, evidence supports that air pollution components may be misinterpreted as pathogens by microglia, resulting in chronic inflammation, oxidative stress, neurotoxicity, and cerebral vascular damage.
The Blood Brain Barrier
Air pollution is known to impact the vascular system, but blood vessels throughout the body display a large range of phenotypes differing in gross structure, function, cellular ultrastructure, and blood-tissue exchange properties 97, which may result in unique responses to air pollution. When compared to most peripherally located “leaky” vessels, cerebral microvessels (3- to 8-μm diameter) are distinct from most of the vasculature in that they are a formidable barrier to macromolecules, various toxins, small organic drugs, and ions 98. Thus, these small vessels within the brain parenchyma constitute the blood-brain barrier (BBB) 99. The BBB is a chemical and physical barrier comprised of multiple cell types, metabolizing enzymes, and transporter proteins that protect the brain from external insult (Figure 3).
Figure 3. Air pollution impacts the BBB at the cellular level.

Brain capillaries form a chemical and physical barrier comprised of multiple cell types, metabolizing enzymes and transporter proteins, protecting the brain from external insult. The circumference of the capillary lumen is surrounded by a single endothelial cell (EC) and the opposing membranes are sealed by tight junctions (TJ). Pericytes (PC) are attached to the abluminal surface of the EC and are thought to regulate BBB function. The basal lamina (BL) is contiguous with the plasma membranes of astrocyte end-feet (AEF) surrounding both the PC and the EC. Black dots depict particulate matter. Chronic exposure to air pollution results in an increase in peripheral circulating cytokines and particulate matter (the particle components of air pollution). Upon chronic exposure to high levels of air pollution, there is a decrease in tight junction proteins, evidence of endothelial cell damage, and upregulation of VCAM/ICAM in the cerebral vasculature, suggesting potential failure of the physical barrier. In addition, particulate matter causes production of cytokines and reactive oxygen species (ROS) in brain capillaries, which signal changes in transporter expression and function (e.g. P-glycoprotein, P-GP and Multidrug Resistance Associated Protein-2, MRP2) and a decrease in expression of various tight junction proteins. Thus, brain capillaries recognize air pollution and respond by regulating the physical and chemical barrier function and producing pro-inflammatory signals. In addition, this response may serve as a pro-inflammatory sensor and ultimately distribute ROS, cytokines, and particulate matter to the brain parenchyma, further contributing to CNS pathology.
Particulate matter has been identified in both human brain capillaries and the brain parenchyma27, suggesting an ability to both interact with cells comprising the BBB and navigate across the BBB through yet unidentified mechanisms. Recent studies report that aluminum nanoparticles reduced human brain microvascular endothelial cell viability, altered mitochondrial potential, increased oxidative stress, and decreased tight junction protein expression, suggesting that nano-size particles have the capacity to injure endothelial cells and damage the BBB 100. Human exposure to air pollution shows endothelial cell damage in the cerebral vasculature, with increases in ICAM and VCAM present 27. In addition, in vitro studies using whole brain rat capillaries reveal that treatment with particulate matter causes production of cytokines and reactive oxygen species (ROS), which signal changes in transporter expression and function (e.g. P-glycoprotein and Multidrug Resistance Associated Protein-2) and a decrease in expression of various tight junction proteins 101. Thus, brain capillaries recognize air pollution and respond to air pollution components by regulating the physical and chemical barrier function and producing pro-inflammatory signals. This response may serve as a pro-inflammatory sensor and ultimately distribute ROS, cytokines, and particulate matter to the brain parenchyma, further contributing to CNS pathology. In addition these findings are also directly relevant to CNS pharmacotherapy in neurodegenerative diseases. Specifically, the PM-induced upregulation of efflux transporters (P-glycoprotein and Multidrug Resistance Associated Protein-2) at the BBB may have significant implications for drug availability in the brain parenchyma for individuals living in heavily polluted cities.
Together, animal, human, and cell culture studies have shown that air pollution causes CNS oxidative stress, neuroinflammation, neuron damage, enhancement of abnormal filamentous proteins (Aβ and α synuclein), BBB changes, and cerebrovascular damage ( Table 1), linking the pathways through which air pollution impacts the CNS disease pathology. While experimental evidence is compelling, given the chronic nature of human exposure to air pollution, CNS effects are likely due to exposure over an entire human lifetime, including critical periods of development. Notably, these chronic effects risk being overlooked by in vitro methods and short term animal exposures. However, these critical experimental studies have provided the foundation necessary to begin to identify the CNS-toxic components of air pollution, providing the opportunity to address their role in CNS disease and paving the way for detailed experimental inquiry at the level of epidemiology.
Table 1.
Research Overview: Effects of Air Pollution on the Brain
| Air Pollution Model | Experimental Model | Neuroinflammation & Pro-inflammatory Markers | Neuropathology & Behavior Changes | References |
|---|---|---|---|---|
| Particulate Matter | Mouse | N/T | DA Neuron Damage in the Substantia Nigra | 41 |
| Mouse | IL1-β, TNFα, and INFγ increase in Olfactory Bulb | Change in Neurotransmitters | 63 | |
| Mouse | Cytokine Production, JNK Activation, Enhanced NFκβ Expression | N/T | 29, 30 | |
| Mouse | N/T | Changes in Neurotransmitters | 31 | |
| Rat | N/T | Lipid Peroxidation, Decrease in Exploratory Behavior | 32 | |
| Cell Culture | Microglial Activation Superoxide Production | DA Neuron Damage | 90 | |
| Cell Culture | Microglial Activation TNFα & IL-6 Production | N/T | 91 | |
| Brain Capillary Culture | TNFα & ROS Production c-Jun Phosphorylation | P-GP & MRP2 Increase Tight Junction Protein Decrease | 101 | |
| Ozone | Rat | N/T | Lipid Peroxidation DA Neuron Damage & Death in Substantia Nigra, Motor Deficits | 75–77 |
| Rat | N/T | Lipid Peroxidation & Impaired Memory | 74, 78 | |
| Rat | Astrocyte IL-6 & TNFα Increase at BBB | Increase in Brainstem VEGF Expression (Adaptive Repair Response) | 80 | |
| Prenatal Rat | N/T | DA, NA, DOPAC and HVA Decrease | 79 | |
| Cell Culture | N/T | Astrocyte Death | 87 | |
| Nanoparticles | Cell Culture | Microglial Activation Superoxide Production | DA Neuron Damage | 66, 102 |
| Cell Culture | N/T | HBMEC Toxicity, Lower Tight Junction Expression | 100 | |
| Mouse | N/T | Oxidative Stress (Brain) Lipid Peroxidation | 103–105 | |
| Chronic Ambient Air Pollution | Human | COX2, IL1-β, iNOS, & CD14 Increase | White Matter Lesions, Diffuse Aβ Plaques, α Synuclein Aggregation, BBB Damage, Cognitive Deficits, & DNA Damage | 10, 14, 27, 28, 52, 100 |
| Dog | iNOS, NFκβ, & CD14 Increase | White Matter Lesions Diffuse Aβ Plaques α Synuclein Aggregation, BBB Damage, & DNA Damage | 11, 12, 28 | |
N/T, Not Tested; DA, Dopamine; IL-1β, Interleukin 1β; TNFα, Tumor Necrosis Factor α; INFγ, Interferon γ; JNK, c-Jun N terminal Kinase; NFκβ, Nuclear Factor κβ, IL-6, Interleukin 6; ROS. Reactive Oxygen Species; PGP, P-Glycoprotein; MRP2 Multidrug Resistance Associated Protein-2; BBB, Blood Brain Barrier; VEGF, Vascular endothelial growth factor; NA, norepinephrine; DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid; HBMEC, Human Brain Microvascular Endothelial Cells; COX2, cycloxygenase 2.
Summary & Conclusions
In summary, air pollution is a complex mixture of environmental toxicants that assault the CNS through several cellular and molecular pathways to cause disease. Reports show that the CNS effects are chronic, beginning in childhood, and may take time (years) to accumulate pathology. Specifically, air pollution has been shown to cause neuroinflammation, oxidative stress, cerebral vascular damage, and neurodegenerative pathology. Air pollution effects cross from the periphery to the brain through systemic inflammation, and translocation of nanoparticles to the brain, where both the physical characteristics of the particle itself and the toxic compounds adsorbed on the particle may cause damage. Astroglia, brain capillaries, and microglia in particular, respond to the components of air pollution with chronic activation, inflammation, and oxidative stress. Given the complex nature of this prevalent environmental toxin, CNS pathology is likely due to the synergistic interaction of multiple pathways and mechanisms, making air pollution a potent, biologically relevant environmental exposure and a significant challenge for mechanistic inquiry. While epidemiology has linked an increased risk of stroke, MS, and PD with exposure to specific types of air pollution, further epidemiological and mechanistic studies into the association between the components of air pollution and the development of CNS diseases are of pressing concern for human health.
Abbreviations
- DEP
Diesel Exhaust Particles
- PM
Particulate Matter
- UFPM
utrafine particulate matter
- PD
Parkinson’s disease
- AD
Alzheimer’s Disease
- ROS
reactive oxygen species
- TNFα
tumor necrosis factor-alpha
- Aβ
Beta amyloid peptide
- LPS
lipopolysaccharide
- PGE2
prostaglandin E2
- CNS
central nervous system
- NFκB
nuclear factor-κB
- GFAP
glial fibrillary acidic protein
- SOD
INFγ, interferon gamma
- BBB
blood-brain barrier
References
- 1.Block ML, et al. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 2.Craig L, et al. Air pollution and public health: a guidance document for risk managers. J Toxicol Environ Health A. 2008;71:588–698. doi: 10.1080/15287390801997732. [DOI] [PubMed] [Google Scholar]
- 3.Riedl MA. The effect of air pollution on asthma and allergy. Curr Allergy Asthma Rep. 2008;8:139–146. doi: 10.1007/s11882-008-0024-8. [DOI] [PubMed] [Google Scholar]
- 4.Mills NL, et al. Adverse cardiovascular effects of air pollution. Nat Clin Pract Cardiovasc Med. 2009;6:36–44. doi: 10.1038/ncpcardio1399. [DOI] [PubMed] [Google Scholar]
- 5.Akimoto H. Global air quality and pollution. Science. 2003;302:1716–1719. doi: 10.1126/science.1092666. [DOI] [PubMed] [Google Scholar]
- 6.Muhlfeld C, et al. Interactions of nanoparticles with pulmonary structures and cellular responses. Am J Physiol Lung Cell Mol Physiol. 2008;294:L817–829. doi: 10.1152/ajplung.00442.2007. [DOI] [PubMed] [Google Scholar]
- 7.Valavanidis A, et al. Airborne particulate matter and human health: toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2008;26:339–362. doi: 10.1080/10590500802494538. [DOI] [PubMed] [Google Scholar]
- 8.Rothen-Rutishauser B, et al. A newly developed in vitro model of the human epithelial airway barrier to study the toxic potential of nanoparticles. ALTEX. 2008;25:191–196. doi: 10.14573/altex.2008.3.191. [DOI] [PubMed] [Google Scholar]
- 9.Simkhovich BZ, et al. Air pollution and cardiovascular injury epidemiology, toxicology, and mechanisms. J Am Coll Cardiol. 2008;52:719–726. doi: 10.1016/j.jacc.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 10.Thomson EM, et al. Air pollution alters brain and pituitary endothelin-1 and inducible nitric oxide synthase gene expression. Environ Res. 2007;105:224–233. doi: 10.1016/j.envres.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Calderon-Garciduenas L, et al. Air pollution and brain damage. Toxicol Pathol. 2002;30:373–389. doi: 10.1080/01926230252929954. [DOI] [PubMed] [Google Scholar]
- 12.Calderon-Garciduenas L, et al. DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol Pathol. 2003;31:524–538. doi: 10.1080/01926230390226645. [DOI] [PubMed] [Google Scholar]
- 13.Peters A, et al. Translocation and potential neurological effects of fine and ultrafine particles a critical update. Part Fibre Toxicol. 2006;3:13. doi: 10.1186/1743-8977-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calderon-Garciduenas L, et al. Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol Pathol. 2004;32:650–658. doi: 10.1080/01926230490520232. [DOI] [PubMed] [Google Scholar]
- 15.Oberdorster G, et al. Translocation of inhaled ultrafine particles to the brain. Inhal Toxicol. 2004;16:437–445. doi: 10.1080/08958370490439597. [DOI] [PubMed] [Google Scholar]
- 16.Zhang ZF, et al. Indoor air pollution of coal fumes as a risk factor of stroke, Shanghai. Am J Public Health. 1988;78:975–977. doi: 10.2105/ajph.78.8.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lloyd-Jones D, et al. Heart Disease and Stroke Statistics--2009 Update. A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008 doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 18.Lokken PR, et al. Air Pollution and Risk of Stroke: Underestimation of Effect Due to Misclassification of Time of Event Onset. Epidemiology. 2008 doi: 10.1097/ede.0b013e31818ef34a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lisabeth LD, et al. Ambient air pollution and risk for ischemic stroke and transient ischemic attack. Ann Neurol. 2008;64:53–59. doi: 10.1002/ana.21403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong YC, et al. Air pollution: a new risk factor in ischemic stroke mortality. Stroke. 2002;33:2165–2169. doi: 10.1161/01.str.0000026865.52610.5b. [DOI] [PubMed] [Google Scholar]
- 21.Kettunen J, et al. Associations of fine and ultrafine particulate air pollution with stroke mortality in an area of low air pollution levels. Stroke. 2007;38:918–922. doi: 10.1161/01.STR.0000257999.49706.3b. [DOI] [PubMed] [Google Scholar]
- 22.Seagrave J, et al. Lung toxicity of ambient particulate matter from southeastern U.S. sites with different contributing sources: relationships between composition and effects. Environ Health Perspect. 2006;114:1387–1393. doi: 10.1289/ehp.9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirtz D, et al. How common are the “common” neurologic disorders? Neurology. 2007;68:326–337. doi: 10.1212/01.wnl.0000252807.38124.a3. [DOI] [PubMed] [Google Scholar]
- 24.Wimo A, et al. An estimate of the worldwide prevalence and direct costs of dementia in 2003. Dement Geriatr Cogn Disord. 2006;21:175–181. doi: 10.1159/000090733. [DOI] [PubMed] [Google Scholar]
- 25.Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(2):R183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 26.Doty RL. The olfactory vector hypothesis of neurodegenerative disease: is it viable? Ann Neurol. 2008;63:7–15. doi: 10.1002/ana.21327. [DOI] [PubMed] [Google Scholar]
- 27.Calderon-Garciduenas L, et al. Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol Pathol. 2008;36:289–310. doi: 10.1177/0192623307313011. [DOI] [PubMed] [Google Scholar]
- 28.Calderon-Garciduenas L, et al. Air pollution, cognitive deficits and brain abnormalities: a pilot study with children and dogs. Brain Cogn. 2008;68:117–127. doi: 10.1016/j.bandc.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 29.Campbell A, et al. Particulate matter in polluted air may increase biomarkers of inflammation in mouse brain. Neurotoxicology. 2005;26:133–140. doi: 10.1016/j.neuro.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Kleinman MT, et al. Inhaled ultrafine particulate matter affects CNS inflammatory processes and may act via MAP kinase signaling pathways. Toxicol Lett. 2008;178:127–130. doi: 10.1016/j.toxlet.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sirivelu MP, et al. Activation of the stress axis and neurochemical alterations in specific brain areas by concentrated ambient particle exposure with concomitant allergic airway disease. Environ Health Perspect. 2006;114:870–874. doi: 10.1289/ehp.8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zanchi AC, et al. Chronic nasal instillation of residual-oil fly ash (ROFA) induces brain lipid peroxidation and behavioral changes in rats. Inhal Toxicol. 2008;20:795–800. doi: 10.1080/08958370802009060. [DOI] [PubMed] [Google Scholar]
- 33.Colvin VL, Kulinowski KM. Nanoparticles as catalysts for protein fibrillation. Proc Natl Acad Sci U S A. 2007;104:8679–8680. doi: 10.1073/pnas.0703194104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cedervall T, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci U S A. 2007;104:2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lynch I, et al. The nanoparticle-protein complex as a biological entity; a complex fluids and surface science challenge for the 21st century. Adv Colloid Interface Sci. 2007;134–135:167–174. doi: 10.1016/j.cis.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 36.Liu L, et al. Promotion of amyloid beta protein misfolding and fibrillogenesis by a lipid oxidation product. J Mol Biol. 2008;377:1236–1250. doi: 10.1016/j.jmb.2008.01.057. [DOI] [PubMed] [Google Scholar]
- 37.Qin Z, et al. Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. J Biol Chem. 2007;282:5862–5870. doi: 10.1074/jbc.M608126200. [DOI] [PubMed] [Google Scholar]
- 38.Carvey PM, et al. Progressive dopamine neuron loss in Parkinson’s disease: the multiple hit hypothesis. Cell Transplant. 2006;15:239–250. doi: 10.3727/000000006783981990. [DOI] [PubMed] [Google Scholar]
- 39.Mikaeloff Y, et al. Parental smoking at home and the risk of childhood-onset multiple sclerosis in children. Brain. 2007;130:2589–2595. doi: 10.1093/brain/awm198. [DOI] [PubMed] [Google Scholar]
- 40.Finkelstein M, Jerrett M. A study of the relationships between Parkinson’s disease and markers of traffic-derived environmental maganese air pollution in two Canadian cities. Evironmental Research. 2007;104:420–432. doi: 10.1016/j.envres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Veronesi B, et al. Effects of subchronic exposures to concentrated ambient particles. VII. Degeneration of dopaminergic neurons in Apo E-/- mice. Inhal Toxicol. 2005;17:235–241. doi: 10.1080/08958370590912888. [DOI] [PubMed] [Google Scholar]
- 42.McColl BW, et al. Systemic infection, inflammation and acute ischemic stroke. Neuroscience. 2009;158:1049–1061. doi: 10.1016/j.neuroscience.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 43.Castillo J, et al. Inflammation markers and prediction of post-stroke vascular disease recurrence: the MITICO study. J Neurol. 2009;256:217–224. doi: 10.1007/s00415-009-0058-4. [DOI] [PubMed] [Google Scholar]
- 44.Cunningham C, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65:304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Mello C, et al. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dantzer R, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–428. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamagawa E, van Eeden SF. Impaired lung function and risk for stroke: role of the systemic inflammation response? Chest. 2006;130:1631–1633. doi: 10.1378/chest.130.6.1631. [DOI] [PubMed] [Google Scholar]
- 49.Folkmann JK, et al. Oxidatively damaged DNA and inflammation in the liver of dyslipidemic ApoE-/- mice exposed to diesel exhaust particles. Toxicology. 2007;237:134–144. doi: 10.1016/j.tox.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 50.Swiston JR, et al. Wood smoke exposure induces a pulmonary and systemic inflammatory response in firefighters. Eur Respir J. 2008;32:129–138. doi: 10.1183/09031936.00097707. [DOI] [PubMed] [Google Scholar]
- 51.Ruckerl R, et al. Air pollution and inflammation (interleukin-6, C-reactive protein, fibrinogen) in myocardial infarction survivors. Environ Health Perspect. 2007;115:1072–1080. doi: 10.1289/ehp.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calderon-Garciduenas L, et al. Systemic inflammation, endothelial dysfunction, and activation in clinically healthy children exposed to air pollutants. Inhal Toxicol. 2008;20:499–506. doi: 10.1080/08958370701864797. [DOI] [PubMed] [Google Scholar]
- 53.Qin L, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rivest S, et al. How the blood talks to the brain parenchyma and the paraventricular nucleus of the hypothalamus during systemic inflammatory and infectious stimuli. Proc Soc Exp Biol Med. 2000;223:22–38. doi: 10.1046/j.1525-1373.2000.22304.x. [DOI] [PubMed] [Google Scholar]
- 55.Perry VH, et al. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- 56.Ling Z, et al. Progressive dopamine neuron loss following supra-nigral lipopolysaccharide (LPS) infusion into rats exposed to LPS prenatally. Exp Neurol. 2006 doi: 10.1016/j.expneurol.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 57.Manousakis G, et al. The interface between stroke and infectious disease: infectious diseases leading to stroke and infections complicating stroke. Curr Neurol Neurosci Rep. 2009;9:28–34. doi: 10.1007/s11910-009-0005-x. [DOI] [PubMed] [Google Scholar]
- 58.Nemmar A, Inuwa IM. Diesel exhaust particles in blood trigger systemic and pulmonary morphological alterations. Toxicol Lett. 2008;176:20–30. doi: 10.1016/j.toxlet.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 59.Wang J, et al. Time-dependent translocation and potential impairment on central nervous system by intranasally instilled TiO(2) nanoparticles. Toxicology. 2008;254:82–90. doi: 10.1016/j.tox.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 60.Wang B, et al. Transport of intranasally instilled fine Fe2O3 particles into the brain: micro-distribution, chemical states, and histopathological observation. Biol Trace Elem Res. 2007;118:233–243. doi: 10.1007/s12011-007-0028-6. [DOI] [PubMed] [Google Scholar]
- 61.Geiser M, et al. Ultrafine particles cross cellular membranes by nonphagocytic mechanisms in lungs and in cultured cells. Environ Health Perspect. 2005;113:1555–1560. doi: 10.1289/ehp.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Niwa Y, et al. Inhalation exposure to carbon black induces inflammatory response in rats. Circ J. 2008;72:144–149. doi: 10.1253/circj.72.144. [DOI] [PubMed] [Google Scholar]
- 63.Tin Tin Win S, et al. Changes in neurotransmitter levels and proinflammatory cytokine mRNA expressions in the mice olfactory bulb following nanoparticle exposure. Toxicol Appl Pharmacol. 2008;226:192–198. doi: 10.1016/j.taap.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 64.Veronesi B, et al. The surface charge of visible particulate matter predicts biological activation in human bronchial epithelial cells. Toxicol Appl Pharmacol. 2002;178:144–154. doi: 10.1006/taap.2001.9341. [DOI] [PubMed] [Google Scholar]
- 65.Veronesi B, et al. Electrostatic charge activates inflammatory vanilloid (VR1) receptors. Neurotoxicology. 2003;24:463–473. doi: 10.1016/S0161-813X(03)00022-6. [DOI] [PubMed] [Google Scholar]
- 66.Long TC, et al. Nanosize titanium dioxide stimulates reactive oxygen species in brain microglia and damages neurons in vitro. Environ Health Perspect. 2007;115:1631–1637. doi: 10.1289/ehp.10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silva GA. Neuroscience nanotechnology: progress, opportunities and challenges. Nat Rev Neurosci. 2006;7:65–74. doi: 10.1038/nrn1827. [DOI] [PubMed] [Google Scholar]
- 68.Ma JY, Ma JK. The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. J Environ Sci Health Part C Environ Carcinog Ecotoxicol Rev. 2002;20:117–147. doi: 10.1081/GNC-120016202. [DOI] [PubMed] [Google Scholar]
- 69.Burton NC, Guilarte TR. Manganese neurotoxicity: lessons learned from longitudinal studies in nonhuman primates. Environ Health Perspect. 2009;117:325–332. doi: 10.1289/ehp.0800035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lucchini R, et al. Exposure to neurotoxic metals and prevalence of parkinsonian syndrome in the area of Brescia. G Ital Med Lav Ergon. 2003;25(Suppl):88–89. [PubMed] [Google Scholar]
- 71.Pryor WA, et al. A new mechanism for the toxicity of ozone. Toxicol Lett. 1995;82–83:287–293. doi: 10.1016/0378-4274(95)03563-x. [DOI] [PubMed] [Google Scholar]
- 72.Pryor WA. Mechanisms of radical formation from reactions of ozone with target molecules in the lung. Free Radic Biol Med. 1994;17:451–465. doi: 10.1016/0891-5849(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 73.Hollingsworth JW, et al. Ozone and pulmonary innate immunity. Proc Am Thorac Soc. 2007;4:240–246. doi: 10.1513/pats.200701-023AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guevara-Guzman R, et al. Estradiol prevents ozone-induced increases in brain lipid peroxidation and impaired social recognition memory in female rats. Neuroscience. 2009;159:940–950. doi: 10.1016/j.neuroscience.2009.01.047. [DOI] [PubMed] [Google Scholar]
- 75.Pereyra-Munoz N, et al. Oxidative damage in substantia nigra and striatum of rats chronically exposed to ozone. J Chem Neuroanat. 2006;31:114–123. doi: 10.1016/j.jchemneu.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 76.Angoa-Perez M, et al. Estrogen counteracts ozone-induced oxidative stress and nigral neuronal death. Neuroreport. 2006;17:629–633. doi: 10.1097/00001756-200604240-00014. [DOI] [PubMed] [Google Scholar]
- 77.Rivas-Arancibia S, et al. Effect of acute ozone exposure on locomotor behavior and striatal function. Pharmacol Biochem Behav. 2003;74:891–900. doi: 10.1016/s0091-3057(03)00011-x. [DOI] [PubMed] [Google Scholar]
- 78.Avila-Costa MR, et al. Memory deterioration in an oxidative stress model and its correlation with cytological changes on rat hippocampus CA1. Neurosci Lett. 1999;270:107–109. doi: 10.1016/s0304-3940(99)00458-9. [DOI] [PubMed] [Google Scholar]
- 79.Gonzalez-Pina R, et al. Prenatal exposure to ozone disrupts cerebellar monoamine contents in newborn rats. Neurochem Res. 2008;33:912–918. doi: 10.1007/s11064-007-9534-3. [DOI] [PubMed] [Google Scholar]
- 80.Araneda S, et al. VEGF overexpression in the astroglial cells of rat brainstem following ozone exposure. Neurotoxicology. 2008;29:920–927. doi: 10.1016/j.neuro.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 81.Gutner UA, et al. [The influence of tumor necrosis factor alpha on the processes of sphingomyelin cycle and lipid peroxidation in brain] Zh Nevrol Psikhiatr Im S S Korsakova. 2005;105:48–54. [PubMed] [Google Scholar]
- 82.Hermans C, et al. Effects of ambient ozone on the procoagulant status and systemic inflammatory response. J Thromb Haemost. 2005;3:2102–2103. doi: 10.1111/j.1538-7836.2005.01547.x. [DOI] [PubMed] [Google Scholar]
- 83.Staal JB, et al. Injection therapy for subacute and chronic low back pain: an updated Cochrane review. Spine. 2009;34:49–59. doi: 10.1097/BRS.0b013e3181909558. [DOI] [PubMed] [Google Scholar]
- 84.Fuccio C, et al. A single subcutaneous injection of ozone prevents allodynia and decreases the over-expression of pro-inflammatory caspases in the orbito-frontal cortex of neuropathic mice. Eur J Pharmacol. 2009;603:42–49. doi: 10.1016/j.ejphar.2008.11.060. [DOI] [PubMed] [Google Scholar]
- 85.Seifert G, et al. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- 86.Damiani CL, O’Callaghan JP. Recapitulation of cell signaling events associated with astrogliosis using the brain slice preparation. J Neurochem. 2007;100:720–726. doi: 10.1111/j.1471-4159.2006.04321.x. [DOI] [PubMed] [Google Scholar]
- 87.Zhou NB, et al. Effects of different concentrations of oxygen-ozone on rats’ astrocytes in vitro. Neurosci Lett. 2008;441:178–182. doi: 10.1016/j.neulet.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 88.Nimmerjahn A, et al. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science. 2005 doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 89.McGeer PL, et al. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 90.Block ML, et al. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. Faseb J. 2004;18:1618–1620. doi: 10.1096/fj.04-1945fje. [DOI] [PubMed] [Google Scholar]
- 91.Sama P, et al. The cellular and genomic response of an immortalized microglia cell line (BV2) to concentrated ambient particulate matter. Inhal Toxicol. 2007;19:1079–1087. doi: 10.1080/08958370701628721. [DOI] [PubMed] [Google Scholar]
- 92.Blasko I, et al. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: the role of microglia and astrocytes. Aging Cell. 2004;3:169–176. doi: 10.1111/j.1474-9728.2004.00101.x. [DOI] [PubMed] [Google Scholar]
- 93.Long TC, et al. Titanium dioxide (P25) produces reactive oxygen species in immortalized brain microglia (BV2): implications for nanoparticle neurotoxicity. Environ Sci Technol. 2006;40:4346–4352. doi: 10.1021/es060589n. [DOI] [PubMed] [Google Scholar]
- 94.Zhang P, et al. Manganese chloride stimulates rat microglia to release hydrogen peroxide. Toxicol Lett. 2007;173:88–100. doi: 10.1016/j.toxlet.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang P, et al. Microglia enhance manganese chloride-induced dopaminergic neurodegeneration: role of free radical generation. Exp Neurol. 2009;217:219–230. doi: 10.1016/j.expneurol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McColl BW, et al. Systemic infection, inflammation and acute ischemic stroke. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 97.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 98.Dallas S, et al. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58:140–161. doi: 10.1124/pr.58.2.3. [DOI] [PubMed] [Google Scholar]
- 99.Miller DS, et al. Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev. 2008;60:196–209. doi: 10.1124/pr.107.07109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen L, et al. Manufactured aluminum oxide nanoparticles decrease expression of tight junction proteins in brain vasculature. J Neuroimmune Pharmacol. 2008;3:286–295. doi: 10.1007/s11481-008-9131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hartz AM, et al. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J. 2008;22:2723–2733. doi: 10.1096/fj.08-106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lovell MA, Markesbery WR. Oxidative damage in mild cognitive impairment and early Alzheimer’s disease. J Neurosci Res. 2007 doi: 10.1002/jnr.21346. [DOI] [PubMed] [Google Scholar]
- 103.Rahman MF, et al. Expression of genes related to oxidative stress in the mouse brain after exposure to silver-25 nanoparticles. Toxicol Lett. 2009;187:15–21. doi: 10.1016/j.toxlet.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 104.Wang J, et al. Potential neurological lesion after nasal instillation of TiO(2) nanoparticles in the anatase and rutile crystal phases. Toxicol Lett. 2008;183:72–80. doi: 10.1016/j.toxlet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 105.Alekseenko AV, et al. Ferritin, a protein containing iron nanoparticles, induces reactive oxygen species formation and inhibits glutamate uptake in rat brain synaptosomes. Brain Res. 2008;1241:193–200. doi: 10.1016/j.brainres.2008.09.012. [DOI] [PubMed] [Google Scholar]