

Abstract
Although ascorbic acid is an important water-soluble antioxidant and enzyme cofactor in plants and animals, humans and some other species do not synthesize ascorbate due to the lack of the enzyme catalyzing the final step of the biosynthetic pathway, and for them it has become a vitamin. This review focuses on the role of ascorbate in various hydroxylation reactions and in the redox homeostasis of subcellular compartments including mitochondria and endoplasmic reticulum. Recently discovered functions of ascorbate in nucleic acid and histone dealkylation and proteoglycan deglycanation are also summarized. These new findings might delineate a role for ascorbate in the modulation of both pro- and anti-carcinogenic mechanisms. Recent advances and perspectives in therapeutic applications are also reviewed. On the basis of new and earlier observations, the advantages of the lost ability to synthesize ascorbate are pondered. The increasing knowledge of the functions of ascorbate and of its molecular sites of action can mechanistically substantiate a place for ascorbate in the treatment of various diseases.
Keywords: vitamin C, ascorbate, dehydroascorbic acid, L-gulonolactone-oxidase, antioxidant, pro-oxidant, mitochondrium, endoplasmic reticulum, redox, scurvy
Introduction
The dominant role of oxidoreductions in the metabolic and energetic exchange between the living organism and its environment is well known. Ascorbate has special functions in this redox interrelationship, as an antioxidant and enzyme cofactor. Generally, ascorbate is regarded as a reducing agent; it is able to serve as an antioxidant in free radical-mediated oxidation processes. However, as a reducing agent it is also able to reduce redox-active metals such as copper and iron, thereby increasing the pro-oxidant chemistry of these metals. Thus ascorbate can act as both a pro-oxidant and an antioxidant. In general, at low ascorbate concentrations, ascorbate is prone to be a pro-oxidant, and at high concentrations, it will tend to be an antioxidant (Buettner and Jurkiewicz, 1996).
In spite of the fact that humans are unable to produce ascorbate due to the absence of the enzyme, gulonolactone oxidase (GLO), which catalyses the last enzymic step in ascorbate synthesis, it is very instructive to understand the redox, metabolic and regulatory aspects of ascorbate production. Ascorbate is formed in the course of the uronic pathway starting from UDP-glucuronate in animals. Thus, its synthesis can be considered as an integral part of carbohydrate metabolism. It has been shown that the UDP-glucose supply is dependent on carbohydrate reserves (Bánhegyi et al., 1988; Braun et al., 1994; Mandl et al., 1995; Bánhegyi and Mandl, 2001). Connection between the ascorbate supply and glycogenolysis, the actual process of glycogen metabolism, has been demonstrated through a redox-dependent metabolic mechanism (Bánhegyi et al., 1996; 1997; Braun et al., 1996a,b; Puskás et al., 1998). However, regulation of vitamin C synthesis by glutathione (GSH) is not universally admitted (Linster and Van Schaftingen, 2003).
Principal questions concerning the transport, metabolism and antioxidant functions of ascorbate have been detailed in several recent reviews (Bánhegyi et al., 1997; 1998a; 2007; Padayatty and Levine, 2001; Duarte and Lunec, 2005; Li and Schellhorn, 2007; Linster and Van Schaftingen, 2007). Our review focuses on new functions of ascorbate as a cofactor for nucleic acid and histone demethylation, proteoglycan deglycanation and on the role of ascorbate in the redox homeostasis of subcellular compartments. Some possible therapeutic applications are also discussed.
It has been known for a long time that the absence of fresh fruits and vegetables in the human diet leads to scurvy – a fatal disease affecting sailors (Lind, 1753) and soldiers (Kramer, 1739) in the past. Thus, for humans to survive, their diet must contain ascorbate; that is why ascorbate is a vitamin in humans. This raises the following questions: which are the most important features of ascorbate, and why does its deficiency have fatal consequences?
Ascorbate as a cofactor in various hydroxylation reactions
Ascorbate plays a crucial role in various hydroxylation reactions. There are several ascorbate- dependent mono- and dioxygenations in various neurotransmitter and hormone formation processes (for review, see Englard and Seifter, 1986), and ascorbate is also required for the hydroxylation of carnitine. It has been suggested that carnitine deficiency is responsible for the early symptoms of scurvy (Rebouche, 1991). In several cases, hydroxylation is catalysed by Fe(II)-dependent non-haeme oxygenases belonging to the family of α-ketoglutarate (2OG)-dependent dioxygenases. Dioxygenases include prolyl, asparaginyl and lysyl hydroxylases (Hewitson et al., 2003; Myllyharju and Kivirikko, 2004). Within this group, ABH2 and ABH3, two human DNA dioxygenases functioning as novel DNA repair enzymes have also been described (Sedgwick, 2004). Oxygen and 2OG are required for oxygenation, which also needs cofactors: Fe2+ and ascorbate. Iron in these enzymes is maintained in the active Fe(II) form by ascorbate (Myllyla et al., 1984; Tschank et al., 1994).
Several proteins with various functions can serve as potential substrates for these oxygenases. However, it is difficult to prove the physiological/pathological significance of these oxidoreductions. The role of the ascorbate-dependent hydroxylation of proline and lysine residues in the stabilization of the collagen molecule has been explored; 30% of cellular protein mass is made by collagen. The role of ascorbate in collagen synthesis and its connection to the development of scurvy has been extensively reviewed in several studies (Englard and Seifter, 1986; Peterkofsky, 1991). At present, at least 27 collagen types with at least 42 distinct polypeptide chains and more than 20 additional proteins having collagen-like domains have been described. In proteins with collagen-like domains, 4-hydroxyprolines are ubiquitous; therefore, proline hydroxylation has a crucial role in interactions between these proteins (Myllyharju and Kivirikko, 2004).
Hypoxia-inducible transcription factor-1α (HIF-1α) is also a substrate of these dioxygenases. Activity of HIF-1α can be regulated by hydroxylation, which needs ascorbate (Figure 1). In hypoxia, HIFs are expressed in all nucleated cells, serving as ‘global’ regulators in a complicated network (Semenza, 2003). More than 100 HIF target genes have already been identified as part of the network. The protein products of these genes are involved in, among other processes, angiogenesis, cell proliferation, glucose uptake, glycolysis, erythropoiesis and iron homeostasis. HIFs are heterodimers, consisting of oxygen-sensitive HIF-α and HIF-β. In hypoxia, the induced HIF-α is translocated to the nucleus, where the heterodimer transcription factor is formed with HIF-β, which is constitutively present there. In normoxia, HIFs are inactive because of the oxygen- (and ascorbate-) dependent hydroxylation and subsequent degradation of the α-subunit. Most of our knowledge is based on observations concerning HIF-1α. Two separate dioxygenations of HIF-1α have been identified: proline hydroxylation and asparagine hydroxylation. Three HIF-1α prolyl hydroxylases (PHD1, PHD2, PHD3) recognize a conserved amino acid sequence, which is present twice in HIF-1α. Thus, two distinct proline residues are hydroxylated, Pro402 and Pro564 by PHD. Hydroxylated HIF-1α binds von Hippel–Lindau tumour promoter protein (pVHL) leading to ubiquitination and rapid 26S proteasomal degradation of the protein. In addition to prolyl hydroxylation, asparaginyl hydroxylation of HIF-1α by asparaginyl hydroxylase (factor inhibiting HIF-1, FIH-1) inhibits the function of its C-terminal transactivation domain (Metzen, 2007). Thus, ascorbate is connected to the ‘oxygen sensor’ role of these hydroxylases and probably functions to reduce the catalytic iron (Schofield and Ratcliffe, 2004; Kaelin and Ratcliffe, 2008).
Figure 1.
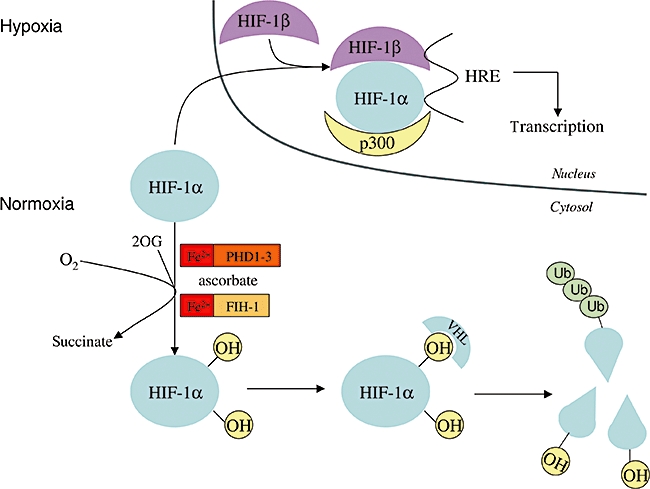
Role of ascorbate in the destruction of HIF-1α in normoxia. During hypoxia, HIF-1α enters the nucleus, forms a dimer with HIF-1β and the dimer binds to DNA through the hypoxia response element (HRE). In this way, transcription of oxygen-dependent genes is initiated. In normoxia, in the presence of oxygen and ascorbate, two proline residues and one asparagine residue of HIF-1α are hydroxylated by Fe(II) and 2-oxo-glutarate (2OG)-dependent dioxygenases, as prolyl hydroxylase domain family members, and by asparaginyl hydroxylase factor inhibiting HIF-1, respectively. Hydroxylation of prolyl residues results in the binding of the von Hippel–Lindau protein, which is part of the E3 ubiquitine ligase complex. This is the precondition for ubiquitination, which leads to the degradation of HIF-1α. Alternatively, Asn-hydroxylation prevents the binding of p300 transcription coactivator protein to HIF-1α. Thus, it inhibits transactivation of HIF-1α and the transcription of oxygen-dependent genes.
However, HIF production can also be induced in certain cases of normoxia. In cell cultures, hypoxia-like responses can be provoked by Co2+ and Ni2+ (Maxwell and Salnikow, 2004). Moreover, transition metals catalyse the oxidation of ascorbate (Buettner and Jurkiewicz, 1996). Ni2+ depletes intracellular ascorbate, and through this action it inhibits hydroxylation of proteins such as HIF-1α and −2α, and causes hypoxia-like stress (Salnikow and Kasprzak, 2005).
The family of 2OG/Fe(II)-dependent dioxygenases has recently been expanded to include new ascorbate-dependent members. An aspartyl/asparaginyl β-hydroxylase has been purified from bovine liver (Gronke et al., 1990; Wang et al., 1991), which post-translationally hydroxylates specific Asp and Asn residues within epidermal growth factor (EGF) and EGF-like domains. The presence of ascorbate was not an absolute requirement for this activity, but ascorbate enhanced the rate of the reaction several times. Experiments on the ascorbate-induced hydroxylation of EGF have typically been performed at low millimolar (1–2 mM) ascorbate concentrations. This is slightly higher than the intracellular ascorbate concentration of a hepatocyte; however, it should be noted that ascorbate is compartmentalized within the cell and in vivo subcellular concentrations are only estimates.
The physiological role of EGF hydroxylation is yet to be determined; it has been proposed that it is involved in Notch pathway signalling and its lack may promote tumour formation in mice (Dinchuk et al., 2002). β-Hydroxylated Asp and Asn residues are present in the EGF-like domains of several vitamin K-dependent clotting factors (VII, IX and X), and other plasma proteins (protein C, S and Z, and complement C1r) (Rees et al., 1988) involved in the Notch and Jagged pathway. The hydroxylation of the EGF-like domains in plasma proteins can promote Ca2+ binding and stabilizes a conformation necessary for protein–protein interactions, while hydroxylation of the proteins of the Notch signalling pathway can mediate cell motility and differentiation (Lahousse et al., 2006).
Recently, the role of 2OG/Fe(II)-dependent hydroxylases has been postulated in the demethylation of histones and nucleic acids. Histone demethylases belong to the jumonji protein family; they catalyse a typical dioxygenase reaction resulting in the formation of succinate and formaldehyde as end products. Their ascorbate-dependent activity was demonstrated in the demethylation of trimethylated lysines in histone H3. These enzymes might be involved in carcinogenesis (Klose et al., 2006a,b; Tsukada et al., 2006; Frescas et al., 2007). The recently described nucleic acid demethylases –Escherichia coli AlkB (Falnes et al., 2002; Trewick et al., 2002) and its human homologues ABH2 and ABH3 (Duncan et al., 2002; Aas et al., 2003; Lee et al., 2005; Yang et al., 2008) – repair damage caused by DNA/RNA alkylation, through a direct oxidative dealkylation mechanism that is dependent on 2OG, Fe2+ and ascorbate. AlkB and ABH3 preferentially repair single-stranded DNA lesions and can also repair methylated bases in RNA, while ABH2 has a primary role in repairing lesions in double-stranded DNA. A similar enzyme (Fto) has recently been shown to catalyse the dealkylation of 3-methylthymine in single-stranded DNA; it also affects the regulation of energy balance in mammals through an unknown mechanism (Gerken et al., 2007). The modulating role of ascorbate in nucleic acid repair and histone demethylation might expose a new function of ascorbate both in cancer prevention and carcinogenesis.
Glypicans and ascorbate
Glypicans constitute a family of glycosylphosphatidylinositol-anchored, cell-surface heparan sulphate proteoglycans that are present in lipid rafts and caveolae. They are selective regulators of ligand–receptor interactions and may thereby control growth and development. They can also migrate between the plasma membrane and endomembranes, and can import basic compounds bound to polyanionic heparan sulphate side chains (Fransson et al., 2004). A member of the family, glypican-1 migrates between the plasma membrane and the Golgi, and vice versa. Cysteines in glypican-1 can be nitrosylated by nitric oxide in a copper-dependent reaction (Cheng et al., 2002). When glypican-1 is exposed to ascorbate, nitric oxide is released from the intrinsic S-nitroso groups of the glypican-1 core protein (Ding et al., 2002). Nitric oxide participates in the deaminative cleavage of heparan sulphate by heparanase at sites where glucosamines possess a free amino group (Ding et al., 2001). The NO-catalysed degradation of heparan sulphate begins in early endosomes but mainly takes place in late endosomes (Mani et al., 2006b). Although these findings are convincing, whether the process is physiologically significant in vivo is still questionable. Further work is needed to clarify the relative contribution of ascorbate-dependent and ascorbate-independent NO formation to the NO-catalysed degradation of heparan sulphate.
Migrating glypicans can thus act as potential vehicles for basic compounds (e.g. spermine, basic peptides, etc.), which may be carried bound to heparan sulphate chains and released from glypican when heparan sulphate is degraded (Belting et al., 2003). The mechanism whereby the cargo, glypican and its degradation products are discharged from transporting endosomes is unknown; however, heparan sulphate fragments could potentially form membrane-penetrating complexes with the cargo molecules.
The deaminative degradation of glypican-1 heparan sulphate is defective in fibroblasts from patients suffering from Niemann–Pick type C1 disease (Mani et al., 2006a). Niemann–Pick type C1 disease is a rare neurovisceral degenerative disorder characterized by the accumulation of unesterified cholesterol, sphingolipids and other lipids within the endosomal/lysosomal system. Ascorbate supplementation of the fibroblast restored the deaminative degradation of glypican-1 heparan sulphate. These results suggest that defective processing of glypican-1 can contribute to cell damage in Niemann–Pick C1 disease.
Ascorbate in the redox homeostasis of certain organelles
Another essential role of ascorbate is related to its functions in redox homeostasis. Because oxidative respiration in mitochondria is a major source of the intracellular production of reactive oxygen species (ROS), the presence of mitochondria and role of vitamin C are of particular interest. Moreover, the possible functions of ascorbate in oxidative protein folding and in the maintenance of the intralumenal oxidative environment suggest it has a particular role in endoplasmic reticulum (ER)-related processes.
Vitamin C and mitochondria
In vivo studies showed that ascorbate concentration in mammalian mitochondria can be increased by dietary vitamin C supplementation (Ingebretsen and Normann, 1982; Li et al., 2001; Ramanathan et al., 2003). However, mitochondrial transport of ascorbate and its intramitochondrial functions are poorly elucidated. Kc et al. (2005) confirmed previous findings that vitamin C enters mitochondria as dehydroascorbic acid (DHA) (Szarka et al., 2004). They found a stereo-selective mitochondrial D-glucose uptake mechanism, which competes with the transport of DHA. Computational analysis of the N-terminal sequences of human GLUT isoforms revealed that GLUT-1 has the highest probability of mitochondrial localization. GLUT-1 in mitochondrial membrane was verified by mitochondrial expression of GLUT1–EGFP, immunoblot analysis and by cellular immunolocalization. Taken together, these observations suggest that, in an analogous fashion to its cellular uptake, vitamin C enters mitochondria in its oxidized state via GLUT-1.
Because DHA is very unstable and only ascorbate possesses antioxidant and free radical scavenger properties, DHA taken up or generated in the matrix must be reduced back to ascorbate, otherwise, in physiological conditions, it is lost within minutes (Winkler, 1987). Various mechanisms have been suggested for intramitochondrial ascorbate recycling. It has been reported that mitochondria are capable of reducing DHA to ascorbate in an α-lipoic acid (LA)-dependent manner (Xu and Wells, 1996). The role of reduced GSH in mitochondrial DHA reduction has also been described (Li et al., 2001). The addition of DHA to mitochondria resulted in a reduction of DHA and a mitochondrial accumulation of ascorbate. Ascorbate levels in mitochondria reached millimolar concentrations. Mitoplasts were also capable of taking up and reducing DHA (Li et al., 2001). Thioredoxin reductase in mitochondria could also reduce DHA. However, as no significant decrease in DHA reduction was detected in mitochondria isolated from selenium-deficient animals, this is likely to be a small component in the mitochondrial DHA reduction machinery (Li et al., 2001). DHA loading caused a significant decrease in mitochondrial GSH concentration. Depletion of mitochondrial GSH content caused significant impairment of DHA reduction to ascorbate. Based on these results, it has been suggested that the GSH-dependent reduction of DHA is one of the major ascorbate-producing reactions in mammalian mitochondria (Li et al., 2001).
The observation, that in the absence of respiratory substrates, liver mitochondria lose their ability to maintain or increase the level of ascorbate (Li et al., 2001; 2002; May et al., 2007), suggests that the respiratory chain contributes to the reduction of DHA. Indeed, it was shown that the formation of ascorbate from DHA could be enhanced by several respiratory substrates (succinate, malate and glycerol-3-phosphate). Using specific inhibitors of the mitochondrial electron transport chain, the site of ascorbate sparing was localized to complex III (Li et al., 2002) (Figure 2). A recent study on guinea pig muscle mitochondria (May et al., 2007) showed that although DHA transport was present, mitochondrial ascorbate accumulation could not be inhibited by GLUT-1 inhibitors, and the depletion of mitochondrial GSH was also absent.
Figure 2.
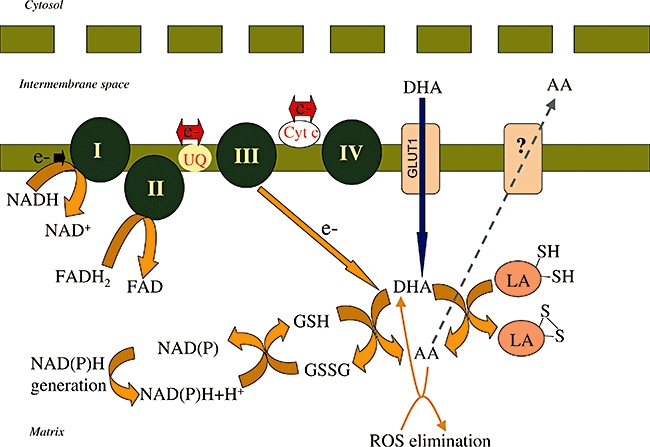
Vitamin C uptake and recycling in mitochondria. Vitamin C in its oxidized form as dehydroascorbic acid (DHA) is transported into mitochondria isolated from human kidney cells via GLUT1. The transported DHA is reduced in the mitochondria. The respiratory chain can donate electrons for the reduction of DHA. The site of DHA reduction is complex III (Li et al., 2002). DHA is also reduced back to ascorbate (AA) by DHA reductase and reduced glutathione (GSH). The oxidized glutathione formed is reduced back by glutathione reductase at the expense of NADPH (Li et al., 2001). DHA reduction can also be mediated by lipoic acid-containing enzyme complexes (Xu and Wells, 1996). AA can leave the mitochondria and this is mediated by an, at present, unidentified transporter. AA quenches reactive oxygen species, in this way protecting the mitochondrial genome and preventing mitochondrial membrane depolarization.
This phenomenon is hard to explain, because the reduction of DHA by GSH is a well-documented chemical reaction (e.g. it also takes place in the absence of any DHA-reducing enzyme) (Wells and Xu, 1994). The lack of the effect of GLUT-1 inhibitors was explained by the differences between the sources of mitochondria in the two studies (human vs. guinea pig, and cell culture vs. tissue).
On the other hand, a marked increase in the rate of DHA reduction upon addition of succinate was observed (May et al., 2007). These data and similar results obtained in plant mitochondria (Szarka et al., 2007) underline the universal role of the mitochondrial electron transfer chain in DHA reduction. Ascorbate can transmigrate between the outer and inner membrane (May et al., 2007), which raises the possibility that mitochondria contribute not only to DHA reduction, but also to the capacity of the whole cell to produce ascorbate.
The involvement of the electron transfer chain in DHA reduction can contribute to the stabilization of the mitochondrial redox balance in at least two ways. Firstly, through the donation of electrons to DHA reduction, it can decrease the reduced state of the electron transfer chain. In this way, the electron leakage of the electron transfer chain and subsequent ROS formation can be also decreased. Secondly, the production of ascorbate provides reduced vitamin C, which can scavenge ROS directly at the site of their generation.
The maintenance of the intramitochondrial ascorbate level seems to have a notable anti-apoptotic effect. The elevated level of ROS can induce the collapse of the mitochondrial membrane potentially leading to apoptosis. In HL-60 cells exposed to hydrogen peroxide, pre-incubation with DHA reduced the intracellular peroxide levels in a dose-dependent manner. In accordance with this phenomenon, the mitochondrial membrane potential was also partially conserved, and the denaturation and mitochondrial release of cytochrome c could also be avoided in DHA-pretreated cells (Gruss-Fischer and Fabian, 2002). In a study involving FAS-induced apoptosis of monocytes, the loss of the mitochondrial membrane potential could be inhibited by pretreatment with vitamin C (Perez-Cruz et al., 2003). During hypoxia and reperfusion, the electron transfer chain, especially complex III, is a primary site of ROS formation and a target of injury in the vascular endothelium (Hashimoto et al., 1994; Therade-Matharan et al., 2004). A dose-dependent reduction of ROS could be observed in response to vitamin C in human umbilical vein and coronary artery endothelial cells subjected to hypoxia–reperfusion (Dhar-Mascareño et al., 2005). The release of cytochrome c was also prevented and the mitochondrial membrane potential was stabilized by vitamin C in cells undergoing hypoxia–reperfusion (Dhar-Mascareño et al., 2005). All these events led to a decreased activation of caspase-9 and caspase-3 with resultant inhibition of apoptosis induced by hypoxia–reperfusion. These findings suggest that mitochondrial vitamin C is a major component in the maintenance of the mitochondrial membrane potential, and that vitamin C exerts its anti-apoptotic effect through its ability to scavenge ROS (Gruss-Fischer and Fabian, 2002; Pearlstein et al., 2002; Perez-Cruz et al., 2003; Dhar-Mascareño et al., 2005; Kc et al., 2005; Lee et al., 2007).
Following oxidative stress, a 3- to 10-fold increase in damage can be found in mitochondrial DNA (mtDNA) compared to that in nuclear DNA (Yakes and Van Houten, 1997; Santos et al., 2003; Jarrett et al., 2006). Kc et al. (2005) showed that mitochondrial ascorbate could protect mtDNA against oxidative damage. It was clearly demonstrated that ascorbate protected mtDNA against the ROS-induced elevation of 8-oxo-dG and apurinic/apyrimidic sites. Furthermore, pretreatment with ascorbate significantly attenuated the hydrogen peroxide-induced shearing of mtDNA (Kc et al., 2005). In accordance with these findings, in cells from the retinal pigment epithelium, vitamin C was found to induce a significant reduction of hydrogen peroxide-induced lesions in mtDNA (Jarrett et al., 2006).
Increased production of free radicals and ROS has been observed in various mitochondrial diseases (Siciliano et al., 2007). Supplementation with vitamin C is part of the treatment of mitochondrial diseases as it alleviates oxidative stress (DiMauro and Mancuso, 2007; Siciliano et al., 2007). Indeed, the elevated level of superoxide production – observed in fibroblasts from patients with electron transport chain deficiencies – could be decreased by ascorbate treatment (Sharma and Mongan, 2001). The activities of I–III and II–III complexes have also been found to be stimulated simultaneously by the addition of vitamin C to the culture media (Sharma and Mongan, 2001). Hence, there have been attempts to use vitamin C as a therapeutic agent in the treatment of various mitochondrial diseases. Vitamin C and menadione treatment of a young woman with mitochondrial myopathy and severe exercise intolerance was expected to bypass the block in complex III (Kennaway et al., 1984; Keightley et al., 2000), because the redox potentials of these electron carriers fit the gap created by the cytochrome c dysfunction (Eleff et al., 1984). However, after an initial improvement documented by 31P nuclear magnetic resonance spectroscopy of muscle (Argov et al., 1986), this state was not sustained [menadione had to be discontinued because of its withdrawal by the Food and Drug Administration (Keightley et al., 2000)]. Unfortunately, the use of vitamin C as an effective treatment for other patients suffering mitochondrial diseases has not been reported.
Ascorbate in the ER
Ascorbate metabolism and the ER are closely related to each other from several aspects. Firstly, the last steps of ascorbate synthesis are localized in the ER. Secondly, because ROS-generating reactions (e.g. oxidative protein folding) reside in this compartment, the antioxidant effect of ascorbate is presumably essential in the ER. Finally, several luminal enzymes of the secretory pathway use ascorbate as a cofactor.
L-Gulonolactone oxidase (GLO), a microsomal enzyme, catalyses the aerobic conversion of gulonolactone to ascorbate, accompanied by the production of hydrogen peroxide (Chatterjee et al., 1960a,b;). The requirement of detergents for the solubilization of GLO from microsomal vesicles (Eliceiri et al., 1969; Nishikimi et al., 1976; Kiuchi et al., 1982) strongly suggests it is located in the membrane. The amino acid sequence of GLO contains several strongly hydrophobic regions forming β-sheets rather than a typical transmembrane helical structure, which are possibly associated with the ER membrane (Koshizaka et al., 1988). The orientation of the catalytic site towards the lumen of the ER is indicated by the intraluminal accumulation of ascorbate and preferential intraluminal GSH oxidation (presumably by hydrogen peroxide) in rat liver microsomes incubated with gulonolactone (Puskás et al., 1998). This observation suggests that ascorbate produced by the liver enters the circulation – at least partly – through this secretion pathway. It should be noted that the mentioned processes are only valid in ascorbate-synthesizing species. However, in GLO-deficient species including humans, effective transport mechanisms facilitate ascorbate influx into the ER lumen.
Although the exact concentration of ascorbate in the ER lumen is unknown, local synthesis (in the case of the liver) and the high demand by luminal ascorbate-dependent enzymes (Fe2+/2OG-dependent dioxygenases: prolyl-3-hydroxylase, prolyl-4-hydroxylase and lysyl hydroxylase) suggest that the concentration of ascorbate is higher here than in plasma or cytosol. Consistent with this assumption, the vesicular structures of the secretory pathway are characterized by high (millimolar) ascorbate concentrations, which are necessary for the functioning of Cu+-dependent monooxygenases such as peptidylglycine α-amidating monooxygenase and dopamine β-hydroxylase (Daniels et al., 1982; von Zastrow et al., 1984; 1986;).
The ER is equipped with powerful electron transfer chains. During the redox cycle of the cytochrome P450 system, ROS can be formed in the ER membrane as a result of ‘chemical accidents’ (Zangar et al., 2004). The terminal oxidase of the oxidative protein folding (Ero1) generates hydrogen peroxide in the lumen (Tu and Weissman, 2002; Gross et al., 2006). A high local concentration of ascorbate is probably important to balance these pro-oxidant events.
The pro-oxidant role of ascorbate
It has been known for a long time that ascorbate can behave as a pro-ooxidant under certain conditions (e.g. at low concentrations and/or in the presence of free ions of redox-active metals such as copper and iron) (Buettner and Jurkiewicz, 1996). Its pro-oxidant behaviour has been regarded as undesirable, as it leads to the formation of ROS (Duarte and Lunec, 2005) or glycated proteins (Birlouez-Aragon and Tessier, 2003). However, certain pro-oxidant effects of ascorbate can also be advantageous. It has been recently reported that ascorbate promotes protein thiol oxidation in rat liver microsomes (Csala et al., 1999). It is thought that a metalloprotein present in the microsomal vesicles oxidizes ascorbate to DHA, which leads to the generation of ROS (Szarka et al., 2002). ROS, directly or indirectly through membrane tocopherol (Csala et al., 2001), oxidizes further ascorbate molecules. DHA formed in or transported into the lumen of the ER (Bánhegyi et al., 1998b; Csala et al., 2000) can be reduced by protein disulphide isomerase oxidizing the active central dithiols of the enzyme. Oxidized protein disulphide isomerase reacts with reduced attendant proteins yielding protein disulphides and catalytically regenerating protein disulphide isomerase (Nardai et al., 2001). Although this scheme fits the results gained in microsomal systems in vitro, whether it correctly describes the in vivo situation is still questionable (Bánhegyi et al., 2003). Further studies are needed to demonstrate the electron transfer in vivo. However, results gained in scorbutic guinea pigs showed that the missing pro-oxidant effect of ascorbate led to ER stress, presumably due to an impairment of oxidative protein folding (Margittai et al., 2005).
Therapeutic role of ascorbate
It is very hard to summarize the therapeutic benefits of vitamin C because: (i) ascorbate is added mainly in different combinations with various other drugs, antioxidants or other (natural) antioxidant diet constituents; (ii) the data obtained and the conclusions reached are sometimes contraversial; (iii) there is disagreement among physicians regarding the therapeutic use of ascorbate in everyday treatment; (iv) in several cases, very small differences are registered and the number of patients participating in these studies is very limited; and (v) there are major differences with respect to the oral and intravenous use of ascorbate. Ascorbate added intravenously is much more effective in elevating serum ascorbate levels (Padayatty and Levine, 2001), while several randomized clinical trials that have utilized oral administration of ascorbate have been unsuccessful.
In humans, the requirement for vitamin C is satisfied by natural sources and vitamin C supplements in the ordinary diet. The two major forms of vitamin C in the diet are L-ascorbic acid and L-DHA. Both ascorbate and DHA are absorbed along the entire length of the human intestine, as shown by the measurement of transport activity in luminal (brush border) membrane vesicles (Malo and Wilson, 2000). The initial rate of uptake of both ascorbate and DHA is saturable with increasing external substrate concentration, reflecting high-affinity ligand–transporter interactions.
The reduced form, L-ascorbic acid, is imported by an active mechanism requiring two sodium-dependent vitamin C transporters (SVCT1 and SVCT2), cloned for the first time in 1999 (Tsukaguchi et al., 1999; Savini et al., 2008). SVCT1 represents a high-capacity, low-affinity (Km: 65–237 µM) ascorbate transporter and is largely present in epithelial tissues, where it is involved in the absorption of dietary ascorbate and renal reabsorption, in order to maintain whole-body homeostasis. Knockout of the SVCT1 transporter gene resulted in 7- to 10-fold higher urinary loss of vitamin C (Corpe et al., 2007). Accordingly, the blood level of vitamin C was 50–70% lower in the homozygote mutants, than in the wild-type littermates.
SVCT2 is a low-capacity, high-affinity transporter (Km: 8–62 µM). It is widely expressed in metabolically active and specialized cells and tissues, in order to maintain the cellular redox state (Savini et al., 2008). Results obtained in SVCT2 knockout mutants showed that SVCT2 is necessary for prenatal transport of the ascorbic acid. It has a major role in the transport of vitamin C across the placenta. Homozygote knockout mutants are characterized by respiratory failure and intracerebral haemorrhage (Sotiriou et al., 2002).
The characteristics of DHA uptake by luminal membranes of the human jejunum clearly differ from those of ascorbate uptake (Wilson, 2005). DHA is taken up by low-affinity (Km∼0.8 mM) sodium-independent facilitated diffusion. Glucose inhibits ascorbate uptake but not that of DHA. However, the transport inhibition profile ruled out the role of sodium-dependent glucose cotransporter in ascorbate transport (Malo and Wilson, 2000). The transport protein responsible for the intestinal absorption of DHA has not yet been identified.
Plasma concentrations of vitamin C are tightly controlled when the vitamin is taken orally. Peak plasma vitamin C concentration seemed to plateau with increasing oral doses (Padayatty et al., 2004). There are two simple reasons for this: on the one hand, as mentioned in the previous paragraph, the capacity of the transporters is limited; on the other hand, the two Na+-dependent transporters can be subjected to fine-tuned regulation by their own substrate. An elevated level of ascorbate in the intestinal lumen led to down-regulation of SVCT1 mRNA in enterocytes (MacDonald et al., 2002). A similar self-regulatory role for ascorbate was demonstrated for SVCT2 in platelets (Savini et al., 2007a). Recently, it was shown that skeletal muscle cells modulated the expression of the SVCT2 carrier according to their redox balance. Both mRNA and protein levels of SVCT2 were up-regulated in hydrogen peroxide-treated myotubes, while antioxidant supplementation with LA lowered the expression of SVCT2 (Savini et al., 2007b). It seems likely that the redox state of the cell can influence the expression of SVCT 2 and in this way, the resulting response regulates the transport and intracellular level of ascorbate.
Plasma levels of vitamin C are not only limited by absorption, but also by reabsorption in the kidneys by SVCT1. Accordingly, the maximum bioavailability of vitamin C is usually attained at lower doses and declines with the elevation of oral supplements: 87% for 30 mg, 80% for 100 mg, 72% for 200 mg, 63% for 500 mg and less than 50% for 1250 mg (Levine et al., 1996; Graumlich et al., 1997). This observation was further confirmed by data from the three-compartment pharmacokinetic model for vitamin C. On the basis of this model, a single oral dose of 3 g – the maximum tolerated single dose – produces a peak plasma concentration of 206 µM, while the 1.25 g oral dose results in just a slightly lower concentration of 187 µM. Finally, for 200 mg – an amount obtained from vitamin C-rich foods – a peak-predicted concentration was approximately 90 µM (Padayatty et al., 2004). Hence, pharmacological plasma concentrations of vitamin C can only be reached through the intravenous administration of the vitamin (Padayatty et al., 2004).
The only clinical benefit of ascorbate addition, with a proven mechanism of action, is the prevention of scurvy. However, intake of as little as 10 mg·day−1 of vitamin C is appropriate for this purpose. This amount results in plasma vitamin C concentrations below 10 µM. In cases of ‘therapeutic’ levels, or even ‘normal’ levels, of vitamin C in the diet, plasma ascorbate concentrations are at least one order of magnitude higher than that necessary to prevent scurvy (Padayatty et al., 2003).
Usually, high doses of ascorbate are administered, as a high vitamin C intake can be toxic (Levine et al., 1999). The adverse effects of vitamin C are summarized by Levine et al. (1999). Excess ascorbate is normally excreted harmlessly in the urine. However, it is also well known that high amounts of ascorbate can be harmful due to oxalate formation. Thus, administration of high doses of vitamin C is contraindicative for patients with oxalate kidney stones or hyperoxaluria (Levine et al., 1999). In patients with renal failure, vitamin C is retained and converted to insoluble oxalate, which can accumulate in various organs. Following kidney transplantation, the administration of vitamin C (self-medication 2 g·day−1 for 3 years while in dialysis) led to the development of renal failure due to the widespread deposition of calcium oxalate crystals (Nankivell and Murali, 2008). Therefore, high-dose vitamin C therapy should be avoided in patients with renal failure or renal insufficiency, and in patients undergoing dialysis (McAllister et al., 1984; Wong et al., 1994; Levine et al., 1999). In patients with a deficiency in glucose-6-phosphate dehydrogenase, intravascular haemolysis occurred after high-dose vitamin C administration. It is also contraindicated in patients with systemic iron overload (Rivers, 1987; Levine et al., 1999).
There are several reasons for administering ascorbate. In addition to solving vitamin C hypo- or avitaminosis, ascorbate can be given as an antioxidant or pro-oxidant. The antioxidant properties of ascorbate have been used to treat various conditions (summarized in previous reviews; Levine et al., 1999; Heitzer et al., 2001), where oxidative stress is involved in the pathogenesis, and it is frequently administered in combination with other antioxidants.
Recently, the significance of oxidative stress in obesity-related diseases, such as type 2 diabetes, has gained further credibility (Robertson, 2004; Scheuner and Kaufman, 2008). In several studies, a decrease in plasma vitamin C levels has been observed in both type I and type II diabetes, and the effects of vitamin C administered in different ways, in addition to various combinations of different antidiabetic drugs and other antioxidants, have been assessed (Szaleczky et al., 1998; Bonnefont-Rousselot, 2004; Dorchy, 2004; Chen et al., 2006; Ratnam et al., 2006; Alamdari et al., 2007; Ceriello et al., 2007a,b;). However, at present, there is no comprehensive agreement regarding its therapeutic effectiveness for these conditions.
The role of ascorbate in the treatment of various infections has been studied for a long time. In patients with sepsis, early enteral pharmaconutrition with several agents, among others vitamin C and other antioxidants, resulted in significantly faster recovery of organ function based on a prospective, randomized, controlled, double-blind clinical trial with 55 patients (Beale et al., 2008).
It has also been suggested that the development of cataract is influenced by ascorbate (Fan et al., 2006). However, it turns out that by itself, ascorbate is unlikely to affect the progression of cataracts in most patients (Fernandez and Afshari, 2008). Vitamin C supplements combined with other antioxidant vitamins and minerals have been found to slow down the progression of advanced age-related macular degeneration and loss of visual acuity in people with signs of this disease (Evans, 2008; Evans and Henshaw, 2008). The effectiveness of vitamin C as a treatment of diabetic retinopathy has also been examined, but further studies are required to prove that it has a significant impact on its progress (Lopes de Jesus et al., 2008).
On the other hand, as shown previously, ascorbate can also act as a pro-oxidant; it can also be applied under circumstances where its pro-oxidant effect is manifested. Conflicting findings have been published on the effects of high-dose vitamin C therapy for patients with terminal cancer (Cameron and Campbell, 1974; Cameron et al., 1975; Cameron and Pauling, 1976; 1978; Creagan et al., 1979; Moertel et al., 1985). The rationale for this therapy is based on its pro-oxidant effects; it has been shown that extracellular ascorbate selectively kills cancer, but not normal cells by generating hydrogen peroxide (Chen et al., 2005; 2008;). Furthermore, it has been demonstrated that for certain tumours (e.g. advanced stages of lung cancer), serum ascorbate, as well as the serum levels of other antioxidants, is decreased (Esme et al., 2008). Consistent with the clinical pharmacokinetics of intravenously administered vitamin C, pharmacological concentrations of ascorbate can only be achieved by intravenous administration (Padayatty et al., 2004; 2006;). At present, ascorbate is used as an alternative cancer therapy (Wittes, 1985; Bernstein and Grasso, 2001; Golde, 2003; Cassileth and Deng, 2004).
Besides affecting redox conditions, it has been suggested that ascorbate can alter the expression of genes. From experiments performed in cell cultures and also in mice, a possible beneficial effect of ascorbate in a hereditary peripheral neuropathy, Charcot–Marie–Tooth disease (CMT-1A), has been suggested with regard to the promotion of myelination; this is based on a gene-inducing action of ascorbate (Passage et al., 2004; Kaya et al., 2008).
Latent scurvy
Hypoascorbinaemia and diabetes mellitus share several clinical symptoms including hypercholesterolaemia, atherosclerosis, microangiopathy, capillary hyperperfusion and haemorrhages. These effects have been observed both in patients with low ascorbate levels and in guinea pigs kept on an ascorbate-deficient diet (Turley et al., 1976; Price et al., 2001). The pathological symptoms of these conditions can be alleviated by the administration of vitamin C (Juhl et al., 2004). Based on clinical and experimental observations, and on the structural similarity between vitamin C and glucose, the theory of ‘latent scurvy’ was formulated (Price et al., 1996); the supposition is that hyperglycaemia competitively inhibits the cellular uptake of ascorbate, inducing intracellular vitamin C deficiency.
In fact, ascorbate (more precisely its oxidated form, DHA) is transported into the majority of cells by glucose transporters (Kónya and Ferdinándy, 2006). This coincidence could mean a further risk of ascorbate deficiency in hyperglycaemia. However, as a recent study pointed out, humans and certain other mammals incapable of de novo vitamin C synthesis have evolved an effective compensatory mechanism. In these species, the affinity of glucose and DHA for the transporter GLUT1 is altered by the membrane protein stomatin. Immature human erythrocytes express GLUT1, and the uptake of glucose is preferred over that of DHA. During red blood cell differentiation, expression of stomatin increases. Stomatin binds to GLUT1 and changes its substrate preference from glucose to DHA (Zhang et al., 2001; Montel-Hagen et al., 2008). Because ascorbate does not accumulate in erythrocytes, red blood cells can be regarded as ascorbate recyclers supporting the antioxidant capacity of the serum and providing vitamin C for other cell types. As GLUT transporters (Joost and Thorens, 2001) and stomatin (Stewart et al., 1992) are ubiquitously distributed in different human cell types and tissues, similar interactions can be hypothesized to occur in human cells other than erythrocytes.
Advantage of losing the ability to synthesize ascorbate
To summarize these data, ascorbate seems to be essential for antioxidant homeostasis and as a cofactor in a series of post-translational events. Consequently, this raises the basic question: what would be the advantage of losing the ability to produce ascorbate? Ascorbic acid-synthesizing capacity appeared in early terrestrial vertebrates as an answer to global hyperoxia. The concentration of atmospheric oxygen elevated dramatically during the mid- to late Paleozoic. The unusual and extreme hyperoxia was toxic for contemporary organisms as witnessed by the Permian mass extinction. Only those tetrapods which developed a powerful antioxidant system against oxygen toxicity survived the era. GLO expression emerged during that time (Chatterjee, 1973; Nandi et al., 1997).
Ascorbate synthesis finally became widespread among mammals. GLO was also present in primitive primates, but interestingly it was lost owing to mutations in the GLO gene (Nishikimi and Yagi, 1991) in the primate lineage leading to monkeys and apes in the Eocene era (55–35 million years ago). This genetic disorder generated a need to obtain ascorbate from diet in humans and other higher primates; ascorbate became vitamin C.
Nucleotide sequence alignment of one exon of the GLO gene from rat with the corresponding exon in the highly mutated, non-functional GLO gene of primates revealed that random nucleotide substitutions occurred throughout the primate sequence, as expected for a gene that got inactivated during evolution and subsequently evolved without functional constraint (Ohta and Nishikimi, 1999). Because other ascorbate-deficient species also evolved through several other lineages, it appears that the inactivation of the GLO gene occurred several times during evolution. These circumstances and the fact that the mutation did not remain a polymorphism affecting only a minority of the population, but after a positive selection mechanism only the mutant type survived, should mean that this change was advantageous. Several theories have been forwarded to explain this phenomenon.
One can regard the loss of GLO as an evolutionary accident, which was not apparent in an ascorbate-rich environment (Pauling, 1970; Szent-Györgyi, 1978; Eaton and Konner, 1985). In fact, the tropical jungle supplied our ancestors with ascorbate, because ‘not only does the monkey go through the jungle, but jungle goes through the monkey’ (Szent-Györgyi, 1978).
Another possible explanation is that vitamin C may have a role as a human fertility factor (Millar, 1992). The higher requirement of the older members of society for vitamin C led to their increased mortality in times of food shortages. This merciless system would reduce the median age of the population by favouring the survival of the youngest, thus enabling the population to regrow rapidly when food resources were restored.
An alternative hypothesis is based on the enzymology of GLO. The enzyme generates not only ascorbate, but also hydrogen peroxide as a by-product, and therefore the net reaction is redox neutral, whereas the dietary intake of ascorbate selectively increases the antioxidant capacity. Hydrogen peroxide formation is accompanied by GSH oxidation during ascorbate synthesis (Bánhegyi et al., 1996). The loss of GLO activity spared the reduced GSH, the main defence system against oxidants, while the access to ascorbate by diet was not hindered. Later, even in an environment less abundant in ascorbate, the evolutionary gains of these periods allowed the conservation of the genetic disorder (Bánhegyi et al., 1997). As a compensation for the lost ascorbate synthesis, primates use end products of different catabolic pathways (e.g. bilirubin, uric acid) as antioxidants. Most animals readily excrete soluble allantoin and biliverdin as end products of purine and porphyrin catabolism. As antioxidants can replace each other in different reactions, these metabolic changes can be regarded as compensatory mechanisms (Proctor, 1970; Ames et al., 1981; Sevanian et al., 1985; Stocker et al., 1990).
Challem and Taylor proposed a different hypothesis (Challem, 1997; Challem and Taylor, 1998). These authors suggested that a retroviral infection inserted silencing Alu elements in the gene for GLO. The lower ascorbate concentration probably was not fully compensated for by other antioxidants, allowing an increased level of free radicals. The increased frequency of free radical-induced mutations could accelerate the rate of evolution of early primates.
Very recently, Johnson et al. (2008) have put forward another original theory. They suppose that the loss of ascorbate synthesis (and rise in uric acid) had a role in maintaining blood pressure during periods of dietary restriction and environmental stress. During climatic changes, the availability of dietary ascorbate could decrease with the concomitant falling of serum ascorbate levels in GLO-deficient species. Oxidative stress (e.g. in the absence of a sufficient amount of ascorbate) results in elevated blood pressure (Vaziri and Rodriguez-Iturbe, 2006). Supporting this hypothesis, epidemiological studies have confirmed an inverse relationship between serum ascorbate levels and blood pressure (Ness et al., 1997; Bates et al., 1998), and some studies have reported that ascorbate supplements reduce blood pressure in patients with hypertension (e.g. Duffy et al., 1999). On the basis of these results, it has been suggested that those species that cannot synthesize ascorbate suffer from oxidative stress and have a higher blood pressure, and this might have provided them with a superior survival advantage in periods of environmental stress.
Perspectives
This review is an attempt to summarize recent advances at the molecular level in ascorbate research and clinical implications. A more comprehensive approach including plant and ascorbate-synthesizing animals might help us to understand the role of vitamin C and its potential effects in the treatment of various diseases. Based on the putative roles of ascorbate in metabolic and redox homeostasis, and distinctive enzyme reactions, therapeutic applications and empirical phenomena are either successfully or unsuccessfully explained by vitamin C deficiency/administration. Recently, this situation has undergone a change. New in vitro data have been obtained on the functions of ascorbate, but further investigations are needed to interpret their physiological or pathological significance and therapeutic implications. The compartmentalization of ascorbate within a cell suggests it has different functions in particular organelles. The redox state of its microenvironment determines its vital metabolism and other functions, and also its signalling role. Further research is required to explore the exact effects of ascorbate in physiological systems and in the pathology of diseases connected to mitochondria and ER. These new findings could also lead to new therapeutic implications regarding ascorbate. Recent publications raise some new aspects and suggest new mechanisms of ascorbate at the molecular level. However, it should be noted that some well-known beneficial effects of ascorbate administration are still only understood at the phenomenological level.
Acknowledgments
This work was supported by NKTH RET05/2004.
Glossary
Abbreviations:
- 2OG
α-ketoglutarate
- AMD
age-related macular degeneration
- CMT
Charcot–Marie–Tooth disease
- DHA
dehydroascorbic acid
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- GLO
gulonolactone oxidase
- GSH
glutathione
- HIF-1α
hypoxia-inducible factor-1α
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- PHD
prolyl hydroxylase
- pVHL
von Hippel–Lindau tumour promoter protein
- ROS
reactive oxygen species
- SGLT
sodium-dependent glucose cotransporter
- SVCT
sodium-dependent vitamin C transporter
References
- Aas PA, Otterlei M, Falnes PO, Vågbø CB, Skorpen F, Akbari M, et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- Alamdari DH, Paletas K, Pegiou T, Sarigianni M, Befani C, Koliakos G. A novel assay for the evaluation of the prooxidant–antioxidant balance, before and after antioxidant vitamin administration in type II diabetes patients. Clin Biochem. 2007;40:248–254. doi: 10.1016/j.clinbiochem.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci USA. 1981;78:6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argov Z, Bank WJ, Maris J, Eleff S, Kennaway NG, Olson RE, et al. Treatment of mitochondrial myopathy due to complex III deficiency with vitamins K3 and C: a 31P-NMR follow-up study. Ann Neurol. 1986;19:598–602. doi: 10.1002/ana.410190615. [DOI] [PubMed] [Google Scholar]
- Bánhegyi G, Mandl J. The hepatic glycogenoreticular system. Pathol Oncol Res. 2001;7:107–110. doi: 10.1007/BF03032575. [DOI] [PubMed] [Google Scholar]
- Bánhegyi G, Garzó T, Antoni F, Mandl J. Glycogenolysis – and not gluconeogenesis – is the source of UDP-glucuronic acid for glucuronidation. Biochim Biophys Acta. 1988;967:429–435. doi: 10.1016/0304-4165(88)90106-7. [DOI] [PubMed] [Google Scholar]
- Bánhegyi G, Csala M, Braun L, Garzó T, Mandl J. Ascorbate synthesis-dependent glutathione consumption in mouse liver. FEBS Lett. 1996;381:39–41. doi: 10.1016/0014-5793(96)00077-4. [DOI] [PubMed] [Google Scholar]
- Bánhegyi G, Braun L, Csala M, Puskás F, Mandl J. Ascorbate metabolism and its regulation in animals. Free Radic Biol Med. 1997;23:793–803. doi: 10.1016/s0891-5849(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Bánhegyi G, Braun L, Csala M, Puskás F, Somogyi A, Kardon T, et al. Ascorbate and environmental stress. Ann N Y Acad Sci. 1998a;851:292–303. [Google Scholar]
- Bánhegyi G, Marcolongo P, Puskás F, Fulceri R, Mandl J, Benedetti A. Dehydroascorbate and ascorbate transport in rat liver microsomal vesicles. J Biol Chem. 1998b;273:2758–2762. doi: 10.1074/jbc.273.5.2758. [DOI] [PubMed] [Google Scholar]
- Bánhegyi G, Csala M, Szarka A, Varsányi M, Benedetti A, Mandl J. Role of ascorbate in oxidative protein folding. Biofactors. 2003;17:37–46. doi: 10.1002/biof.5520170105. [DOI] [PubMed] [Google Scholar]
- Bánhegyi G, Benedetti A, Csala M, Mandl J. Stress on redox. FEBS Lett. 2007;581:3634–3640. doi: 10.1016/j.febslet.2007.04.028. [DOI] [PubMed] [Google Scholar]
- Bates CJ, Walmsley CM, Prentice A, Finch S. Does vitamin C reduce blood pressure? Results of a large study of people aged 65 or older. J Hypertens. 1998;16:925–932. doi: 10.1097/00004872-199816070-00005. [DOI] [PubMed] [Google Scholar]
- Beale RJ, Sherry T, Lei K, Campbell-Stephen L, McCook J, Smith J, et al. Early enteral supplementation with key pharmaconutrients improves Sequential Organ Failure Assessment score in critically ill patients with sepsis: outcome of a randomized, controlled, double-blind trial. Crit Care Med. 2008;36:131–144. doi: 10.1097/01.CCM.0000297954.45251.A9. [DOI] [PubMed] [Google Scholar]
- Belting M, Mani K, Jönsson M, Cheng F, Sandgren S, Jonsson S, et al. Glypican-1 is a vehicle for polyamine uptake in mammalian cells: a pivotal role for nitrosothiol-derived nitric oxide. J Biol Chem. 2003;278:47181–47189. doi: 10.1074/jbc.M308325200. [DOI] [PubMed] [Google Scholar]
- Bernstein BJ, Grasso T. Prevalence of complementary and alternative medicine use in cancer patients. J Clin Oncol. 2001;15:1267–1272. [PubMed] [Google Scholar]
- Birlouez-Aragon I, Tessier FJ. Antioxidant vitamins and degenerative pathologies. A review of vitamin C. J Nutr Health Aging. 2003;7:103–109. [PubMed] [Google Scholar]
- Bonnefont-Rousselot D. The role of antioxidant micronutrients in the prevention of diabetic complications. Treat Endocrinol. 2004;3:41–52. doi: 10.2165/00024677-200403010-00005. [DOI] [PubMed] [Google Scholar]
- Braun L, Garzó T, Mandl J, Bánhegyi G. Ascorbic acid synthesis is stimulated by enhanced glycogenolysis in murine liver. FEBS Lett. 1994;352:4–6. doi: 10.1016/0014-5793(94)00905-8. [DOI] [PubMed] [Google Scholar]
- Braun L, Csala M, Poussu A, Garzó T, Mandl J, Bánhegyi G. Glutathione depletion induces glycogenolysis dependent ascorbate synthesis in isolated murine hepatocytes. FEBS Lett. 1996a;388:173–176. doi: 10.1016/0014-5793(96)00548-0. [DOI] [PubMed] [Google Scholar]
- Braun L, Puskás F, Csala M, Györffy E, Garzó T, Mandl J, et al. Gluconeogenesis from ascorbic acid: ascorbate recycling in isolated murine hepatocytes. FEBS Lett. 1996b;390:183–186. doi: 10.1016/0014-5793(96)00654-0. [DOI] [PubMed] [Google Scholar]
- Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996;145:532–541. [PubMed] [Google Scholar]
- Cameron E, Campbell A. The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem Biol Interact. 1974;9:285–315. doi: 10.1016/0009-2797(74)90019-2. [DOI] [PubMed] [Google Scholar]
- Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: prolongation of survival times in terminal human cancer. Proc Natl Acad Sci USA. 1976;73:3685–3689. doi: 10.1073/pnas.73.10.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc Natl Acad Sci USA. 1978;75:4538–4542. doi: 10.1073/pnas.75.9.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron E, Campbell A, Jack T. The orthomolecular treatment of cancer. III. Reticulum cell sarcoma: double complete regression induced by high-dose ascorbic acid therapy. Chem Biol Interact. 1975;11:387–393. doi: 10.1016/0009-2797(75)90007-1. [DOI] [PubMed] [Google Scholar]
- Cassileth BR, Deng G. Complementary and alternative therapies for cancer. Oncologist. 2004;9:80–89. doi: 10.1634/theoncologist.9-1-80. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Kumar S, Piconi L, Esposito K, Giugliano D. Simultaneous control of hyperglycemia and oxidative stress normalizes endothelial function in type 1 diabetes. Diabetes Care. 2007a;30:649–654. doi: 10.2337/dc06-2048. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Piconi L, Esposito K, Giugliano D. Telmisartan shows an equivalent effect of vitamin C in further improving endothelial dysfunction after glycemia normalization in type 1 diabetes. Diabetes Care. 2007b;30:1694–1698. doi: 10.2337/dc07-0318. [DOI] [PubMed] [Google Scholar]
- Challem JJ. Did the loss of endogenous ascorbate propel the evolution of anthropoidea and Homo sapiens? Med Hypotheses. 1997;48:387–392. doi: 10.1016/s0306-9877(97)90033-5. [DOI] [PubMed] [Google Scholar]
- Challem JJ, Taylor EW. Retroviruses, ascorbate, and mutations, in the evolution of Homo sapiens. Free Radical Biol Med. 1998;25:130–132. doi: 10.1016/s0891-5849(98)00034-3. [DOI] [PubMed] [Google Scholar]
- Chatterjee IB. Evolution and the biosynthesis of ascorbic acid. Science. 1973;182:1271–1272. doi: 10.1126/science.182.4118.1271. [DOI] [PubMed] [Google Scholar]
- Chatterjee IB, Chatterjee GC, Ghosh NC, Ghosh JJ, Guha BC. Biological synthesis of l-ascorbic acid in animal tissues: conversion of l-gulonolactone into l-ascorbic acid. Biochem J. 1960a;74:193–203. doi: 10.1042/bj0740193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee IB, Chatterjee GC, Ghosh NC, Ghosh JJ, Guha BC. Biological synthesis of l-ascorbic acid in animal tissues: conversion of d-glucuronolactone and l-gulonolactone into L-ascorbic acid. Biochem J. 1960b;76:279–292. doi: 10.1042/bj0760279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci USA. 2005;120:13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Karne RJ, Hall G, Campia U, Panza JA, Cannon RO, 3rd, et al. High-dose oral vitamin C partially replenishes vitamin C levels in patients with type 2 diabetes and low vitamin C levels but does not improve endothelial dysfunction or insulin resistance. Am J Physiol Heart Circ Physiol. 2006;290:H137–H145. doi: 10.1152/ajpheart.00768.2005. [DOI] [PubMed] [Google Scholar]
- Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci USA. 2008;105:11105–11109. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Mani K, van den Born J, Ding K, Belting M, Fransson LA. Nitric oxide-dependent processing of heparan sulfate in recycling S-nitrosylated glypican-1 takes place in caveolin-1-containing endosomes. J Biol Chem. 2002;277:44431–44439. doi: 10.1074/jbc.M205241200. [DOI] [PubMed] [Google Scholar]
- Corpe C, Tu H, Wang J, Eck P, Wang Y, Schnermann J, et al. SVCT1 (Slc23a1) knock out mice: Slc23a1 as the vitamin C kidney reabsorptive transporter. FASEB J. 2007;21:lb520. [Google Scholar]
- Creagan ET, Moertel CG, O'Fallon JR, Schutt AJ, O'Connell MJ, Rubin J, et al. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N Engl J Med. 1979;301:687–690. doi: 10.1056/NEJM197909273011303. [DOI] [PubMed] [Google Scholar]
- Csala M, Braun L, Mile V, Kardon T, Szarka A, Kupcsulik P, et al. Ascorbate-mediated electron transfer in protein thiol oxidation in the endoplasmic reticulum. FEBS Lett. 1999;460:539–543. doi: 10.1016/s0014-5793(99)01412-x. [DOI] [PubMed] [Google Scholar]
- Csala M, Mile V, Benedetti A, Mandl J, Bánhegyi G. Ascorbate oxidation is a prerequisite for its transport into rat liver microsomal vesicles. Biochem J. 2000;349:413–415. doi: 10.1042/0264-6021:3490413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csala M, Szarka A, Margittai E, Mile V, Kardon T, Braun L, et al. Role of vitamin E in ascorbate-dependent protein thiol oxidation in rat liver endoplasmic reticulum. Arch Biochem Biophys. 2001;388:55–59. doi: 10.1006/abbi.2000.2260. [DOI] [PubMed] [Google Scholar]
- Daniels AJ, Dean G, Viveros OH, Diliberto EJ., Jr Secretion of newly taken-up ascorbic acid by adrenomedullary chromaffin cells. Science. 1982;216:737–739. doi: 10.1126/science.7079733. [DOI] [PubMed] [Google Scholar]
- Dhar-Mascareño M, Cárcamo JM, Golde DW. Hypoxia-reoxygenation-induced mitochondrial damage and apoptosis in human endothelial cells are inhibited by vitamin C. Free Radic Biol Med. 2005;38:1311–1322. doi: 10.1016/j.freeradbiomed.2005.01.017. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Mancuso M. Mitochondrial diseases: therapeutic approaches. Biosci Rep. 2007;27:125–137. doi: 10.1007/s10540-007-9041-4. [DOI] [PubMed] [Google Scholar]
- Dinchuk JE, Focht RJ, Kelley JA, Henderson NL, Zolotarjova NI, Wynn R, et al. Absence of post-translational aspartyl β-hydroxylation of epidermal growth factor domains in mice leads to developmental defects and an increased incidence of intestinal neoplasia. J Biol Chem. 2002;277:12970–12977. doi: 10.1074/jbc.M110389200. [DOI] [PubMed] [Google Scholar]
- Ding K, Sandgren S, Mani K, Belting M, Fransson LA. Modulations of glypican-1 heparan sulfate structure by inhibition of endogenous polyamine synthesis. Mapping of spermine-binding sites and heparanase, heparin lyase, and nitric oxide/nitrite cleavage sites. J Biol Chem. 2001;276:46779–46791. doi: 10.1074/jbc.M105419200. [DOI] [PubMed] [Google Scholar]
- Ding K, Mani K, Cheng F, Belting M, Fransson LA. Copper-dependent autocleavage of glypican-1 heparan sulfate by nitric oxide derived from intrinsic nitrosothiols. J Biol Chem. 2002;277:33353–33360. doi: 10.1074/jbc.M203383200. [DOI] [PubMed] [Google Scholar]
- Dorchy H. Screening for subclinical complications in young type 1 diabetic patients: experience acquired in Brussels. Pediatr Endocrinol Rev. 2004;1:380–403. [PubMed] [Google Scholar]
- Duarte TL, Lunec J. Review: when is an antioxidant not an antioxidant? A review of novel actions and reactions of vitamin C. Free Radic Res. 2005;39:671–686. doi: 10.1080/10715760500104025. [DOI] [PubMed] [Google Scholar]
- Duffy SJ, Gokce N, Holbrook M, Huang A, Frei B, Keaney JF, Jr, et al. Treatment of hypertension with ascorbic acid. Lancet. 1999;354:2048–2049. doi: 10.1016/s0140-6736(99)04410-4. [DOI] [PubMed] [Google Scholar]
- Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci USA. 2002;99:16660–16665. doi: 10.1073/pnas.262589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med. 1985;312:283–289. doi: 10.1056/NEJM198501313120505. [DOI] [PubMed] [Google Scholar]
- Eleff S, Kennaway NG, Buist NR, Darley-Usmar VM, Capaldi RA, Bank WJ, et al. 31P NMR study of improvement in oxidative phosphorylation by vitamins K3 and C in a patient with a defect in electron transport at complex III in skeletal muscle. Proc Natl Acad Sci USA. 1984;81:3529–3533. doi: 10.1073/pnas.81.11.3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri GL, Lai EK, McCay PB. Gulonolactone oxidase. Solubilization, properties, and partial purification. J Biol Chem. 1969;244:2641–2645. [PubMed] [Google Scholar]
- Englard S, Seifter S. The biochemical functions of ascorbic acid. Annu Rev Nutr. 1986;6:365–406. doi: 10.1146/annurev.nu.06.070186.002053. [DOI] [PubMed] [Google Scholar]
- Esme H, Cemek M, Sezer M, Saglam H, Demir A, Melek H, et al. High levels of oxidative stress in patients with advanced lung cancer. Respirology. 2008;13:112–116. doi: 10.1111/j.1440-1843.2007.01212.x. [DOI] [PubMed] [Google Scholar]
- Evans J. Antioxidant supplements to prevent or slow down the progression of AMD: a systematic review and meta-analysis. Eye. 2008;22:751–760. doi: 10.1038/eye.2008.100. [DOI] [PubMed] [Google Scholar]
- Evans JR, Henshaw K. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Syst Rev. 2008;1:CD000253. doi: 10.1002/14651858.CD000253.pub2. [DOI] [PubMed] [Google Scholar]
- Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- Fan X, Reneker LW, Obrenovich ME, Strauch C, Cheng R, Jarvis SM, et al. Vitamin C mediates chemical aging of lens crystallins by the Maillard reaction in a humanized mouse model. Proc Natl Acad Sci USA. 2006;103:16912–16917. doi: 10.1073/pnas.0605101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MM, Afshari NA. Nutrition and the prevention of cataracts. Curr Opin Ophthalmol. 2008;19:66–70. doi: 10.1097/ICU.0b013e3282f2d7b6. [DOI] [PubMed] [Google Scholar]
- Fransson LA, Belting M, Cheng F, Jönsson M, Mani K, Sandgren S. Novel aspects of glypican glycobiology. Cell Mol Life Sci. 2004;61:1016–1024. doi: 10.1007/s00018-004-3445-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D, Guardavaccaro D, Bassermann F, Koyama-Nasu R, Pagano M. JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature. 2007;450:309–313. doi: 10.1038/nature06255. [DOI] [PubMed] [Google Scholar]
- Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde DW. Vitamin C in cancer. Integr Cancer Ther. 2003;2:158–159. doi: 10.1177/1534735403002002009. [DOI] [PubMed] [Google Scholar]
- Graumlich JF, Ludden TM, Conry-Cantilena C, Cantilena LR, Jr, Wang Y, Levine M. Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm Res. 1997;14:1133–1139. doi: 10.1023/a:1012186203165. [DOI] [PubMed] [Google Scholar]
- Gronke RS, Welsch DJ, VanDusen WJ, Garsky VM, Sardana MK, Stern AM, et al. Partial purification and characterization of bovine liver aspartyl β-hydroxylase. J Biol Chem. 1990;265:8558–8565. [PubMed] [Google Scholar]
- Gross E, Sevier CS, Heldman N, Vitu E, Bentzur M, Kaiser CA, et al. Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc Natl Acad Sci USA. 2006;103:299–304. doi: 10.1073/pnas.0506448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruss-Fischer T, Fabian I. Protection by ascorbic acid from denaturation and release of cytochrome c, alteration of mitochondrial membrane potential and activation of multiple caspases induced by H2O2, in human leukemia cells. Biochem Pharmacol. 2002;63:1325–1335. doi: 10.1016/s0006-2952(02)00863-8. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Itoh K, Nishida K, Okano T, Miyazawa Y, Okinaga K. Rapid superoxide production by endothelial cells and their injury upon reperfusion. J Surg Res. 1994;57:693–697. doi: 10.1006/jsre.1994.1203. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Schlinzig T, Krohn K, Meinertz T, Münzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104:2673–2678. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- Hewitson KS, McNeill LA, Elkins JM, Schofield CJ. The role of iron and 2-oxoglutarate oxygenases in signalling. Biochem Soc Trans. 2003;31:510–515. doi: 10.1042/bst0310510. [DOI] [PubMed] [Google Scholar]
- Ingebretsen OC, Normann PT. Transport of ascorbate into guinea pig liver mitochondria. Biochim Biophys Acta. 1982;684:21–26. doi: 10.1016/0005-2736(82)90044-x. [DOI] [PubMed] [Google Scholar]
- Jarrett SG, Cuenco J, Boulton M. Dietary antioxidants provide differential subcellular protection in epithelial cells. Redox Rep. 2006;11:144–152. doi: 10.1179/135100006X116646. [DOI] [PubMed] [Google Scholar]
- Johnson RJ, Gaucher EA, Sautin YY, Henderson GN, Angerhofer AJ, Benner SA. The planetary biology of ascorbate and uric acid and their relationship with the epidemic of obesity and cardiovascular disease. Med Hypotheses. 2008;71:22–31. doi: 10.1016/j.mehy.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost HG, Thorens B. The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review) Mol Membr Biol. 2001;18:247–256. doi: 10.1080/09687680110090456. [DOI] [PubMed] [Google Scholar]
- Juhl B, Klein F, Christiansen JS. Vitamin C treatment reduces transcapillary escape rate of albumin in type 1 diabetes. Eur J Intern Med. 2004;15:428–435. doi: 10.1016/j.ejim.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Kaelin W, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kaya F, Belin S, Micallef J, Blin O, Fontés M. Analysis of the benefits of vitamin cocktails in treating Charcot–Marie–Tooth disease type 1A. Muscle Nerve. 2008;38:1052–1054. doi: 10.1002/mus.21071. [DOI] [PubMed] [Google Scholar]
- Kc S, Cárcamo JM, Golde DW. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005;19:1657–1667. doi: 10.1096/fj.05-4107com. [DOI] [PubMed] [Google Scholar]
- Keightley JA, Anitori R, Burton MD, Quan F, Buist NR, Kennaway NG. Mitochondrial encephalomyopathy and complex III deficiency associated with a stop-codon mutation in the cytochrome b gene. Am J Hum Genet. 2000;67:1400–1410. doi: 10.1086/316900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennaway NG, Buist NR, Darley-Usmar VM, Papadimitriou A, Dimauro S, Kelley RI, et al. Lactic acidosis and mitochondrial myopathy associated with deficiency of several components of complex III of the respiratory chain. Pediatr Res. 1984;18:991–999. doi: 10.1203/00006450-198410000-00017. [DOI] [PubMed] [Google Scholar]
- Kiuchi K, Nishikimi M, Yagi K. Purification and characterization of l-gulonolactone oxidase from chicken kidney microsomes. Biochemistry. 1982;21:5076–5082. doi: 10.1021/bi00263a035. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006a;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006b;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- Kónya C, Ferdinándy P. Vitamin C: new role of the old vitamin in the cardiovascular system. Br J Pharmacol. 2006;147:125–127. doi: 10.1038/sj.bjp.0706494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshizaka T, Nishikimi M, Ozawa T, Yagi K. Isolation and sequence analysis of a complementary DNA encoding rat liver L-gulono-gamma-lactone oxidase, a key enzyme for l-ascorbic acid biosynthesis. J Biol Chem. 1988;263:1619–1621. [PubMed] [Google Scholar]
- Kramer JGH. Medicina Castrensis. Vienna: Peter Konrad Monath; 1739. [Google Scholar]
- Lahousse SA, Carter JJ, Xu XJ, Wands JR, de la Monte SM. Differential growth factor regulation of aspartyl-(asparaginyl)-β-hydroxylase family genes in SH-Sy5y human neuroblastoma cells. BMC Cell Biol. 2006;7:41. doi: 10.1186/1471-2121-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Jin SG, Cai S, Chen Y, Pfeifer GP, O'Connor TR. Repair of methylation damage in DNA and RNA by mammalian AlkB homologues. J Biol Chem. 2005;280:39448–39459. doi: 10.1074/jbc.M509881200. [DOI] [PubMed] [Google Scholar]
- Lee WY, Lee JS, Lee SM. Protective effects of combined ischemic preconditioning and ascorbic acid on mitochondrial injury in hepatic ischemia/reperfusion. J Surg Res. 2007;142:45–52. doi: 10.1016/j.jss.2006.08.043. [DOI] [PubMed] [Google Scholar]
- Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93:3704–3709. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Rumsey SC, Daruwala R, Park JB, Wang Y. Criteria and recommendations for vitamin intake. JAMA. 1999;281:1415–1423. doi: 10.1001/jama.281.15.1415. [DOI] [PubMed] [Google Scholar]
- Li Y, Schellhorn HE. New developments and novel therapeutic perspectives for vitamin C. J Nutr. 2007;137:2171–2184. doi: 10.1093/jn/137.10.2171. [DOI] [PubMed] [Google Scholar]
- Li X, Cobb CE, Hill KE, Burk RF, May JM. Mitochondrial uptake and recycling of ascorbic acid. Arch Biochem Biophys. 2001;387:143–153. doi: 10.1006/abbi.2000.2245. [DOI] [PubMed] [Google Scholar]
- Li X, Cobb CE, May JM. Mitochondrial recycling of ascorbic acid from dehydroascorbic acid: dependence on the electron transport chain. Arch Biochem Biophys. 2002;403:103–110. doi: 10.1016/S0003-9861(02)00205-9. [DOI] [PubMed] [Google Scholar]
- Lind J. Treatise of the Scurvy. London: A. Millar; 1753. [Google Scholar]
- Linster CL, Van Schaftingen E. Rapid stimulation of free glucuronate formation by non-glucuronidable xenobiotics in isolated rat hepatocytes. J Biol Chem. 2003;278:36328–36333. doi: 10.1074/jbc.M306593200. [DOI] [PubMed] [Google Scholar]
- Linster CL, Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- Lopes de Jesus CC, Atallah AN, Valente O, Moça Trevisani VF. Vitamin C and superoxide dismutase (SOD) for diabetic retinopathy. Cochrane Database Syst Rev. 2008;1:CD006695. doi: 10.1002/14651858.CD006695.pub2. [DOI] [PubMed] [Google Scholar]
- McAllister CJ, Scowden EB, Dewberry FL, Richman A. Renal failure secondary to massive infusion of vitamin C. JAMA. 1984;252:1684. [PubMed] [Google Scholar]
- MacDonald L, Thumser AE, Sharp P. Decreased expression of the vitamin C transporter SVCT1 by ascorbic acid in a human intestinal epithelial cell line. Br J Nutr. 2002;87:97–100. doi: 10.1079/BJN2001492. [DOI] [PubMed] [Google Scholar]
- Malo C, Wilson JX. Glucose modulates vitamin C transport in adult human small intestinal brush border membrane vesicles. J Nutr. 2000;130:63–69. doi: 10.1093/jn/130.1.63. [DOI] [PubMed] [Google Scholar]
- Mandl J, Bánhegyi G, Kalapos MP, Garzó T. Increased oxidation and decreased conjugation of drugs in the liver caused by starvation. Altered metabolism of certain aromatic compounds and acetone. Chem Biol Interact. 1995;96:87–101. doi: 10.1016/0009-2797(94)03587-x. [DOI] [PubMed] [Google Scholar]
- Mani K, Cheng F, Fransson LA. Defective nitric oxide-dependent, deaminative cleavage of glypican-1 heparan sulfate in Niemann-Pick C1 fibroblasts. Glycobiology. 2006a;16:711–718. doi: 10.1093/glycob/cwj121. [DOI] [PubMed] [Google Scholar]
- Mani K, Cheng F, Fransson LA. Constitutive and vitamin C-induced, NO-catalyzed release of heparan sulfate from recycling glypican-1 in late endosomes. Glycobiology. 2006b;16:1251–1261. doi: 10.1093/glycob/cwl045. [DOI] [PubMed] [Google Scholar]
- Margittai E, Bánhegyi G, Kiss A, Nagy G, Mandl J, Schaff Z, et al. Scurvy leads to endoplasmic reticulum stress and apoptosis in the liver of Guinea pigs. J Nutr. 2005;135:2530–2534. doi: 10.1093/jn/135.11.2530. [DOI] [PubMed] [Google Scholar]
- Maxwell P, Salnikow K. HIF-1: an oxygen and metal responsive transcription factor. Cancer Biol Ther. 2004;3:29–35. doi: 10.4161/cbt.3.1.547. [DOI] [PubMed] [Google Scholar]
- May JM, Li L, Qu ZC, Cobb CE. Mitochondrial recycling of ascorbic acid as a mechanism for regenerating cellular ascorbate. Biofactors. 2007;30:35–48. doi: 10.1002/biof.5520300105. [DOI] [PubMed] [Google Scholar]
- Metzen E. Enzyme substrate recognition in oxygen sensing: how the HIF trap snaps. Biochem J. 2007;408:5–6. doi: 10.1042/BJ20071306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar J. Vitamin C – the primate fertility factor? Med Hypotheses. 1992;38:292–295. doi: 10.1016/0306-9877(92)90019-9. [DOI] [PubMed] [Google Scholar]
- Moertel CG, Fleming TR, Creagan ET, Rubin J, O'Connell MJ, Ames MM. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N Engl J Med. 1985;312:137–141. doi: 10.1056/NEJM198501173120301. [DOI] [PubMed] [Google Scholar]
- Montel-Hagen A, Kinet S, Manel N, Mongellaz C, Prohaska R, Battini JL, et al. Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell. 2008;132:1039–1048. doi: 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Myllyla R, Majamaa K, Gunzler V, Hanauske-Abel HM, Kivirikko KI. Ascorbate is consumed stoichiometrically in the uncoupled reactions catalyzed by prolyl 4-hydroxylase and lysyl hydroxylase. J Biol Chem. 1984;259:5403–5405. [PubMed] [Google Scholar]
- Nandi A, Mukhopadhyay CK, Ghosh MK, Chattopadhyay DJ, Chatterjee IB. Evolutionary significance of vitamin C biosynthesis in terrestrial vertebrates. Free Radical Biol Med. 1997;22:1047–1054. doi: 10.1016/s0891-5849(96)00491-1. [DOI] [PubMed] [Google Scholar]
- Nankivell BJ, Murali KM. Images in clinical medicine. Renal failure from vitamin C after transplantation. N Engl J Med. 2008;358:e4. doi: 10.1056/NEJMicm070984. [DOI] [PubMed] [Google Scholar]
- Nardai G, Braun L, Csala M, Mile V, Csermely P, Benedetti A, et al. Protein-disulfide isomerase- and protein thiol-dependent dehydroascorbate reduction and ascorbate accumulation in the lumen of the endoplasmic reticulum. J Biol Chem. 2001;276:8825–8828. doi: 10.1074/jbc.M010563200. [DOI] [PubMed] [Google Scholar]
- Ness AR, Chee D, Elliott P. Vitamin C and blood pressure – an overview. J Hum Hypertens. 1997;11:343–350. doi: 10.1038/sj.jhh.1000423. [DOI] [PubMed] [Google Scholar]
- Nishikimi M, Yagi K. Molecular basis for the deficiency in humans of gulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am J Clin Nutr. 1991;54:1203S–1208S. doi: 10.1093/ajcn/54.6.1203s. [DOI] [PubMed] [Google Scholar]
- Nishikimi M, Tolbert BM, Udenfriend S. Purification and characterization of l-gulono-gamma-lactone oxidase from rat and goat liver. Arch Biochem Biophys. 1976;175:427–435. doi: 10.1016/0003-9861(76)90530-0. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Nishikimi M. Random nucleotide substitutions in primate nonfunctional gene for l-gulono-gamma-lactone oxidase, the missing enzyme in l-ascorbic acid biosynthesis. Biochim Biophys Acta. 1999;1472:408–411. doi: 10.1016/s0304-4165(99)00123-3. [DOI] [PubMed] [Google Scholar]
- Padayatty SJ, Levine M. New insights into the physiology and pharmacology of vitamin C. CMAJ. 2001;164:353–355. [PMC free article] [PubMed] [Google Scholar]
- Padayatty SJ, Katz A, Wang Y, Eck P, Kwon O, Lee JH, et al. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J Am Coll Nutr. 2003;22:18–35. doi: 10.1080/07315724.2003.10719272. [DOI] [PubMed] [Google Scholar]
- Padayatty SJ, Sun H, Wang Y, Riordan HD, Hewitt SM, Katz A, et al. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004;140:533–537. doi: 10.7326/0003-4819-140-7-200404060-00010. [DOI] [PubMed] [Google Scholar]
- Padayatty SJ, Riordan HD, Hewitt SM, Katz A, Hoffer LJ, Levine M. Intravenously administered vitamin C as cancer therapy: three cases. CMAJ. 2006;174:937–942. doi: 10.1503/cmaj.050346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10:396–401. doi: 10.1038/nm1023. [DOI] [PubMed] [Google Scholar]
- Pauling L. Evolution and the need for ascorbic acid. Proc Natl Acad Sci USA. 1970;67:1643–1648. doi: 10.1073/pnas.67.4.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlstein DP, Ali MH, Mungai PT, Hynes KL, Gewertz BL, Schumacker PT. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb Vasc Biol. 2002;22:566–573. doi: 10.1161/01.atv.0000012262.76205.6a. [DOI] [PubMed] [Google Scholar]
- Perez-Cruz I, Carcamo JM, Golde DW. Vitamin C inhibits FAS-induced apoptosis in monocytes and U937 cells. Blood. 2003;102:336–343. doi: 10.1182/blood-2002-11-3559. [DOI] [PubMed] [Google Scholar]
- Peterkofsky B. Ascorbate requirement for hydroxylation and secretion of procollagen: relationship to inhibition of collagen synthesis in scurvy. Am J Clin Nutr. 1991;54:1135S–1140S. doi: 10.1093/ajcn/54.6.1135s. [DOI] [PubMed] [Google Scholar]
- Price KD, Price CS, Reynolds RD. Hyperglycemia-induced latent scurvy and atherosclerosis: the scorbutic-metaplasia hypothesis. Med Hypotheses. 1996;46:119–129. doi: 10.1016/s0306-9877(96)90011-0. [DOI] [PubMed] [Google Scholar]
- Price KD, Price CS, Reynolds RD. Hyperglycemia-induced ascorbic acid deficiency promotes endothelial dysfunction and the development of atherosclerosis. Atherosclerosis. 2001;158:1–12. doi: 10.1016/s0021-9150(01)00569-x. [DOI] [PubMed] [Google Scholar]
- Proctor P. Similar functions of uric acid and ascorbate in man? Nature. 1970;228:868. doi: 10.1038/228868a0. [DOI] [PubMed] [Google Scholar]
- Puskás F, Braun L, Csala M, Kardon T, Marcolongo P, Benedetti A, et al. Gulonolactone oxidase activity-dependent intravesicular glutathione oxidation in rat liver microsomes. FEBS Lett. 1998;430:293–296. doi: 10.1016/s0014-5793(98)00678-4. [DOI] [PubMed] [Google Scholar]
- Ramanathan K, Shila S, Kumaran S, Panneerselvam C. Ascorbic acid and α-tocopherol as potent modulators on arsenic induced toxicity in mitochondria. J Nutr Biochem. 2003;14:416–420. doi: 10.1016/s0955-2863(03)00076-7. [DOI] [PubMed] [Google Scholar]
- Ratnam DV, Ankola DD, Bhardwaj V, Sahana DK, Kumar MN. Role of antioxidants in prophylaxis and therapy: a pharmaceutical perspective. J Control Release. 2006;113:189–207. doi: 10.1016/j.jconrel.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Rebouche CJ. Ascorbic acid and carnitine biosynthesis. Am J Clin Nutr. 1991;54:1147S–1152S. doi: 10.1093/ajcn/54.6.1147s. [DOI] [PubMed] [Google Scholar]
- Rees DJ, Jones IM, Handford PA, Walter SJ, Esnouf MP, Smith KJ, et al. The role of β-hydroxyaspartate and adjacent carboxylate residues in the first EGF domain of human factor IX. EMBO J. 1988;7:2053–2061. doi: 10.1002/j.1460-2075.1988.tb03045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers JM. Safety of high-level vitamin C ingestion. Ann N Y Acad Sci. 1987;498:445–454. doi: 10.1111/j.1749-6632.1987.tb23780.x. [DOI] [PubMed] [Google Scholar]
- Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279:42351–42354. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- Salnikow K, Kasprzak KS. Ascorbate depletion: a critical step in nickel carcinogenesis? Environ Health Perspect. 2005;113:577–584. doi: 10.1289/ehp.7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem. 2003;278:1728–1734. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- Savini I, Rossi A, Catani MV, Ceci R, Avigliano L. Redox regulation of vitamin C transporter SVCT2 in C2C12 myotubes. Biochem Biophys Res Commun. 2007a;361:385–390. doi: 10.1016/j.bbrc.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Savini I, Catani MV, Arnone R, Rossi A, Frega G, Del Principe D, et al. Translational control of the ascorbic acid transporter SVCT2 in human platelets. Free Radic Biol Med. 2007b;42:608–616. doi: 10.1016/j.freeradbiomed.2006.11.028. [DOI] [PubMed] [Google Scholar]
- Savini I, Rossi A, Pierro C, Avigliano L, Catani MV. SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids. 2008;34:347–355. doi: 10.1007/s00726-007-0555-7. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocrine Reviews. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Sedgwick B. Repairing DNA-methylation damage. Nat Rev Mol Cell Biol. 2004;5:148–157. doi: 10.1038/nrm1312. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Sevanian A, Davies KJ, Hochstein P. Conservation of vitamin C by uric acid in blood. J Free Radic Biol Med. 1985;1:117–124. doi: 10.1016/0748-5514(85)90015-7. [DOI] [PubMed] [Google Scholar]
- Sharma P, Mongan PD. Ascorbate reduces superoxide production and improves mitochondrial respiratory chain function in human fibroblasts with electron transport chain deficiencies. Mitochondrion. 2001;1:191–198. doi: 10.1016/s1567-7249(01)00016-2. [DOI] [PubMed] [Google Scholar]
- Siciliano G, Volpi L, Piazza S, Ricci G, Mancuso M, Murri L. Functional diagnostics in mitochondrial diseases. Biosci Rep. 2007;27:53–67. doi: 10.1007/s10540-007-9037-0. [DOI] [PubMed] [Google Scholar]
- Sotiriou S, Gispert S, Cheng J, Wang Y, Chen A, Hoogstraten-Miller S, et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- Stewart GW, Hepworth-Jones BE, Keen JN, Dash BC, Argent AC, Casimir CM. Isolation of cDNA coding for an ubiquitous membrane protein deficient in high Na+, low K+ stomatocytic erythrocytes. Blood. 1992;79:1593–1601. [PubMed] [Google Scholar]
- Stocker R, McDonagh AF, Glazer AN, Ames BN. Antioxidant activities of bile pigments: biliverdin and bilirubin. Methods Enzymol. 1990;186:301–309. doi: 10.1016/0076-6879(90)86123-d. [DOI] [PubMed] [Google Scholar]
- Szaleczky E, Prechl J, Ruzicska É, Fehér J, Braun L, Bánhegyi G, et al. Reduction of glycated hemoglobin levels by long term, high dose ascorbic acid supplementation in healthy and diabetic patients. Med Sci Monit. 1998;4:241–244. [Google Scholar]
- Szarka A, Stadler K, Jenei V, Margittai E, Csala M, Jakus J, et al. Ascorbyl free radical and dehydroascorbate formation in rat liver endoplasmic reticulum. J Bioenerg Biomembr. 2002;34:317–323. doi: 10.1023/a:1020212720330. [DOI] [PubMed] [Google Scholar]
- Szarka A, Horemans N, Bánhegyi G, Asard H. Facilitated glucose and dehydroascorbate transport in plant mitochondria. Arch Biochem Biophys. 2004;428:73–80. doi: 10.1016/j.abb.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Szarka A, Horemans N, Kovács Z, Gróf P, Mayer M, Bánhegyi G. Dehydroascorbate reduction is coupled to the respiratory electron transfer chain. Physiol Plant. 2007;129:225–232. [Google Scholar]
- Szent-Györgyi A. The Living State and Cancer. New York: Marcel Dekker; 1978. [Google Scholar]
- Therade-Matharan S, Laemmel E, Duranteau J, Vicaut E. Reoxygenation after hypoxia and glucose depletion causes reactive oxygen species production by mitochondria in HUVEC. Am J Physiol Regul Integr Comp Physiol. 2004;287:1037–1043. doi: 10.1152/ajpregu.00048.2004. [DOI] [PubMed] [Google Scholar]
- Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- Tschank G, Sanders J, Baringhaus KH, Dallacker F, Kivirikko KI, Gunzler V. Structural requirements for the utilization of ascorbate analogues in the prolyl 4-hydroxylase reaction. Biochem J. 1994;300:75–79. doi: 10.1042/bj3000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang Y, et al. A family of mammalian Na+-dependent l-ascorbic acid transporters. Nature (London) 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- Tu BP, Weissman JS. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell. 2002;10:983–994. doi: 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- Turley SD, West CE, Horton BJ. The role of ascorbic acid in the regulation of cholesterol metabolism and in the pathogenesis of artherosclerosis. Atherosclerosis. 1976;24:1–18. doi: 10.1016/0021-9150(76)90060-5. [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:582–593. doi: 10.1038/ncpneph0283. [DOI] [PubMed] [Google Scholar]
- Wang QP, VanDusen WJ, Petroski CJ, Garsky VM, Stern AM, Friedman PA. Bovine liver aspartyl β-hydroxylase. Purification and characterization. J Biol Chem. 1991;266:14004–14010. [PubMed] [Google Scholar]
- Wells WW, Xu DP. Dehydroascorbate reduction. J Bioenerg Biomembr. 1994;26:369–377. doi: 10.1007/BF00762777. [DOI] [PubMed] [Google Scholar]
- Wilson JX. Regulation of vitamin C transport. Annu Rev Nutr. 2005;25:105–125. doi: 10.1146/annurev.nutr.25.050304.092647. [DOI] [PubMed] [Google Scholar]
- Winkler BS. In vitro oxidation of ascorbic acid and its prevention by GSH. Biochim Biophys Acta. 1987;925:258–264. doi: 10.1016/0304-4165(87)90190-5. [DOI] [PubMed] [Google Scholar]
- Wittes RE. Vitamin C and cancer. N Engl J Med. 1985;312:178–179. doi: 10.1056/NEJM198501173120310. [DOI] [PubMed] [Google Scholar]
- Wong K, Thomson C, Bailey RR, McDiarmid S, Gardner J. Acute oxalate nephropathy after a massive intravenous dose of vitamin C. Aust N Z J Med. 1994;24:410–411. doi: 10.1111/j.1445-5994.1994.tb01477.x. [DOI] [PubMed] [Google Scholar]
- Xu DP, Wells WW. Lipoic acid dependent regeneration of ascorbic acid from dehydroascorbic acid in rat liver mitochondria. J Bioenerg Biomembr. 1996;28:77–85. [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CG, Yi C, Duguid EM, Sullivan CT, Jian X, Rice PA, et al. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangar RC, Davydov DR, Verma S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol. 2004;199:316–331. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- von Zastrow M, Tritton TR, Castle JD. Identification of l-ascorbic acid in secretion granules of the rat parotid gland. J Biol Chem. 1984;259:11746–11750. [PubMed] [Google Scholar]
- von Zastrow M, Tritton TR, Castle JD. Exocrine secretion granules contain peptide amidation activity. Proc Natl Acad Sci USA. 1986;83:3297–3301. doi: 10.1073/pnas.83.10.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Abbud W, Prohaska R, Ismail-Beigi F. Overexpression of stomatin depresses GLUT-1 glucose transporter activity. Am J Physiol Cell Physiol. 2001;280:C1277–C1283. doi: 10.1152/ajpcell.2001.280.5.C1277. [DOI] [PubMed] [Google Scholar]