

Abstract
The extracellular matrix (ECM) plays a significant role in the structure and function of the lung. The ECM is a three-dimensional fibre mesh, comprised of various interconnected and intercalated macromolecules, among which are the glycosaminoglycans (GAG). GAG are long, linear and highly charged, heterogeneous polysaccharides that are composed of a variable number of repeating disaccharide units (macromolecular sugars) and most of them, as their name implies, have a sweet taste. In the lung, GAG support the structure of the interstitium, the subepithelial tissue and the bronchial walls, and are secreted in the airway secretions. Besides maintaining lung tissue structure, GAG also play an important role in lung function as they regulate hydration and water homeostasis, modulate the inflammatory response and influence lung tissue repair and remodelling. However, depending on their size and/or degree of sulphation, and their immobilization or solubilization in the ECM, specific GAG in the lung either live up to their sweet taste/name, supporting normal lung physiology, or they are associated to ‘bitter’ effects, related to lung pathology. The present review discusses the biological role of GAG in the lung as well as the involvement of these molecules in various respiratory diseases. Given the great structural diversity of GAG, understanding the changes in GAG expression that occur in lung diseases may lead to novel targets for pharmacological intervention in order to prevent and/or to treat a range of lung diseases.
Keywords: glycosaminoglycans, pulmonary fibrosis, pulmonary hypertension, asthma, COPD, hyaluronic acid, hyaluronic acid synthases, hyaluronidases, lung diseases, hyaluronic acid receptors
Introduction
The structure of the extracellular matrix (ECM) in the lung plays several important roles in lung function: (i) it offers a low resistant pathway, allowing effective exchange of gases; (ii) it provides tensile and compressive strength and elasticity, with a strong and expandable framework that supports the fragile alveolar-capillary intersection; (iii) it controls the behaviour of cells by binding of growth factors and by interaction with cell surface receptors; and (iv) it acts as a buffer against retention of water (Negrini et al., 2000; Suki et al., 2005). The alveolar wall is composed of an epithelial cell layer and its basement membrane, the capillary basement membrane and endothelial cells, and the ECM, which is a thin layer of interstitial space lying between the capillary endothelium and the alveolar epithelium (West and Mathieu-Costello, 1999). In some areas, the two basement membranes are physically fused to reduce the diffusion distance. In the segments where the two basement membranes are not fused, the interstitium is composed of cells, a macromolecular fibrous component and the fluid phase of the ECM. In these parts, the ECM functions as a three-dimensional mechanical scaffold characterized by a fibrous mesh consisting mainly of collagen type I and III, providing tensile strength, and elastin (Suki et al., 2005). The three-dimensional fibre mesh is filled mainly with glycosaminoglycans (GAG) (macromolecular sugars), which are the major components of the nonfibrillar compartment of the interstitium (Negrini et al., 2000). The majority of GAG, as their name implies, have a sweet taste (glycos in Greek means sweet).
Although many studies have described the role of proteins, proteoglycans, growth factors and other type of molecules in a wide range of pulmonary diseases (Turino and Cantor, 2003; Bai et al., 2005), the actions of GAG in lung pathophysiology are much less well understood. GAG have overall attracted less attention by researchers, due to the fact that they are abundant in almost all tissues, they have very high molecular mass, immense size versatility and they are not directly encoded by a single gene. However, the improvement of techniques for the isolation, purification and characterization of GAG, the discovery of specific GAG synthases and GAG-degrading enzymes and, most importantly, the recent key technologies regarding carbohydrate sequencing and the synthesis of defined oligosaccharides have contributed to progress in glycomics research. Synthetic tools and high-throughput experiments, such as carbohydrate arrays, are beginning to affect biological research. These techniques are now being applied to the development of carbohydrate-based diagnostics, vaccines and therapeutics (Peter et al., 2007). With respect to lung structure and function, there is an accumulation of evidence for the involvement of GAG in lung physiology and pathology. The comprehensive study of GAG will improve our knowledge on the development of lung diseases, may allow early diagnosis of ECM alterations and lung remodelling processes, and may promote development of novel targets for pharmacological interventions.
Glycosaminoglycans
GAG represent major components of the ECM that undergo significant alterations in content, synthesis and distribution during neonatal organ growth (Schmid et al., 1982), acute injury (Cantor et al., 1980), degenerative diseases or age-related tissue modifications (Kumar et al., 2005; Vitanzo and Sennett, 2006; Blewis et al., 2007; Ito et al., 2007; Wang et al., 2007).
With respect to their structure, GAG are long, linear and heterogeneous polysaccharides, which consist of repeating disaccharide units, with sequences that vary in the basic composition of the disaccharide unit, type of linkage and degree of acetylation and sulphation. The disaccharide units are galactose, N-acetylglucosamine, N-acetylgalactosamine, glucuronic acid or iduronic acid (Figure 1). The chain length of GAG can range from several hundred to several thousand disaccharide units, and thus their molecular mass may vary over three orders of magnitude, implying that the polymer chains have great variability in size and structure (Scott, 1992). There are two main types of GAG: the non-sulphated GAG hyaluronic acid (HA) and the sulphated GAG (heparan sulphate and heparin, chondroitin sulphate, dermatan sulphate and keratan sulphate). With the exception of HA, GAG are usually covalently attached to a protein core (Figure 1), forming overall structures referred to as proteoglycans (Johnson et al., 2005).
Figure 1.
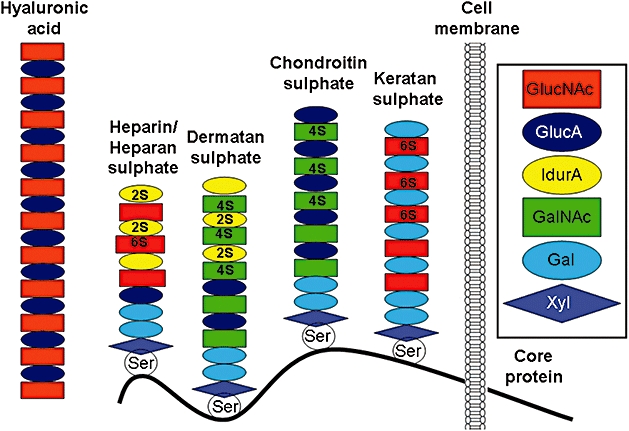
Schematic structure of glycosaminoglycans and proteoglycans. In the extracellular matrix, heparin, heparan sulphate, dermatan sulphate, chondroitin sulphate and keratan sulphate are connected to a transmembrane protein core via a serine residue. Hyaluronic acid is not linked to a protein core. GlucNAc, N-acetylglucosamine; GlucA, D-glucuronic acid; IdurA, L-iduronic acid; GalNAc, N-acetylgalactosamine; Gal, galactose; Xyl, xylose; Ser, serine; 2S, 4S, 6S, position of sulphate residue on the sugar moiety.
GAG are either an integral part of the ECM or they are located directly on the cellular membrane, where they can function as protein receptors or activators. The turnover of these local GAG and their composition is essential for organ function and homeostasis. In order to understand the biological function of GAG, one must be aware that GAG molecules are not encoded by a single gene, as already mentioned, but are synthesized by specific synthases, which are regulated by tissue or cell type local microenvironments. Moreover, GAG are not synthesized by the action of a single synthase, but result from the as yet poorly understood interactions of several independently regulated synthases (Funderburgh, 2002; Itano and Kimata, 2002; Kusche-Gullberg and Kjellen, 2003). Furthermore, there is evidence that during the assembly of GAG the addition of sulphate residues to the GAG chain determines their function. For example, it has been established that the anticoagulant activity of heparin depends on specific binding to antithrombin and that the antithrombin binding site on heparin is a pentasaccharide with specific modifications by sulphation (Sugahara, 1992).
Biological function of GAG in the lung
Hyaluronic acid
HA is the most abundant non-sulphated GAG in the lung ECM. It differs from the other GAG because it is spun out from the cell membrane, rather than being secreted through the Golgi apparatus. HA is a linear polysaccharide composed of up to 10 000 disaccharides, constituted by a uronic acid residue covalently linked to (1-b-3) N-acetyl-D-glucosamine, with a flexible and coiled configuration. Thus, HA has a high molecular mass (up to 107 Da), which is considerably larger than other GAG. In solution, the molecular chain behaves as an extended random coil, with a diameter of approximately 500 nm (Laurent, 1970). At concentrations as low as 0.1%, the molecular chains were entangled, resulting in an extremely high, shear-dependent viscosity. These properties enable HA to regulate water balance, osmotic pressure and flow resistance, to interact with proteins, and also to act as a sieve, lubricant, and to stabilize structures by virtue of its electrostatic interactions (Laurent, 1970). It is a ubiquitous molecule of the connective tissue matrix and is necessary for its assembly and stabilization (Gerdin and Hallgren, 1997). A unique characteristic of HA that relates to its various functions is its high anion charge, which attracts many water molecules offering tissue hydration (Turino and Cantor, 2003). Therefore, accumulation of HA in the interstitial tissue may immobilize water and regulate the water content in the interstitium (Gerdin and Hallgren, 1997). HA is also involved in several lung functions, such as tissue repair, which is achieved via regulation of HA synthase 2 and hyaluronidase 2 in response to mediators released during the healing process after lung injury, thus leading to the fibroproliferative response (Li et al., 2000b). Furthermore, HA enhances cellular host defence mechanism by stimulating the activity of blood neutrophils, such as phagocytosis and free oxygen radical formation and migration (Turino and Cantor, 2003). In this respect, it has been shown that subcutaneous administration of HA reduced the number of bacterial infections in patients with an increased susceptibility to such infections. Patients with chronic bronchitis and recurrent acute exacerbation of their disease, who were treated with HA, had significantly fewer acute exacerbations than did placebo-treated patients (Venge et al., 1996). In addition, HA seems to protect against proteolytic enzymes as it has been reported that aerosol preparation of streptococcal HA prevents experimental emphysema induced by intratracheal administration of porcine pancreatic elastase (Cantor et al., 2000). In the pulmonary alveolus, HA interacts with surfactant phospholipids, contributing to the stability of the surfactant layer (Lu et al., 2005).
In the lungs, the content of HA is 15–150 mg·g−1 dry weight (species specificity), which is mainly localized in the peri-bronchial and interalveolar/perialveolar tissue (Petrigni and Allegra, 2006). The quantity of HA in human lung secretions was found to be approximately 66 ng·mL−1 with values ranging from 34 to 423 ng·mL−1 (Dentener et al., 2005).
Polymerization of HA occurs by the action of one or more of the three HA synthases (termed HAS 1, HAS 2 and HAS 3) (Itano et al., 1999), through the joining of the glycosidic residues to the reducing chain extremity, and is extruded while still elongating into the ECM (Stern, 2003). HAS isoforms have distinct catalytic rates, resulting to HA products of different molecular mass (Itano and Kimata, 2002). HAS 1 and HAS 2 synthesize relatively long stretches (2–4 × 106 Da). HAS 1 is the least active, while HAS 2 is more catalytically active, and therefore might be the main enzyme responsible for increased synthesis found in stress-induced conditions and wound healing (Stern, 2005). HAS 3 is the most active isoform, it polymerizes short stretches of disaccharide chains (0.4–2.5 × 105 Da) (Itano et al., 1999; Weigel et al., 1997) and may be involved in activation of signal transduction (Stern, 2005). HA is metabolized by hyaluronidases (HYAL), mainly by HYAL 1 and HYAL 2, present in various tissues, including the lung (Dentener et al., 2005).
Two cell-associated receptors, CD44 and RHAMM (receptor for HA-mediated motility), mediate the biological effects of HA (Slevin et al., 2007). CD44 and RHAMM are abundant in different cell types. For example, CD44 is expressed in fibroblasts, epithelial and smooth muscle cells and immune cells, such as neutrophils, macrophages and lymphocytes (Sherman et al., 1994), while RHAMM is expressed in cells following injury (Savani et al., 1995), in tumours and in some mutant active Ras-transformed cell lines (Li et al., 2000a; Ahrens et al., 2001). CD44 is a type I integral cell surface membrane glycoprotein that participates in cell–cell and cell–matrix adhesion (Naor et al., 2002). HA binds to the extracellular domain of CD44 and subsequently the cytoplasmic tail of CD44 activates intracellular signalling pathways that regulate the interactions of signalling complexes with actin cytoskeleton (Toole, 2004). The interaction of HA with CD44 regulates leucocyte rolling and activation, as well as tumour metastasis. Furthermore, CD44-dependent clearance of HA fragments is crucial in resolving lung inflammation in the bleomycin model of lung injury (Teder et al., 2002), demonstrating an essential role for CD44 in the resolution phase of inflammation.
HA also binds to RHAMM, which controls the effect of HA on cell migration, proliferation and motility, apparently via RHAMM interaction with the cytoskeleton (Savani et al., 1995; Turley et al., 2002). Cell surface RHAMM occurs in cell lamellae and podosomes (Turley, 1980) and exogenous HA has been reported to promote spreading of cell lamellae and turnover of focal adhesions in fibroblasts transfected with oncogenic Ras that express cell surface RHAMM (Hall et al., 1994). Agonist RHAMM antibodies mimic, whereas blocking RHAMM antibodies inhibit, this effect (Bourguignon et al., 2001). The rapid formation and then disassembly of focal adhesions precede the HA-induced increase in cell motility (Hall et al., 1994). It has been reported that these effects are possibly mediated through the integrin fibronectin receptors, α4β1 or α5β1 (Gares and Pilarski, 2000). Evidence of increased focal adhesion turnover following addition of HA to migrating cells highlights the dynamic role of the pericellular matrix (Hall et al., 1994). Rapid uptake of HA and translocation to the nucleus, with signalling mediated through RHAMM, was associated with increased motility. With respect to the effects of HA on proliferation via HA-RHAMM interaction, it has been reported that intracellular RHAMM associates with the actin cytoskeleton and the mitotic spindle microtubules (Assmann et al., 1999). HA is also internalized in premitotic cells and co-distributes around the mitotic spindle with microtubule-associated RHAMM (Evanko et al., 2004). Microtubules are compression bearing elements of the cytoskeleton that target focal adhesions for dissolution (Kaverina et al., 1999). This raises the possibility that HA internalized from the pericellular matrix may also regulate or facilitate events involving microtubules during cell shape changes. The ability of RHAMM to form coiled coils as well as its partial homology with proteins such as D-CLIP (Akhmanova et al., 2001) that link microtubules and actin filaments suggest that intracellular RHAMM proteins may connect actin and microtubule cytoskeleton (Assmann et al., 2001). As RHAMM has been associated with several kinases, it is likely that intracellular RHAMM proteins act as adapter molecules (Bustelo, 2000) that connect the cytoskeleton to signalling complexes. Transcriptional profiling of the cell cycle progression reveals enhanced expression of both RHAMM and HAS 2 at the G2M boundary (Cho et al., 2001). These results attribute a possible role of intracellular HA/RHAMM interactions in proliferation.
Several studies have demonstrated that the biological effects of HA appear to vary depending on its average molecular mass (Uchiyama et al., 1990; McKee et al., 1996; Ohkawara et al., 2000; Horton et al., 2002; Tammi et al., 2002; Turino and Cantor, 2003; Mascarenhas et al., 2004; Bai et al., 2005) (Figure 2). In physiological conditions, HA has a high average molecular mass, in excess of 106 Da. However, following tissue injury, degradation of HA may occur intracellularly (Rodén et al., 1989) or extracellularly (Lin et al., 1994) leading to the formation of HA fragments of lower molecular mass, of about 0.25 × 106 Da, which can induce the expression of inflammatory genes (Tammi et al., 2002). Low-molecular-weight fragments can stimulate activated macrophages to express RNAs of numerous chemokines and cytokines (McKee et al., 1996; Horton et al., 2002), whereas fragments of higher molecular weight have an opposite effect and suppressed chemokine expression (Turino and Cantor, 2003). It has been shown that HA of low molecular mass stimulated the maturation of dentritic cells and the synthesis of proinflammatory interleukin (IL)-1β, IL-12, and tumour necrosis factor (TNF)-α (Taylor et al., 2004). The latter effect seems to be restricted to an interaction of HA fragments with the toll-like receptor 4 (Termeer et al., 2002; Taylor et al., 2004). HA of low molecular mass has also been shown to stimulate macrophages to produce important mediators of tissue injury and repair, such as inducible nitric oxide synthase, plasminogen activator inhibitor-1, macrophage inflammatory protein-1α and -1β, TNF-α and macrophage metalloelastase (McKee et al., 1996; 1997; Horton et al., 1998a; 1998b; 1999; 2000;). HA fragments can synergize with interferon-γ to induce the antifibrotic chemokines monokine induced by interferon-γ and interferon-inducible protein-10 (Horton et al., 1998b). The transcriptional mechanism involved in mediating the synergy between HA and interferon-γ on monokine induced by interferon-γ gene expression was identified to be activation of nuclear factor (NF)-kappaB (Horton et al., 2002). There are no available data regarding the molecular mechanisms via which HA of high molecular mass suppress chemokine expression.
Figure 2.
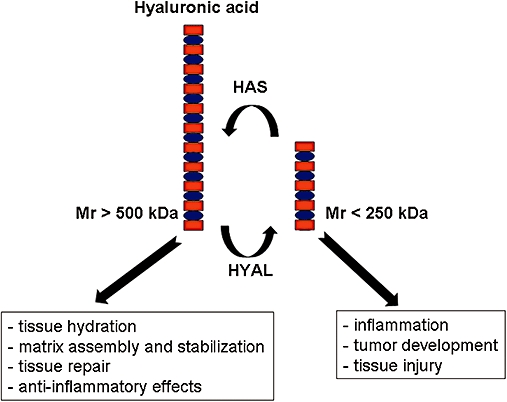
The biological functions of hyaluronic acid of low and high molecular mass in human cells and tissues. HAS, hyaluronic acid synthases; HYAL, hyaluronidases.
Furthermore, it has also been shown that fragmented HA, which induces inflammatory gene expression in vitro, is in the same size range as HA that accumulates under inflammatory conditions in vivo (McKee et al., 1996). It seems that under conditions of inflammation, tumour development or tissue injury, HYAL activity or oxidation generate HA fragments that subsequently can initiate gene transcription, influencing cell proliferation and migration (Uchiyama et al., 1990).
Heparan sulphate
Heparan sulphate is the most abundant sulphated GAG within the lung parenchyma with a size ranging from 0.005 to 0.07 × 106 Da. It is expressed on virtually every cell in the body and comprises 50–90% of the total endothelial proteoglycans (Handel et al., 2005). Heparan sulphate has the most variable structure, mainly due to variations in the sulphation patterns of its chains. It is initially produced in a cell surface-bound form, but it can also be secreted as a soluble GAG.
It has been shown that heparan sulphate interacts with various proteins affecting the topography, half-life and bioactivity of the protein. Furthermore, heparan sulphate is involved in a variety of biological processes, including cell–matrix interactions and activation of chemokines, enzymes and growth factors (Nader et al., 1987; Whitelock and Iozzo, 2005). In this context, the interaction between fibroblast growth factors (FGFs) and their tyrosine kinase receptors depends on the sequence of the heparan sulphate chain (Handel et al., 2005). Heparan sulphate plays a critical role in FGF signalling by facilitating and/or stabilizing the formation of FGF–FGF receptor complexes and enhancing FGF oligomerization (Raman et al., 2005). In addition, heparan sulphate binds FGF in the ECM, storing it in an inactive form until needed, thereby allowing rapid response to stimuli (Aviezer et al., 1999).
Heparan sulphate interacts with cytokines such as IL-5, IL-6, IL-8, IL-10, TNF-α and platelet factor-4 (Lipscombe et al., 1998; Mummery and Rider, 2000; Menart et al., 2002). Binding of heparan sulphate to cytokines is due to noncovalent and reversible interactions; however, the precise mechanism of these interactions at the molecular level is not known. This binding is likely to retain cytokines, which are otherwise small, readily diffusible proteins, close to their sites of release in the tissues, thus favouring paracrine rather than endocrine activity (Mummery and Rider, 2000). Because many cytokines are highly pleiotropic, interaction with heparan sulphate in the vicinity of their secretion represents a mechanism of regulation, as the cytokine would be mostly available only to neighbouring target cells. However, some release of the cytokine into the circulatory system could still occur, either because the binding interactions are noncovalent and reversible, or because of the fragmentation of the proteoglycan macromolecule involved by the action of proteases or heparinases. In the latter case, the cytokine could remain bound to heparan sulphate-peptide fragments or heparan sulphate oligosaccharides. Beyond such a general role in compartmentalizing cytokines within the tissues, the binding of cytokines to heparan sulphate has been shown to have particular roles for individual cytokines (Mummery and Rider, 2000). Thus, the interaction of heparan sulphate with IL-8 promotes the activity of the cytokine, whereas in the case of platelet factor-4, the interaction inhibits the activity. The binding of FGF-2 to heparan sulphate has been shown to have an essential role in the signalling pathway of the growth factor, because it is a prerequisite for subsequent engagement to the high-affinity cell surface FGF-2 receptor (Rapraeger et al., 1991; Yayon et al., 1991). These examples illustrate that the binding of a cytokine to heparan sulphate may have major consequences on cytokine activity, and that these may be quite different from one cytokine to another.
Heparan sulphate has also been shown to interact with various ECM proteins, including fibronectin, laminin, thrombospondin, collagen types I, II, IV, V, VI, XIII and XVIII, and endostatin. The binding of endostatin by heparan sulphate is important for its antiangiogenic function (Kreuger et al., 2002). The interaction between heparan sulphate and laminin could be important in determining the integrity of basement membranes (Parthasarathy, 1998). The interaction between heparan sulphate and collagen type V plays an important role in the modulation of cell adhesion to the substratum (LeBaron et al., 1989).
Heparin
Heparin is an over-sulphated variant of heparan sulphate, commonly used as an anticoagulant drug (Whitelock and Iozzo, 2005). Heparin has a molecular mass ranging from 0.006–0.05 × 106 Da and shares many structural and functional activities with heparan sulphate. The lung is a rich source of heparin as the lung is abundant in mast cells, which are responsible for the bulk synthesis of heparin (Poole, 1986). Mast cell heparin resides in secretory granules, where most of the GAG chains are linked to a core protein (serglycin), forming macromolecular proteoglycans that are much larger than commercial heparin.
Endothelial cells may also produce a highly sulphated heparin that is capable of binding antithrombin III directly on the vascular wall, preventing adherence of platelets and clotting factors to the vessel wall (Merry et al., 1999). In respect to vessel integrity and tissue remodelling, as well as tumour development, the interaction of heparins with specific isoforms of the vascular endothelium-derived growth factor (VEGF) and its receptors is of interest. It has been shown that heparin binds and activates VEGF-A145 and -A165, but not -A121; it binds to VEGF-B167, but not to -B186; it binds to VEGF-E, -F1 and F2, but not to VEGF-C or -D (Soker et al., 1994). The capacity of heparin to bind specific VEGF isoforms depends on the number of sulphate groups, and the localization of VEGF in the tissue is assumed to be directed by heparins (Springer et al., 2007). The effect of heparins on the function of VEGF is highly controversial. In some studies, heparins enhanced the VEGF function, which is of specific interest as heparin is used in patients with neoplasia (Pyda et al., 2006), while in other conditions heparins counteracted VEGF actions (Moy et al., 2006; Wijelath et al., 2006).
Many studies have shown that heparin plays a beneficial role in the regulation of the inflammatory response (Page, 1997). In this context it has been shown that heparin can inhibit the influx of neutrophils into certain tissues and inhibit T cell trafficking, partly by an inhibitory effect on the heparinase enzyme secreted by T cells (Tyrell et al., 1995). Furthermore, heparin has been shown to be released from human lung mast cells in response to allergen exposure, and increased levels of a heparin-like substance have been reported in the plasma of asthmatic individuals (Green et al., 1993). Heparin can also inhibit allergen-induced eosinophil infiltration into the airways of experimental animals (Ahmed et al., 1993). After the inflammatory cells have passed through the lung tissue, it is recognized that there is a number of stages involved, including adhesion to the vascular endothelium, diapedesis across the endothelial cells and chemotaxis within tissues. It is clear that heparin can inhibit all stages of cell migration, including the carbohydrate–selectin interactions between endothelial cells and leucocytes, the presentation of specific chemoattractants to activated leucocytes and leucocyte trafficking. Endothelial heparan sulphate has also been reported to act as a ligand for L-selectin during neutrophil rolling (Wang et al., 2005). Although the above mechanisms underlie the effect of heparin on neutrophil migration, the ability of heparin to interfere with eosinophil adherence is less well understood. Nonetheless, heparin is able to inhibit the actions of several important eosinophil chemoattractants, such as platelet factor-4 (Tyrell et al., 1995).
Although the precise mechanism of the anti-inflammatory effects of heparin has not been clarified, it has been suggested that inhibition of the interaction between pro-inflammatory cytokines and membrane-associated GAG may provide a mechanism for inducing clinically useful immunosuppression. Whereas immobilized heparin is essential for the biological activity of chemokines, soluble heparin has been shown to inhibit the biological effects of chemokines (Kuschert et al., 1999). This may be explained as follows: chemokines can form complexes with either cell surface or soluble GAG and these interactions lead to different functions. Immune modulators, such as chemokines and growth factors, exert their biological activity through high-affinity interactions with cell surface receptors, thereby activating specific signalling pathways. Many of these molecules also participate in low-affinity interactions with another class of molecules, referred to as proteoglycans, which consist of a protein core to which GAG chains are attached. Certain chemokines require interactions with GAG for their in vivo function (Handel et al., 2005). The GAG interaction is thought to provide a mechanism for retaining chemokines on cell surfaces, facilitating the formation of chemokine gradients. These gradients serve as directional cues to guide the migration of the appropriate cells in the context of their inflammatory, developmental and homeostatic functions. In contrast, soluble GAG chemokines complexes are unable to bind the receptor, resulting in a block of the biological activity. It is therefore likely that the anti-inflammatory effects of heparin are mediated, at least in part, by interference with the chemokine system (Handel et al., 2005).
Chondroitin sulphate
Chondroitin sulphate is a key regulator for ECM protein activation and degradation. The function of chondroitin sulphate depends on the location of its sulphate groups, which determines its structure and binding characteristics. It has been shown that chondroitin sulphate activates ECM-degrading enzymes, such as matrix metalloproteases (MMP). MMP constitute a heterogeneous family of 28 proteolytic enzymes (Visse and Nagase, 2003; Tu et al., 2008). Their common features include the presence of a Zn atom in their active site and their involvement, after activation, in basement membrane remodelling, wound contraction, neovascularization, tissue repair and generally ECM homeostasis (Visse and Nagase, 2003; Tu et al., 2008). MMP are also involved in lung disorders (Greenlee et al., 2007) and hypoxia-associated lung diseases (Karakiulakis et al., 2007). The MMP gene family encodes for several proenzymes (proMMP), with common and distinctive structural and functional properties, classified as: (i) collagenases 1–4 (MMP-1, -8, -13 and -18 respectively); (ii) gelatinases A and B (MMP-2 and -9 respectively); (iii) stromelysins 1–3 (MMP-3, -10 and -11 respectively); (iv) matrilysins 1 and 2 (MMP-7 and -26 respectively); (v) membrane types 1–6 (MT-MMP: MMP-14, -15, -16, -17, -24 and -25 respectively); and (vi) various others (MMP-12, -19, -20, -21, -23, -27 and -28) (Visse and Nagase, 2003). ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) -4 and -7B have also been implicated in a number of connective tissue pathologies (Jones, 2006). The activity of MMP is tightly regulated during proMMP expression (Stetler-Stevenson et al., 1993) and by proteolytic activation of secreted latent MMP (Cao et al., 1995). Most MMP are not constitutively expressed in vivo but can be rapidly induced in response to signals, such as growth factors, cytokines, phorbol esters and cell–cell or cell–ECM interactions (Tu et al., 2008). MMP, both in latent and activated form, are also regulated by the formation of stoichiometric 1:1 complexes with specific inhibitors, known as tissue inhibitors of MMP (Stetler-Stevenson et al., 1993). Chondroitin sulphate has been reported to be involved in the degradation of aggrecan, which is a chondroitin sulphate-rich proteoglycan, by MMP-2 (Rodriguez et al., 2006; Iida et al., 2007), MMP-13 (Miwa et al., 2006), ADAMTS4 (Miwa et al., 2006) and ADAMTS7B (Somerville et al., 2004). MMP-2 is activated by direct binding of chondroitin sulphate to the C-domain of proMMP-2, which is essential to present the inactive enzyme to its activator, the MT-MMP-3 (Iida et al., 2007).
Chondroitin sulphate has also been reported to have anti-inflammatory properties in osteoarthritis, preventing joint space narrowing and reducing joint swelling and effusion. These effects are produced by reduction of NF-kappaB nuclear translocation, probably by diminishing extracellular signal-regulated kinase, p38 mitogen-activated protein kinase and c-Jun N-terminal kinase activation (Iovu et al., 2008).
Dermatan sulphate
Dermatan sulphate binds to a variety of proteoglycans, affects the function of growth factors, modifies the action of the heparin cofactors (Sugahara et al., 2003; Kinsella et al., 2004; Villena and Brandan, 2004) and influences proliferation in a cell type-specific manner. Dermatan sulphate also occurs as a circulating GAG in the blood and binds to several proteins that are part of the coagulation cascade, including activated protein C, heparin cofactor II, fibrinogen, fibronectin, apolipoprotein B, kininogen, trypsin inhibitor A and factor H. All these proteins are sensitive to serine protease. Dermatan sulphate can activate serine protease with similar capacity to heparan sulphate, but the mechanism of action of dermatan sulphate activation of these proteases is unclear (Saito and Munakata, 2007). Binding of dermatan sulphate to activated protein C and heparin cofactor II accounts for its antithrombotic activity (Du et al., 2007).
Furthermore, dermatan sulphate functions as a docking molecule for a range of human pathogenic microorganisms. It has been shown that dermatan sulphate mediates the adhesion of Penicillium marneffei to the host's ECM (Penc et al., 1999), of pneumococci to nasopharyngeal epithelial cells (Srinoulprasert et al., 2006) and of spirochetes to fibronectin (Tonnaer et al., 2006). P. marneffei, an endemic fungus in Southeast Asia and southern China, is the cause of opportunistic infection in HIV-infected patients who may present with symptoms and signs of the lungs, and abnormal chest radiographs. P. marneffei pneumonia does not have specific clinical manifestations, but should be included during differential diagnosis, especially in immunocompromized patients (Deesomchok and Tanprawate, 2006). Streptococcus pneumoniae has been recognized as a major cause of pneumonia (Neralla and Meyer, 2004). The pathogenesis of pneumococcal infection is a complex interplay between pneumococcal virulence determinants and the host immune response (Gillespie and Balakrishnan, 2000). Pulmonary hemorrhage represents the main cause of death in severe forms of leptospiral pneumonias due to infection with spirochetes. The pathogenesis of the disease involves novel outer membrane proteins and dysregulation of sodium transporters of alveolar epithelial cells, and innate immunity in the development of severe disease (Dolhnikoff et al., 2007).
Keratan sulphate
Keratan sulphate is among the most abundant GAG in airway secretions (Monzon et al., 2006). It is also highly expressed in the cornea, bones, brain and in the ECM. Keratan sulphate is composed by galactose and N-acetylglucosamine linked together by the action of glycosyl transferases in an alternate pattern (Christner et al., 1979). Sulphation of keratan sulphate, which occurs while a chain is elongated, regulates the organ or cell type-specific function of this GAG (Krusius et al., 1986; Nieduszynski et al., 1990; Degroote et al., 1997; Uchimura et al., 1998). In the ECM, keratan sulphate is linked to the large proteoglycan aggrecan and its length and sulphation increases with age (Praus and Brettschneider, 1975; Brown et al., 1998). Despite the abundance of keratan sulphate in lung secretions, there are no available data regarding its functional role in the lung.
Role of GAG in lung diseases
Altered ECM turnover is a hallmark of several pulmonary diseases, including idiopathic pulmonary arterial hypertension (IPAH), pulmonary fibrosis, asthma or chronic obstructive pulmonary disease (COPD), which underlines the importance of ECM homeostasis for proper lung function (Rabinovitch, 2001; Noble and Jiang, 2006; Laurent et al., 2007). In the lung, the ECM is subjected to a daily turnover of about 10% of total ECM, indicating that subtle changes in turnover rates accumulate to large changes in total ECM composition with time (McAnulty and Laurent, 1987). As GAG are main constituents of the lung ECM, alterations in their synthesis and degradation may reflect to lung pathology.
Idiopathic pulmonary arterial hypertension
IPAH is a rare but fatal disease characterized by elevated blood pressure in the pulmonary circulation, due to increased vascular resistance of pulmonary arterioles (Gaine and Rubin, 1998; Humbert et al., 2004a; Rabinovitch, 2008). If untreated, IPAH leads to right ventricular hypertrophy, failure and subsequent death. Despite major recent advances in the pathophysiology, diagnosis and therapy of IPAH, the molecular mechanisms underlying endothelial and smooth muscle cell dysfunction in this disease remain to be fully elucidated (Humbert et al., 2004b). Early in disease pathogenesis, endothelial cell dysfunction triggers increased vasoconstriction and in situ thrombosis (Rabinovitch, 2008). This is followed by pathologic vascular remodelling, a process characterized by intimal fibrosis and thickening of the medial and adventitial layers, due to uncontrolled proliferation of pulmonary arterial smooth muscle cells (PASMC) and perivascular fibroblasts (Olschewski et al., 2001; Humbert et al., 2004b; Eickelberg and Morty, 2007; Rabinovitch, 2008). In parallel, enhanced cellular activation of PASMC and fibroblasts leads to excessive ECM deposition, which potentiates the increased stiffness of pulmonary arteries in IPAH (Rabinovitch, 2001; Hassoun, 2005).
In addition to the pathological changes discussed above, there is increasing interest on the proinflammatory state of the vessel wall in the progression of IPAH (Rabinovitch, 2008). In this respect, additional features of IPAH include the thickening of the pulmonary adventitia and venous hypertrophy (Chazova et al., 1995), increased expression of transforming growth factor (TGF)-β, matrix proteins (such as collagen, elastin, fibronectin, tenascin-C, and GAG) (Jones et al., 1997), macrophages and T cells (Liptay et al., 1993), as well as inflammatory cytokines, such as S100A4 (also known as metastasin 1) (Greenway et al., 2004) and fractalkine (Balabanian et al., 2002). Furthermore, it has been shown that the loss of function of the receptor of bone morphogenic protein (BMP), BMPRII leads to up-regulation of the proinflammatory cytokine IL-6 (Hagen et al., 2007) and that increased expression of the nuclear transcription factor NFATc2, which is associated with inflammatory cells, is observed in T cells from IPAH patients (Bonnet et al., 2007).
It has recently been shown that GAG play a significant role in inflammatory and non-inflammatory lung diseases, exhibiting spatio-temporally distinct effects on epithelial or mesenchymal cell types (Noble and Jiang, 2006; Laurent et al., 2007). As already mentioned, GAG regulate hydration and water homeostasis, maintain cell and tissue structure and function, modulate inflammatory responses and influence tissue repair and remodelling (Noble and Jiang, 2006; Souza-Fernandes et al., 2006). We have recently investigated GAG expression in lungs of patients with IPAH or control transplant donors, and analysed expression and localization of GAG metabolizing enzymes in vivo and in vitro (Papakonstantinou et al., 2008). We found that IPAH lung tissues exhibit a significantly increased content of HA, the major GAG produced by PASMC. While the relative amount of HA was significantly increased, the content of the sulphated GAG heparan, dermatan or chondroitin sulphate was decreased, indicating an increased ratio of non-sulphated to sulphated GAG. Immunohistochemical staining in tissue sections from lungs of IPAH patients revealed increased HA deposition in areas associated with remodelled pulmonary arteries. The increased HA content of IPAH lung tissues was associated with increased gene expression of HAS 1 and decreased gene expression of HYAL 1. Similar changes were observed in primary human PASMC cultured from the lungs of IPAH patients, which demonstrated a significant decrease in HYAL 1 mRNA levels, compared with PASMC obtained from transplant donor lungs (Papakonstantinou et al., 2008). In addition, stimulation of PASMC with TGF-β1, a growth factor involved in the pathogenesis of IPAH (Humbert et al., 2004b; Eickelberg and Morty, 2007), led to increased HAS 1 gene expression as early as 2 h after stimulation. As increased HA staining was observed in remodeled pulmonary arteries (Papakonstantinou et al., 2008), it is tempting to speculate that HA secretion by PASMC directly influences PASMC proliferation, and may control vasoreactive responses in IPAH. We have previously shown that HA inhibits proliferation of human PASMC (Papakonstantinou et al., 1995; 1998;). This effect of HA is mediated via interaction with specific cell surface receptors CD44 or RHAMM (Toole, 2004; Jiang et al., 2007), but also via interaction with toll-like receptors TLR2 and 4 (Jiang et al., 2005). This is substantiated by the fact that selective overexpression of HAS 2 in smooth muscle cells of large and small vessels of transgenic mice resulted in significantly increased HA content in the tunica media of the aorta, enhanced mechanical stiffness and strength, and accelerated development of atherosclerosis (Chai et al., 2005), and that CD44 expression promoted susceptibility to atherosclerosis, recruitment of macrophages and smooth muscle cell activation in CD44 heterozygote and wild-type mice as compared with CD44-null littermates (Cuff et al., 2001).
It is of interest to note that the biological effects of HA vary depending on its average molecular mass (Turino and Cantor, 2003; Jiang et al., 2007). As mentioned above, under physiological conditions, HA is a polymer of average molecular mass above of 106 Da, exerting its ‘sweet’ beneficiary effects for normal lung function. In contrast, HA fragments of lower molecular mass are associated to ‘bitter’ effects, leading to malfunction of the lung (Figure 2). Available evidence indicates that HA fragments accumulate following tissue injury. HA fragments (0.3–0.5 × 106 Da) have been reported to prolong the survival of eosinophils in vitro (Ohkawara et al., 2000), while HA fragments (<2 × 105 Da) induced the expression of cytokines, chemokines or inducible NO synthase by macrophages (McKee et al., 1996), affected ECM turnover in murine alveolar macrophages (McKee et al., 1996) and stimulated TGF-β1 synthesis (Ohkawara et al., 2000). Furthermore, it has been shown that fragmented HA with an average molecular mass of 0.25 × 106 Da induced the expression of inflammatory genes (Tammi et al., 2002), while HA of higher molecular weight exhibited the opposite effect and suppressed chemokine expression (Turino and Cantor, 2003). In this respect, it would be of interest to elucidate whether HA expressed in the vascular system of control donor lung tissue specimens is of different average molecular weight than in IPAH lung tissue specimens.
Our in vivo observations on IPAH lung tissues were also supported by experiments investigating the effects of BMP and TGF-β superfamily on GAG synthesis and HA secretion in human primary cultures of PASMC. Among the growth factors involved in vascular pathophysiology, BMP and TGF-β isoforms are pleiotropic mediators that, due to abundant expression of their respective receptors BMPRs and TβRs, exhibit multiple effects on all cells within the vascular system (Pepper, 1997; Blobe et al., 2000; Topper, 2000). Recently, the TGF-β and BMP receptor systems have gained significant attention, as mutations in the gene encoding BMPRII (BMPR2) have been linked to familial and sporadic primary pulmonary hypertension (Deng et al., 2000; Thomson et al., 2000; Machado et al., 2001; Newman et al., 2001). In addition to BMPR2, mutations in the gene encoding ALK1, a TβRI isoform, have been associated with the appearance of pulmonary hypertension in hereditary hemorrhagic telangiectasia types I and II respectively (Osler-Rendu-Weber syndrome) (McAllister et al., 1994; Johnson et al., 1996; McDonald et al., 2000). We found that TGF-β1, but not BMP-2, significantly stimulated total GAG synthesis and HA secretion, an effect that coincided with an increase in HAS 1 gene expression. The TGF-β1-dependent increase in GAG synthesis was mediated via the Smad and p38 MAP kinase pathways (Papakonstantinou et al., 2008). As TGF-β1 is a potent stimulus for PASMC migration (Eickelberg and Morty, 2007), HAS 1-mediated HA synthesis may represent a necessary step for PASMC activation and migration. Dysregulation of HAS/HYAL expression and/or activity may lead to the generation of HA of different molecular masses with distinct biological effects that potentially contribute to the pathogenesis of IPAH. The aforementioned changes in HAS/HYAL expression, along with the change in HA synthesis and content in IPAH, may also result from tissue hypoxia observed in IPAH. We have previously reported that hypoxia potentiates GAG synthesis induced by TGF-β or platelet-derived growth factor-BB by human primary lung fibroblasts from normal lungs (Papakonstantinou et al., 2000; 2002;), suggesting that hypoxia is a synergistic regulator of increased GAG deposition observed in IPAH. In conclusion, our studies demonstrate that synergistic regulation of GAG metabolizing enzymes in favour of accumulation may regulate pathologic remodelling in IPAH by favouring the activation state of PASMC.
Pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a chronic interstitial lung disease that usually results from injury to the lung parenchyma, increased proliferation of mesenchymal cells and excessive accumulation of connective tissue matrix in the interstitium and intraalveolar space of the lung. After injury, resident alveolar cells, including endothelial/epithelial cells and alveolar macrophages, secrete cytokines, chemokines, adhesion molecules and tissue factor. Under normal conditions, this reaction leads to temporary inflammation, tissue formation, remodelling and, finally, normal tissue repair. However, persistent injury and chronic inflammation may accelerate the fibroproliferative response by promoting the secretion of growth factors from resident lung cells, including alveolar macrophages. These growth factors stimulate the proliferation of fibroblasts and smooth muscle cells and the secretion of ECM components, such as collagen, fibronectin and GAG in the lung (Souza-Fernandes et al., 2006). Further to the increased deposition of ECM molecules in the lung, the relative insufficiency of metalloproteinases, the increased secretion of tissue inhibitors of metalloproteinases and the abnormal function of the fibrinolysis system also play critical roles in the fibroproliferative processes in the lung (Suzuki et al., 2004).
Lung fibrosis is mainly studied using the animal model of bleomycin-induced lung injury (Snider et al., 1978). Such studies have demonstrated a critical role for the HA receptor CD44 in the inflammatory processes (Teder et al., 2002). Clearance of HA fragments is crucial in resolving lung inflammation and is dependent on CD44, which is expressed on haematopoietic cells. CD44 deficiency leads to continuous inflammation, increases mortality rate, results in accumulation of HA, prolongs inflammatory gene expression, decreases clearance of apoptotic neutrophils and impairs the ability to generate active TGF-β1 (Teder et al., 2002). CD44 can also mediate wound microvascular endothelial cell migration on fibrogen and invasion into fibrin matrix (Henke et al., 1996).
We have recently investigated the differential expression of GAG in lung tissue specimens from patients with IPF, as compared with controls (lung donors) (Papakonstantinou et al., 2006). We identified the presence of HA, heparan sulphate, dermatan sulphate and chondroitin sulphate both in IPF and control lung tissue specimens. Electrophoresis of total GAG showed that there was a significant increase in the relative amount of HA in IPF, as compared with controls. This increase was about 300%, as also confirmed by ELISA. The observed increase of HA was also associated with changes in gene expression of HAS 2 and 3 and HYAL 1–3, indicating that there is a differential turnover of HA in IPF. As HA is a key molecule mediating important cellular functions, our results indicate that the increased expression of HA in IPF may be associated with the pathology of the disease.
Asthma – chronic obstractive pulmonary disease
Recent reports on asthma and COPD research provided clear evidence that the pathologies of both diseases cannot be solely explained on the basis of a deregulated immune-response. Malfunction of structure-forming cells and disturbance of the homeostasis of ECM molecules contribute significantly to the pathology of both diseases and reflect to airway remodelling (Johnson et al., 2001; Postma and Timens, 2006). Tissue remodelling describes the structural alterations that occur in the lung due to prolonged chronic inflammation within the airways, and involves qualitatitive or quantitative changes in cell density and the composition of the ECM in the pulmonary epithelium, the basement membrane and the submucosa. Consequently, this modification in the ECM affects airway resistance, compliance and elasticity, leading eventually to loss of lung function (Johnson et al., 2001; Postma and Timens, 2006).
It has been suggested that HA plays a protective role in the bronchial tissue as it could block bronchial obstruction induced by aerosol administration of pancreatic elastase in sheep (Scuri et al., 2001). HA also interacts with tissue kallikreins, which is a multiple family of serine proteinases that are secreted by serous cells of tracheobronchial submucosal glands (Forteza et al., 1999) and generate kallidin (lysyl-bradykinin) from low- and high-molecular-weight kininogen (Lauredo et al., 2004). In the lung, the generation of kallidin and its cleavage product bradykinin can contribute to the pathophysiology of asthma (Baumgarten et al., 1986; Christiansen et al., 1992). Both peptides cause vasodilatation (Tadjkarimi et al., 1992) and increase in vascular permeability (Bell and Wainer, 1983) and bronchoconstriction (Scuri et al., 2000). In vitro experiments have demonstrated that HA can inactivate tissue kallikrein, suggesting that the protective effects of HA against elastase-induced bronchoconstriction are mediated through the inactivation of tissue kallikrein (Forteza et al., 1999).
In this context, inhaled HA has been studied in humans as a protective agent against exercise-induced bronchoconstriction (Kunz et al., 2006; Petrigni and Allegra, 2006). However, the results of such studies are contradictory. Petrigni and Allegra (2006) in a randomized, cross-over, single-blind study design have shown that administration of HA (0.4–4 × 106 Da) in aerosol form significantly reduces the bronchial hyper-reactivity to muscular exercise in asthmatics. The authors concluded that such effect could be attributed to the unique physicochemical properties of HA in the remodelling process. At the same time, it was demonstrated that a single dose of HA administered before exercise did not protect against exercise-induced bronchoconstriction in asthmatic patients. The authors suggested that this may be due to the small size (0.15 × 106 Da) of HA that they have used, which actually increased the degree of airway inflammation in asthmatic patients (Kunz et al., 2006).
The argument for a ‘sweet’ protective role of HA of high molecular mass in asthma is further supported by reports that HA of high but not of low molecular mass inhibited the function of alveolar (Negrini et al., 2006) and peritoneal macrophages (Farias et al., 2005). In contrast, a ‘bitter’ role for HA of low molecular mass is indicated by reports that goblet cell metaplasia induced by reactive oxygen species in normal human bronchial epithelial cells was associated with HA depolymerization (Iozzo, 1998). Recent experiments in our laboratory using primary airway smooth muscle cells from healthy lung tissue (control) and from patients with asthma or COPD have shown that there is decreased expression of HA in airway smooth muscle cells from patients with asthma and COPD, as compared with controls. Furthermore, HA in asthma and COPD is of lower molecular mass than HA in controls. Of great interest is our observation that the use of corticosteroids and β2-agonists enhanced synthesis of HA to control levels (Klagas et al., 2009).
Heparin has been shown to protect against exercise-induced bronchoconstriction (Ahmed et al., 1993; Garrigo et al., 1996). The authors suggested that this effect may be due to prevention of mediator release rather than a direct effect on smooth muscle. Furthermore, heparin may be capable of modulating the extent of remodelling of the airway wall seen in asthma, by modulating the actions of a range of proteins, including ECM proteins, growth factors and certain enzymes, and by inhibiting the proliferation of lung fibroblasts and airway smooth muscle cells (Tyrell et al., 1995). Additionally, heparin is released in the airways physiologically as a homeostatic mechanism to limit the extent of the cellular adhesion and diapedesis. Thus, the release of heparin provides a plausible homeostatic mechanism to limit tissue damage and remodelling following an inflammatory insult to the mucosal surface (Page, 1997).
The ‘sweet’ and ‘bitter’ effect of GAG in the pathophysiology of organs other than the lung and relevant pharmacotherapy
The ‘sweet’ or ‘bitter’ effect of GAG reported above for lungs appears to transcend to the pathophysiology of other organs. Some of the recent data regarding the effect of GAG in animals or therapeutic uses in humans for diseases other than pulmonary are presented herewith.
Hyaluronic acid
It has been reported that high-molecular-weight HA inhibits neo-intimal macrophage influx after balloon-catheter induced injury in the cholesterol-fed rabbit (Ferns et al., 1995). With respect to humans, it has been reported that HA ophthalmic viscoelastic devices are used in corneal transplantation and vitreoretinal, anterior segment and glaucoma surgery, within the eye and on the surface of the eye to prevent dryness and to facilitate wound healing (Balazs, 2008). Similarly, scleral implants of HA have been reported to reduce the intraocular pressure-lowering effect and complication rate of nonpenetrating deep sclerectomy in primary open angle glaucoma (Russo et al., 2008). In patients with rheumatoid arthritis, intraarticular administration of HA has been reported to alleviate the pain associated with knee (Chou et al., 2008) and ankle joints (Cohen et al., 2008) and to significantly decrease the rate of deterioration of joint structure (Migliore and Granata, 2008). In patients with interstitial cystitis it has been reported that oral or intravesical administration of HA reduced bladder inflammation and ‘replenized’ the GAG layer (Theoharides et al., 2008), and that intravesical administration of HA is regarded as a safe and efficacious method of treatment of patients with interstitial cystitis/painful bladder syndrome (Porru et al., 2008).
Interactions of HA with CD44 induce activation of receptor tyrosine kinases and promote ‘bitter’ effects, such as drug resistance in a variety of cancer cell types, including breast, lung and head and neck carcinomas, and lymphoma (Misra et al., 2005; Wang and Bourguignon, 2006; Cordo Russo et al., 2008). An anticipated ‘sweet’ effect of small HA oligomers, acting as antagonists to high-molecular-weight HA–CD44 interactions, may be proved to be useful in therapeutic strategies aimed at preventing tumour recurrence from therapy-resistant sub-populations within malignant cancers (Toole and Slomiany, 2008).
Heparan sulphate
Intraperitoneal administration of heparanase, an endo-beta-D-glucuronidase that cleaves heparan sulphate saccharide chains, ameliorated the clinical signs in a murine non-obese diabetic model (T cell-dependent disease) of type 1 diabetes by inducing a shift from a Th1- to Th2-phenotype (Bitan et al., 2008). Heparan sulphate also exerts anti-inflammatory effects. Syndecan-1 is a major heparan sulphate proteoglycan. Syndecan-1-null mice showed significantly increased liver injury, vascular permeability and death in response to staphylococcal enterotoxin B challenge compared with wild-type mice. Administration of heparan sulphate rescued syndecan-1-null mice from staphylococcal enterotoxin B induced lethal toxic shock by suppressing the production of TNF-α and IL-6, and attenuating inflammatory tissue injury (Hayashida et al., 2008). In addition, polysulphated molecules, as the family of heparan mimetics, have been reported to have significant therapeutic potential for prion diseases (Ouidja et al., 2007). In humans, it has been reported that heparanase directly contributes to prostate tumour growth in bone and its ability to metastasize to distant organs and that local in vivo silencing by small interfering RNA of heparanase resulted in a fourfold inhibition of prostate tumour growth, suggesting that anti-heparanase strategy may become an important modality in the treatment of prostate cancer patients, particularly those with bone metastases (Lerner et al., 2008).
Heparin
Administration of enoxaparin, which is a low-molecular-weight heparin, in patients with diabetic nephropathy exerted antiproteinuric effect not via the renin-angiotensin-aldosterone system, but apparently by inducing intrinsic alterations in the glomerular filter (Benck et al., 2006).
Chondroitin sulphate
Administration of chondroitin sulphate rich in the specific epitope GlcUA-GalNAc (4, 6-O-disulphate) or a specific decasaccharide fraction of this epitope inhibited the metastasis of Lewis lung carcinoma-derived LM66-H11 cells to lungs in the mouse (Li et al., 2008). Also, administration of chondroitin-4-sulphate reduced the effects of caerulein-induced acute pancreatitis in the mouse experimental model, by blocking NF-kappaB and caspase activation due to its antioxidant activity (Campo et al., 2008a). With respect to spinal cord injury, chondroitin sulphate proteoglycan plays a key role during the acute recovery stage in mice, and the sugar component of the proteoglycan is responsible for the inhibitory role of these compounds in axonal regeneration. It has been reported that chondroitin sulphate proteoglycan is considered to be a major obstacle for central nervous system recovery after spinal cord injury in mice due to its well-known activity in limiting axonal growth (Rolls et al., 2008). Administration of chondroitinase ABC increased corticospinal axon growth in organotypic cocultures of brain cortex and spinal cord (Nakamae et al., 2008), while intrathecal administration of chondroitinase ABC in a rat model with spinal cord injury caused a slight decrease in chondroitin sulphate proteoglycans in the scar and a minimal increase in sensory axonal regeneration into and across the laceration gap (Shields et al., 2008). On the same line, inhibition of chondroitin sulphate proteoglycan synthesis by xyloside immediately after injury impaired functional motor recovery and increased tissue loss after spinal cord injury in mice (Rolls et al., 2008).
The ‘sweet’ role of chondroitin sulphate proteoglycan during the acute stage and its ‘bitter’ effect at later stages emphasizes the need to retain the endogenous potential of this molecule in repair by controlling its levels at different stages of post-injury repair (Rolls et al., 2008). With respect to arthritis, it has been reported that administration of chondroitin sulphate in a rabbit model of atherosclerosis aggravated by chronic arthritis reduced the concentration of the proinflammatory molecules C-reactive protein, IL-6 and the nuclear translocation of NF-kappaB and inhibited the expression of CCL2/monocyte chemoattractant protein-1 and cyclooxygenase-2. Moreover, chondroitin sulphate decreased the percentage of rabbits with atherosclerosis and chronic arthritis that developed vascular lesions in the aorta (Herrero-Beaumont et al., 2008). Similarly, intraperitoneal administration of chondroitin-4-sulphate in mice with collagen-induced arthritis ameliorated all the symptoms of inflammation in the articular knee and paw joints, limited lipid peroxidation, inhibited NF-kappaB activation, decreased mRNA MMP-13 and caspases expression of and their related protein, restored endogenous antioxidants and reduced PMN accumulation in the damaged cartilage (Campo et al., 2008b).
In patients with osteoarthritis, administration of 800 mg of chondroitin sulphate for 3 month mode, twice a year, has been recommended by the European League Against Rheumatism for the treatment of knee osteoarthritis as evidence 1A and strength of recommendation A, which represents the highest level for a therapeutic strategy (Uebelhart, 2008). However, data provided by meta-analyses published between 1997 and 2007 indicate that chondroitin sulphate has a slight to moderate efficacy in the symptomatic treatment of osteoarthritis, with an excellent safety profile (Monfort et al., 2008).
Further application of chondroitin sulphate in humans refers to eye surgery, where chondroitin sulphate is topically applied for phacoemulsification postoperatively (Praveen et al., 2008). In humans, it has also been reported that instillation of chondroitin sulphate effectively reduced all symptoms in patients with chronic forms of cystitis (Nordling and van Ophoven, 2008), that oral or intravesical administration of chondroitin sulphate reduced bladder inflammation and ‘replenished’ the GAG layer (Theoharides et al., 2008) and that intravesical administration of chondroitin sulphate is regarded as a safe and efficacious method of treatment of patients with interstitial cystitis/painful bladder syndrome (Porru et al., 2008). Further effects of chondroitin sulphate are associated in humans with coagulation. Odiparcil, an orally active beta-d-thioxyloside analoglue, exerts antithrombotic activity apparently by increasing circulating endogenous levels of chondroitin sulphate-related GAG (Myers et al., 2008). However, contaminated heparin with oversulphated chondroitin sulphate has been associated with severe anaphylactoid reactions via activation of the contact system after intravenous heparin administration (Kishimoto et al., 2008).
Dermatan sulphate
It has been reported that administration of dermatan sulphate is an effective anticoagulant in patients on continuous renal replacement therapy (Vitale et al., 2008).
Keratan sulphate
Increase in serum keratan sulphate was associated with progression of osteoarthritis (Tang et al., 2008).
Cancer
With respect to cancer, a growing body of evidence supports crucial roles of GAG at various pathophysiological steps of tumour progression. GAG regulate tumour proliferation, invasion, haematogenous metastasis and angiogenesis, and increased understanding of these roles sets the stage for developing pharmaceutical agents that target these molecules. Such novel agents might be used alone or in combination with operative and/or chemoradiation strategies for treating cancer (Fuster and Esko, 2005). For example, heparan sulphate proteoglycans allow primary tumour growth and angiogenesis through growth factor-dependent signalling (Fuster and Esko, 2005). It might be possible to target tumour cells by developing agents that bind heparan sulphate or influence its synthesis (Bishop et al., 2007).
Vaccines
There are various vaccines used made of viral coat carbohydrates against microorganisms responsible for infectious diseases and other conditions, such as Salmonella typhi– Typhus, Sreptococcus pneumoniae– Pneumonia, Neisseria meningitides– Bacterial meningitis, Haemophilus influenza type b – Influenza or various vaccines under development that use surface glycolipid against Plasmodium falciparum– Malaria and Leishmania– Leishmaniasis, or finally the monosaccharide oseltamivir phosphate against Influenza (Verez-Bencomo et al., 2004; Seeberger and Werz, 2007; Oppenheimer et al., 2008).
Conclusion
There is increasing evidence over the last decade that GAG are involved in the pathophysiology of the lung and other organs. Of particular interest is the fact that the same molecule may exert a beneficiary or a detrimental effect, depending on the size, as is the case of HA, or the degree of sulphation, as is the case of heparin. This reciprocal transition from the ‘sweet’ to the ‘bitter’ involvement of GAG in lung function is apparently mediated by specific enzymes responsible for the synthesis or degradation of GAG, rendering both the end GAG product as well as the liable enzymes as putative targets for treatment of specific lung diseases. Advances in carbohydrate and GAG synthesis, analysis and manipulation through the fields of glycochemistry and glycobiology and new technologies using carbohydrate and lectin microarrays offer improved diagnostic and drug development possibilities that are fascinating for the pharmacologist.
Glossary
Abbreviations:
- COPD
chronic obstructive pulmonary disease
- ECM
extracellular matrix
- GAG
glycosaminoglycans
- HA
hyaluronic acid
- HAS
hyaluronic acid synthase
- HYAL
hyaluronidase
- IL
interleukin
- IPAH
idiopathic pulmonary arterial hypertension
- PASMC
pulmonary arterial smooth muscle cells
- RHAMM
receptor for HA-mediated motility
- TGF
transforming growth factor
- TNF
tumour necrosis factor
- VEGF
vascular endothelium-derived growth factor
References
- Ahmed T, Garrigo J, Danta I. Preventing bronchoconstriction in exercise-induced asthma with inhaled heparin. N Engl J Med. 1993;329:90–95. doi: 10.1056/NEJM199307083290204. [DOI] [PubMed] [Google Scholar]
- Ahrens T, Assmann V, Fieber C, Termeer C, Herrlich P, Hofmann M, et al. CD44 is the principal mediator of hyaluronic-acid-induced melanoma cell proliferation. J Invest Dermatol. 2001;116:93–101. doi: 10.1046/j.1523-1747.2001.00236.x. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, et al. Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell. 2001;104:923–935. doi: 10.1016/s0092-8674(01)00288-4. [DOI] [PubMed] [Google Scholar]
- Assmann V, Jenkinson D, Marshall JF, Hart IR. The intracellular hyaluronan receptor RHAMM/IHABP interacts with microtubules and actin filaments. J Cell Sci. 1999;112:3943–3954. doi: 10.1242/jcs.112.22.3943. [DOI] [PubMed] [Google Scholar]
- Assmann V, Gillett CE, Pouson R, Ryder K, Hart JR, Hanby AM. The pattern of expression of the microtubule-binding protein RHAMM/IHABP in mammary carcinoma suggests a role in the invasive behaviour of tumour cells. J Pathol. 2001;195:191–196. doi: 10.1002/path.941. [DOI] [PubMed] [Google Scholar]
- Aviezer D, Safran M, Yayon A. Heparin differentially regulates the interaction of fibroblast growth factor-4 with FGF receptors 1 and 2. Biochem Biophys Res Commun. 1999;263:621–626. doi: 10.1006/bbrc.1999.1434. [DOI] [PubMed] [Google Scholar]
- Bai KJ, Spicer AP, Mascarenhas MM, Yu L, Ochoa CD, Garg HG, et al. The role of hyaluronan synthase 3 in ventilator-induced lung injury. Am J Respir Crit Care Med. 2005;172:92–98. doi: 10.1164/rccm.200405-652OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balabanian K, Foussat A, Dorfmüller P, Durand-Gasselin I, Capel F, Bouchet-Delbos L, et al. CX(3)C chemokine fractalkine in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165:1419–1425. doi: 10.1164/rccm.2106007. [DOI] [PubMed] [Google Scholar]
- Balazs EA. Hyaluronan as an ophthalmic viscoelastic device. Curr Pharm Biotechnol. 2008;9:236–238. doi: 10.2174/138920108785161596. [DOI] [PubMed] [Google Scholar]
- Baumgarten CR, Nichols RC, Naclerio RM, Proud D. Concentrations of glandular kallikrein in human nasal secretions during experimentally induced allergic rhinitis. J Immunol. 1986;137:1323–1328. [PubMed] [Google Scholar]
- Bell RD, Wainer BS. Effects of bradykinin on renal lymph flow and composition. Lymphology. 1983;16:38–42. [PubMed] [Google Scholar]
- Benck U, Haeckel S, Clorius JH, van der Woude FJ. Proteinuria-lowering effect of heparin therapy in diabetic nephropathy without affecting the renin-angiotensin-aldosterone system. Clin J Am Soc Nephrol. 2006;2:58–67. doi: 10.2215/CJN.02400706. [DOI] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. Heparan sulfate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Bitan M, Weiss L, Zeira M, Reich S, Pappo O, Vlodavsky I, et al. Heparanase prevents the development of type 1 diabetes in non-obese diabetic mice by regulating T-cell activation and cytokines production. Diabetes Metab Res Rev. 2008;24:413–421. doi: 10.1002/dmrr.868. [DOI] [PubMed] [Google Scholar]
- Blewis ME, Nugent-Derfus GE, Schmidt TA, Schumacher BL, Sah RL. A model of synovial fluid lubricant composition in normal and injured joints. Eur Cell Mater. 2007;13:26–39. doi: 10.22203/ecm.v013a03. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci USA. 2007;104:11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon LY, Zhu H, Shao L, Chen YW. CD44 interaction with c-Src kinase promotes cortactin-mediated cytoskeleton function and hyaluronic acid-dependent ovarian tumor cell migration. J Biol Chem. 2001;276:7327–7333. doi: 10.1074/jbc.M006498200. [DOI] [PubMed] [Google Scholar]
- Brown GM, Huckerby TN, Bayliss MT, Nieduszynski IA. Human aggrecan keratan sulfate undergoes structural changes during adolescent development. J Biol Chem. 1998;273:26408–26414. doi: 10.1074/jbc.273.41.26408. [DOI] [PubMed] [Google Scholar]
- Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo GM, Avenoso A, Campo S, Nastasi G, Traina P, D'Ascola A, et al. Chondroitin-4-sulfate reduced oxidative injury in caerulein-induced pancreatitis in mice: the involvement of NF-kappaB translocation and apoptosis activation. Exp Biol Med (Maywood) 2008a;233:741–752. doi: 10.3181/0711-RM-318. [DOI] [PubMed] [Google Scholar]
- Campo GM, Avenoso A, Campo S, D'Ascola A, Traina P, Calatroni A. Chondroitin-4-sulfate inhibits NF-kB translocation and caspase activation in collagen-induced arthritis in mice. Osteoarthritis Cartilage. 2008b;16:1474–1483. doi: 10.1016/j.joca.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Cantor JO, Bray BA, Ryan SF, Mandl I, Turino GM. Glycosaminoglycan and collagen synthesis in N-nitroso-N-methylurethane induced pulmonary fibrosis. Proc Soc Exp Biol Med. 1980;164:1–8. doi: 10.3181/00379727-164-40814. [DOI] [PubMed] [Google Scholar]
- Cantor JO, Shteyngart B, Cerreta JM, Liu M, Armand G, Turino GM. The effect of hyaluronan on elastic fiber injury in vitro and elastase-induced airspace enlargement in vivo. Proc Soc Exp Biol Med. 2000;225:65–71. doi: 10.1046/j.1525-1373.2000.22508.x. [DOI] [PubMed] [Google Scholar]
- Cao J, Sato H, Takino T, Seiki M. The C-terminal region of membrane type matrix metalloproteinase is a functional trans-membrane domain required for pro-gelatinase A activation. J Biol Chem. 1995;270:801–805. doi: 10.1074/jbc.270.2.801. [DOI] [PubMed] [Google Scholar]
- Chai S, Chai Q, Danielsen CC, Hjorth P, Nyengaard JR, Ledet T, et al. Overexpression of hyaluronan in the tunica media promotes the development of atherosclerosis. Circ Res. 2005;96:583–591. doi: 10.1161/01.RES.0000158963.37132.8b. [DOI] [PubMed] [Google Scholar]
- Chazova I, Loyd JE, Zhdanov VS, Newman JH, Belenkov Y, Meyrick B. Pulmonary artery adventitial changes and venous involvement in primary pulmonary hypertension. Am J Pathol. 1995;146:389–397. [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, Huang M, Campbell MJ, Dong H, Steinmetz L, Sapinoso L, et al. Nat Genet. 2001;27:48–54. doi: 10.1038/83751. [DOI] [PubMed] [Google Scholar]
- Chou CL, Li HW, Lee SH, Tsai KL, Ling HY. Effect of intra-articular injection of hyaluronic acid in rheumatoid arthritis patients with knee. J Chin Med Assoc. 2008;71:411–415. doi: 10.1016/S1726-4901(08)70092-3. [DOI] [PubMed] [Google Scholar]
- Christiansen SC, Proud D, Sarnoff RB, Juergens U, Cochrane CG, Zuraw BL. Elevation of tissue kallikrein and kinin in the airways of asthmatic subjects after endobronchial allergen challenge. Am Rev Respir Dis. 1992;145:900–905. doi: 10.1164/ajrccm/145.4_Pt_1.900. [DOI] [PubMed] [Google Scholar]
- Christner JE, Distler JJ, Jourdian GW. Biosynthesis of keratan sulfate: purification and properties of a galactosyltransferase from bovine cornea. Arch Biochem Biophys. 1979;192:548–558. doi: 10.1016/0003-9861(79)90125-5. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Altman RD, Hollstrom R, Hollstrom C, Sun C, Gipson B. Safety and efficacy of intra-articular sodium hyaluronate (Hyalgan) in a randomized, double-blind study for osteoarthritis of the ankle. Foot Ankle Int. 2008;29:657–663. doi: 10.3113/FAI.2008.0657. [DOI] [PubMed] [Google Scholar]
- Cordo Russo RI, Garcia MG, Alaniz L, Blanco G, Alvarez E, Hajos SE. Hyaluronan oligosaccharides sensitize lymphoma resistant cell lines to vincristine by modulating P-glycoprotein activity and PI3K/Akt pathway. Int J Cancer. 2008;122:1012–1018. doi: 10.1002/ijc.23122. [DOI] [PubMed] [Google Scholar]
- Cuff CA, Kothapalli D, Azonobi I, Chun S, Zhang Y, Belkin R, et al. The adhesion receptor CD44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. J Clin Invest. 2001;108:1031–1040. doi: 10.1172/JCI12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deesomchok A, Tanprawate S. A 12-case series of penicillium marneffei pneumonia. J Med Assoc Thai. 2006;89:441–447. [PubMed] [Google Scholar]
- Degroote S, Lo-Guidice JM, Strecker G, Ducourouble MP, Roussel P, Lamblin G. Characterization of an N-acetylglucosamine-6-O-sulfotransferase from human respiratory mucosa active on mucin carbohydrate chains. J Biol Chem. 1997;72:29493–29501. doi: 10.1074/jbc.272.47.29493. [DOI] [PubMed] [Google Scholar]
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentener MA, Vernooy JH, Hendriks S, Wouters EF. Enhanced levels of hyaluronan in lungs of patients with COPD: relationship with lung function and local inflammation. Thorax. 2005;60:114–119. doi: 10.1136/thx.2003.020842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolhnikoff M, Mauad T, Bethlem EP, Carvalho CR. Leptospiral pneumonias. Curr Opin Pulm Med. 2007;13:230–235. doi: 10.1097/MCP.0b013e3280f9df74. [DOI] [PubMed] [Google Scholar]
- Du HY, Ji SL, Song HF, Ye QN, Cao JC. The relationship between the structure of dermatan sulfate derivatives and their antithrombotic activities. Thromb Res. 2007;119:377–384. doi: 10.1016/j.thromres.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Morty RE. Transforming growth factor beta/bone morphogenic protein signaling in pulmonary arterial hypertension: remodeling revisited. Trends Cardiovasc Med. 2007;17:263–269. doi: 10.1016/j.tcm.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Evanko PS, Parks TW, Wight NT. Intracellular hyaluronan in arterial smooth muscle cells: association with microtubules, RHAMM, and the mitotic spindle. J Histochem Cytochem. 2004;52:1525–1535. doi: 10.1369/jhc.4A6356.2004. [DOI] [PubMed] [Google Scholar]
- Farias LL, Faffe DS, Xisto DG, Santana MC, Lassance R, Prota LF, et al. Positive endexpiratory pressure prevents lung mechanical stress caused by recruitment/derecruitment. J Appl Physiol. 2005;98:53–61. doi: 10.1152/japplphysiol.00118.2004. [DOI] [PubMed] [Google Scholar]
- Ferns GA, Konneh M, Rutherford C, Woolaghan E, Anggard EE. Hyaluronan (HYAL-BV 5200) inhibits neo-intimal macrophage influx after balloon-catheter induced injury in the cholesterol-fed rabbit. Atherosclerosis. 1995;114:157–164. doi: 10.1016/0021-9150(94)05479-3. [DOI] [PubMed] [Google Scholar]
- Forteza R, Lauredo I, Abraham WM, Conner GE. Bronchial tissue kallikrein activity is regulated by hyaluronic acid binding. Am J Respir Cell Mol Biol. 1999;21:666–674. doi: 10.1165/ajrcmb.21.6.3651. [DOI] [PubMed] [Google Scholar]
- Funderburgh JL. Keratan sulfate biosynthesis. IUBMB Life. 2002;54:187–194. doi: 10.1080/15216540214932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet. 1998;352:719–725. doi: 10.1016/S0140-6736(98)02111-4. [DOI] [PubMed] [Google Scholar]
- Gares SL, Pilarski LM. Balancing thymocyte adhesion and motility: a functional linkage between beta1 integrins and the motility receptor RHAMM. Dev Immunol. 2000;7:209–225. doi: 10.1155/2000/94616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigo J, Danta I, Ahmed T. Time course of the protective effect of inhaled heparin on exercise-induced asthma. Am J Respir Crit Care Med. 1996;153:1702–1707. doi: 10.1164/ajrccm.153.5.8630624. [DOI] [PubMed] [Google Scholar]
- Gerdin B, Hallgren R. Dynamic role of hyaluronan (HYA) in connective tissue activation and inflammation. J Intern Med. 1997;242:49–55. doi: 10.1046/j.1365-2796.1997.00173.x. [DOI] [PubMed] [Google Scholar]
- Gillespie SH, Balakrishnan I. Pathogenesis of pneumococcal infection. J Med Microbiol. 2000;49:1057–1067. doi: 10.1099/0022-1317-49-12-1057. [DOI] [PubMed] [Google Scholar]
- Green WF, Konnaris K, Woolcock AJ. Effect of salbutamol, fenoterol, and sodium cromoglycate on the release of heparin from sensitized human lung fragments challenged with Dermatophagoides pteronyssinus allergen. Am J Respir Cell Mol Biol. 1993;8:518–521. doi: 10.1165/ajrcmb/8.5.518. [DOI] [PubMed] [Google Scholar]
- Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007;7:69–98. doi: 10.1152/physrev.00022.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambartsumian N, Lukanidin E, Rabinovitch M. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol. 2004;164:253–262. doi: 10.1016/S0002-9440(10)63115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen M, Fagan K, Steudel W, Carr M, Lane K, Rodman DM, et al. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1473–L1479. doi: 10.1152/ajplung.00197.2006. [DOI] [PubMed] [Google Scholar]
- Hall CL, Wang C, Lange LA, Turley EA. Hyaluronan and the hyaluronan receptor RHAMM promote focal adhesion turnover and transient tyrosine kinase activity. J Cell Biol. 1994;126:575–588. doi: 10.1083/jcb.126.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE. Regulation of protein function by glycosaminoglycans – as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- Hassoun PM. Deciphering the “matrix” in pulmonary vascular remodelling. Eur Respir J. 2005;25:778–779. doi: 10.1183/09031936.05.00027305. [DOI] [PubMed] [Google Scholar]
- Hayashida K, Chen Y, Bartlett AH, Park PW. Syndecan-1 is an in vivo suppressor of Gram-positive toxic shock. J Biol Chem. 2008;283:19895–19903. doi: 10.1074/jbc.M801614200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke CA, Roongta U, Mickelson DJ, Knutson JR, McCarthy JB. CD44-related chondroitin sulfate proteoglycan, a cell surface receptor implicated with tumor cell invasion, mediates endothelial cell migration on fibrinogen and invasion into a fibrin matrix. J Clin Invest. 1996;97:2541–2552. doi: 10.1172/JCI118702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Beaumont G, Marcos ME, Sánchez-Pernaute O, Granados R, Ortega L, Montell E, et al. Effect of chondroitin sulfate in a rabbit model of atherosclerosis aggravated by chronic arthritis. Br J Pharmacol. 2008;154:843–851. doi: 10.1038/bjp.2008.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton MR, Burdick MD, Strieter RM, Bao C, Noble PW. Regulation of hyaluronan-induced chemokine gene expression by IL-10 and IFN-gamma in mouse macrophages. J Immunol. 1998a;160:3023–3030. [PubMed] [Google Scholar]
- Horton MR, McKee CM, Bao C, Liao F, Farber JM, Hodge-DuFour J, et al. Hyaluronan fragments synergize with interferon-gamma to induce the C-X-C chemokines mig and interferon-inducible protein-10 in mouse macrophages. J Biol Chem. 1998b;273:35088–35094. doi: 10.1074/jbc.273.52.35088. [DOI] [PubMed] [Google Scholar]
- Horton MR, Shapiro S, Bao C, Lowenstein CJ, Noble PW. Induction and regulation of macrophage metalloelastase by hyaluronan fragments in mouse macrophages. J Immunol. 1999;162:4171–4176. [PubMed] [Google Scholar]
- Horton MR, Olman MA, Bao C, White KE, Choi AM, Chin BY, et al. Regulation of plasminogen activator inhibitor-1 and urokinase by hyaluronan fragments in mouse macrophages. Am J Physiol Lung Cell Mol Physiol. 2000;279:L707–L715. doi: 10.1152/ajplung.2000.279.4.L707. [DOI] [PubMed] [Google Scholar]
- Horton MR, Boodoo S, Powell JD. NF-kappa B activation mediates the cross-talk between extracellular matrix and interferon-gamma (IFN-gamma) leading to enhanced monokine induced by IFN-gamma (MIG) expression in macrophages. J Biol Chem. 2002;277:43757–43762. doi: 10.1074/jbc.M206007200. [DOI] [PubMed] [Google Scholar]
- Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004a;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004b;351:1425–1436. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- Iida J, Wilhelmson KL, Ng J, Lee P, Morrison C, Tam E, et al. Cell surface chondroitin sulfate glycosaminoglycan in melanoma: role in the activation of pro-MMP-2 (pro-gelatinase A) Biochem J. 2007;403:553–563. doi: 10.1042/BJ20061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovu M, Dumais G, du Souich P. Anti-inflammatory activity of chondroitin sulfate. Osteoarthritis Cartilage. 2008;16:S14–S18. doi: 10.1016/j.joca.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–652. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- Itano N, Kimata K. Mammalian hyaluronan synthases. IUBMB Life. 2002;54:195–199. doi: 10.1080/15216540214929. [DOI] [PubMed] [Google Scholar]
- Itano N, Sawai T, Yoshida M, Lenas P, Yamada Y, Imagawa M, et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J Biol Chem. 1999;274:25085–25092. doi: 10.1074/jbc.274.35.25085. [DOI] [PubMed] [Google Scholar]
- Ito T, Fraser IP, Yeo Y, Highley CB, Bellas E, Kohane DS. Anti-inflammatory function of an in situ cross-linkable conjugate hydrogel of hyaluronic acid and dexamethasone. Biomaterials. 2007;28:1778–1786. doi: 10.1016/j.biomaterials.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Ann Rev Cell Dev Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- Johnson PR, Roth M, Tamm M, Hughes M, Ge Q, King G, et al. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med. 2001;164:474–477. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005;16:625–636. doi: 10.1016/j.cytogfr.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Jones GC. ADAMTS proteinases: potential therapeutic targets? Curr Pharm Biotechnol. 2006;7:25–31. doi: 10.2174/138920106775789656. [DOI] [PubMed] [Google Scholar]
- Jones P, Cowan K, Rabinovitch M. Progressive pulmonary vascular disease is characterized by a proliferative response related to deposition of tenascin-C and is preceded by subendothelial accumulation of fibronectin. Am J Pathol. 1997;150:1349–1360. [PMC free article] [PubMed] [Google Scholar]
- Karakiulakis G, Papakonstantinou E, Aletras AJ, Tamm M, Roth M. Cell type-specific effect of hypoxia and platelet-derived growth factor-BB on extracellular matrix turnover and its consequences for lung remodeling. J Biol Chem. 2007;282:908–195. doi: 10.1074/jbc.M602178200. [DOI] [PubMed] [Google Scholar]
- Kaverina I, Krylyshkina O, Small VJ. Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J Cell Biol. 1999;146:1033–1044. doi: 10.1083/jcb.146.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella MG, Bressler SL, Wight TN. The regulated synthesis of versican, decorin, and biglycan: extracellular matrix proteoglycans that influence cellular phenotype. Crit Rev Eukaryot Gene Expr. 2004;143:203–234. doi: 10.1615/critreveukaryotgeneexpr.v14.i3.40. [DOI] [PubMed] [Google Scholar]
- Kishimoto TK, Viswanathan K, Ganguly T, Elankumaran S, Smith S, Pelzer K, et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med. 2008;358:2457–2467. doi: 10.1056/NEJMoa0803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klagas I, Goulet S, Karakiulakis G, Zhong J, Baraket M, Black JL, et al. Decreased hyaluronan in airway smooth muscle cells from patients with asthma and COPD. Eur Respir J. 2009 doi: 10.1183/09031936.00070808. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Matsumoto T, Vanwildemeersch M, Sasaki T, Timpl R, Claesson-Welsh L, et al. Role of heparan sulfate domain organization in endostatin inhibition of endothelial cell function. EMBO J. 2002;21:6303–6311. doi: 10.1093/emboj/cdf638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusius T, Finne J, Margolis RK, Margolis RU. Identification of an O-glycosidic mannose-linked sialylated tetrasaccharide and keratan sulfate oligosaccharides in the chondroitin sulfate proteoglycan of brain. J Biol Chem. 1986;261:8237–8242. [PubMed] [Google Scholar]
- Kumar N, Bentolila A, Domb AJ. Structure and biological activity of heparinoid. Mini Rev Med Chem. 2005;5:441–447. doi: 10.2174/1389557053765538. [DOI] [PubMed] [Google Scholar]
- Kunz LI, Rensen EL, Sterk PJ. Inhaled hyaluronic acid against exercise-induced bronchoconstriction in asthma. Pulm Pharmacol Ther. 2006;19:286–291. doi: 10.1016/j.pupt.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Kusche-Gullberg M, Kjellen L. Sulfotransferases in glycosaminoglycan biosynthesis. Curr Opin Struct Biol. 2003;13:605–611. doi: 10.1016/j.sbi.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Kuschert GS, Coulin F, Power CA, Proudfoot AE, Hubbard RE, Hoogewerf AJ, et al. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. 1999;38:12959–12968. doi: 10.1021/bi990711d. [DOI] [PubMed] [Google Scholar]
- Lauredo IT, Forteza RM, Botvinnikova Y, Abraham WM. Leukocytic cell sources of airway tissue kallikrein. Am J Physiol Lung Cell Mol Physiol. 2004;286:L734–L740. doi: 10.1152/ajplung.00129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent GJ, Chambers RC, Hill MR, McAnulty RJ. Regulation of matrix turnover: fibroblasts, forces, factors and fibrosis. Biochem Soc Trans. 2007;35:647–651. doi: 10.1042/BST0350647. [DOI] [PubMed] [Google Scholar]
- Laurent TC. Structure of hyaluronic acid. In: Balazs EA, editor. Chemistry and Molecular Biology of the Intercellular Matrix. New York: Academic Press; 1970. pp. 703–732. [Google Scholar]
- LeBaron RG, Hook A, Esko JD, Gay S, Hook M. Binding of heparan sulfate to type V collagen. A mechanism of cell-substrate adhesion. J Biol Chem. 1989;264:7950–7956. [PubMed] [Google Scholar]
- Lerner I, Baraz L, Pikarsky E, Meirovitz A, Edovitsky E, Peretz T, et al. Function of heparanase in prostate tumorigenesis: potential for therapy. Clin Cancer Res. 2008;14:668–676. doi: 10.1158/1078-0432.CCR-07-1866. [DOI] [PubMed] [Google Scholar]
- Li F, Ten Dam GB, Murugan S, Yamada S, Hashiguchi T, Mizumoto S, et al. Involvement of highly sulfated chondroitin sulfate in the metastasis of the Lewis lung carcinoma cells. J Biol Chem. 2008;283:34294–34304. doi: 10.1074/jbc.M806015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Guo L, Li JW, Liu N, Qi R, Liu J. Expression of hyaluronan receptors CD44 and RHAMM in stomach cancers: relevance with tumor progression. Int J Oncol. 2000a;17:927–932. [PubMed] [Google Scholar]
- Li Y, Rahmanian M, Widstrom C, Lepperdinger G, Frost GI, Heldin P. Irradiation induced expression of hyaluronan (HA) synthase 2 and hyaluronidase 2 genes in rat lung tissue accompanies active turnover of HA and induction of types I and III collagen gene expression. Am J Resp Cell Mol Biol. 2000b;23:411–418. doi: 10.1165/ajrcmb.23.3.4102. [DOI] [PubMed] [Google Scholar]
- Lin Y, Mahan K, Lathrop WE, Myles DG, Primakoff P. A hyaluronidase activity of the sperm plasma membrane protein PH-20 enables sperm to penetrate the cumulus cell layer surrounding the egg. J Cell Biol. 1994;125:1157–1163. doi: 10.1083/jcb.125.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe RJ, Nakhoul AM, Sanderson CJ, Coombe DR. Interleukin-5 binds to heparin/heparan sulfate. A model for an interaction with extracellular matrix. J Leukoc Biol. 1998;63:342–350. doi: 10.1002/jlb.63.3.342. [DOI] [PubMed] [Google Scholar]
- Liptay MJ, Parks WC, Mecham RP, Roby J, Kaiser LR, Cooper JD, et al. Neointimal macrophages colocalize with extracellular matrix gene expression in human atherosclerotic pulmonary arteries. J Clin Invest. 1993;91:588–594. doi: 10.1172/JCI116238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KW, Goerke J, Clements JA, Taeusch HW. Hyaluronan decreases surfactant inactivation in vitro. Pediatr Res. 2005;57:237–241. doi: 10.1203/01.PDR.0000150726.75308.22. [DOI] [PubMed] [Google Scholar]
- Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas MM, Day RM, Ochoa CD, Choi WI, Yu L, Ouyang B, et al. Low molecular weight hyaluronan from stretched lung enhances interleukin-8 expression. Am J Respir Cell Mol Biol. 2004;30:51–60. doi: 10.1165/rcmb.2002-0167OC. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- McAnulty RJ, Laurent GJ. Collagen synthesis and degradation in vivo. Evidence for rapid rates of collagen turnover with extensive degradation of newly synthesized collagen in tissues of the adult rat. Coll Rel Res. 1987;7:93–104. doi: 10.1016/s0174-173x(87)80001-8. [DOI] [PubMed] [Google Scholar]
- McDonald JE, Miller FJ, Hallam SE, Nelson L, Marchuk DA, Ward KJ. Clinical manifestations in a large hereditary hemorrhagic telangiectasia (HHT) type 2 kindred. Am J Med Genet. 2000;93:320–327. doi: 10.1002/1096-8628(20000814)93:4<320::aid-ajmg12>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, et al. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J Clin Invest. 1996;98:2403–2413. doi: 10.1172/JCI119054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee CM, Lowenstein CJ, Horton MR, Wu J, Bao C, Chin BY, et al. Hyaluronan fragments induce nitric-oxide synthase in murine macrophages through a nuclear factor kappaB-dependent mechanism. J Biol Chem. 1997;272:8013–8018. doi: 10.1074/jbc.272.12.8013. [DOI] [PubMed] [Google Scholar]
- Menart V, Fonda I, Kenig M, Porekar VG. Increased in vitro cytotoxicity of TNF-alpha analog LK-805 is based on the interaction with cell surface heparan sulfate proteoglycan. Ann N Y Acad Sci. 2002;973:194–206. doi: 10.1111/j.1749-6632.2002.tb04632.x. [DOI] [PubMed] [Google Scholar]
- Merry CLR, Lyon M, Deakin JA, Hopwood JJ, Gallagher JT. Highly sensitive sequencing of the sulfated domains of Heparan sulfate. J Biol Chem. 1999;274:18455–18462. doi: 10.1074/jbc.274.26.18455. [DOI] [PubMed] [Google Scholar]
- Migliore A, Granata M. Intra-articular use of hyaluronic acid in the treatment of osteoarthritis. Clin Interv Aging. 2008;3:365–369. doi: 10.2147/cia.s778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra S, Ghatak S, Toole BP. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J Biol Chem. 2005;280:20310–20315. doi: 10.1074/jbc.M500737200. [DOI] [PubMed] [Google Scholar]
- Miwa HE, Gerken TA, Huynh TD, Flory DM, Hering TM. Mammalian expression of full-length bovine aggrecan and link protein: formation of recombinant proteoglycan aggregates and analysis of proteolytic cleavage by ADAMTS-4 and MMP-13. Biochim Biophys Acta. 2006;1760:472–486. doi: 10.1016/j.bbagen.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Monfort J, Martel-Pelletier J, Pelletier JP. Chondroitin sulfate for symptomatic osteoarthritis: critical appraisal of meta-analyses. Curr Med Res Opin. 2008;24:1303–1308. doi: 10.1185/030079908x297231. [DOI] [PubMed] [Google Scholar]
- Monzon ME, Casalino-Matsuda SM, Forteza RM. Identification of glycosaminoglycans in human airway secretions. Am J Respir Cell Mol Biol. 2006;34:135–141. doi: 10.1165/rcmb.2005-0256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy AB, Blackwell K, Wu MH, Granger HJ. Growth factor- and heparin-dependent regulation of constitutive and agonist-mediated human endothelial barrier function. Am J Physiol Heart Circ Physiol. 2006;291:H2126–H2135. doi: 10.1152/ajpheart.00185.2006. [DOI] [PubMed] [Google Scholar]
- Mummery RS, Rider CC. Characterization of the heparin binding properties of IL-6. J Immunol. 2000;165:5671–5679. doi: 10.4049/jimmunol.165.10.5671. [DOI] [PubMed] [Google Scholar]
- Myers AL, Upreti VV, Khurana M, Eddington ND. Characterization of total plasma glycosaminoglycan levels in healthy volunteers following oral administration of a novel antithrombotic odiparcil with aspirin or enoxaparin. J Clin Pharmacol. 2008;48:1158–1170. doi: 10.1177/0091270008323751. [DOI] [PubMed] [Google Scholar]
- Nader HB, Dietrich CP, Buonassisi V, Colburn P. Heparin sequences in the heparan sulfate chains of an endothelial cell proteoglycan. Proc Nat Acad Sci U S A. 1987;84:3565–3569. doi: 10.1073/pnas.84.11.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamae T, Tanaka N, Nakanishi K, Kamei N, Sasaki H, Hamasaki T, et al. Chondroitinase ABC promotes corticospinal axon growth in organotypic cocultures. Spinal Cord. 2008;47:161–165. doi: 10.1038/sc.2008.74. [DOI] [PubMed] [Google Scholar]
- Naor D, Nedvetzki S, Golan I, Melnik L, Faitelson Y. CD44 in cancer. Crit Rev Clin Lab Sci. 2002;39:527–579. doi: 10.1080/10408360290795574. [DOI] [PubMed] [Google Scholar]
- Negrini D, Passi A, De Luca G, Miserocchi G. Matrix proteoglycans in development of pulmonary edema. In: Garg HG, Roughley PJ, Hales CA, editors. Proteoglycans In Lung Disease. New York: Marcel Dekker; 2000. pp. 143–168. [Google Scholar]
- Negrini D, Tenstad O, Passi A, Wiig H. Differential degradation of matrix proteoglycans and edema development in rabbit lung. Am J Physiol Lung Cell Mol Physiol. 2006;290:L470–L477. doi: 10.1152/ajplung.00310.2005. [DOI] [PubMed] [Google Scholar]
- Neralla S, Meyer KC. Drug treatment of pneumococcal pneumonia in the elderly. Drugs Aging. 2004;21:851–864. doi: 10.2165/00002512-200421130-00003. [DOI] [PubMed] [Google Scholar]
- Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345:319–324. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- Nieduszynski IA, Huckerby TN, Dickenson JM, Brown GM, Tai GH, Morris HG, et al. There are two major types of skeletal keratan sulfates. Biochem J. 1990;271:243–245. doi: 10.1042/bj2710243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble PW, Jiang D. Matrix regulation of lung injury inflammation and repair: the role of innate immunity. Proc Am Thor Soc. 2006;3:401–404. doi: 10.1513/pats.200604-097AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordling J, van Ophoven A. Intravesical glycosaminoglycan replenishment with chondroitin sulfate in chronic forms of cystitis. A multi-national, multi-centre, prospective observational clinical trial. Arzneimittelforschung. 2008;58:328–335. doi: 10.1055/s-0031-1296515. [DOI] [PubMed] [Google Scholar]
- Ohkawara Y, Tamura G, Iwasaki T, Tanaka A, Kikuchi T, Shirato K. Activation and transforming growth factor-beta production in eosinophils by hyaluronan. Am J Respir Cell Mol Biol. 2000;23:444–451. doi: 10.1165/ajrcmb.23.4.3875. [DOI] [PubMed] [Google Scholar]
- Olschewski H, Rose F, Grunig E, Ghofrani HA, Walmrath D, Schulz R, et al. Cellular pathophysiology and therapy of pulmonary hypertension. J Lab Clin Med. 2001;138:367–377. doi: 10.1067/mlc.2001.119285. [DOI] [PubMed] [Google Scholar]
- Oppenheimer SB, Alvarez M, Nnoli J. Carbohydrate-based experimental therapeutics for cancer, HIV/AIDS and other diseases. Acta Histochemica. 2008;110:6–13. doi: 10.1016/j.acthis.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouidja MO, Petit E, Kerros ME, Ikeda Y, Morin C, Carpentier G, et al. Structure-activity studies of heparan mimetic polyanions for anti-prion therapies. Biochem Biophys Res Commun. 2007;363:95–100. doi: 10.1016/j.bbrc.2007.08.113. [DOI] [PubMed] [Google Scholar]
- Page CP. Proteoglycans: the ‘Teflon’ of the airways? Thorax. 1997;52:924–925. doi: 10.1136/thx.52.10.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papakonstantinou E, Karakiulakis G, Eickelberg O, Perruchoud AP, Block LH, Roth M. A 340 kDa hyaluronic acid secreted by human vascular smooth muscle cells regulates their proliferation and migration. Glycobiology. 1998;8:821–830. doi: 10.1093/glycob/8.8.821. [DOI] [PubMed] [Google Scholar]
- Papakonstantinou E, Karakiulakis G, Roth M, Block LH. Platelet-derived growth factor stimulates the secretion of hyaluronic acid by proliferating human vascular smooth muscle cells. Proc Natl Acad Sci U S A. 1995;92:9881–9885. doi: 10.1073/pnas.92.21.9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papakonstantinou E, Karakiulakis G, Tamm M, Perruchoud AP, Roth M. Hypoxia modifies the effect of PDGF on glycosaminoglycan synthesis by primary human lung cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L825–L834. doi: 10.1152/ajplung.2000.279.5.L825. [DOI] [PubMed] [Google Scholar]
- Papakonstantinou E, Roth M, Tamm M, Eickelberg O, Perruchoud AP, Karakiulakis G. Hypoxia differentially enhances the effects of transforming growth factor-beta isoforms on the synthesis and secretion of glycosaminoglycans by human lung fibroblasts. J Pharmacol Exp Ther. 2002;301:830–837. doi: 10.1124/jpet.301.3.830. [DOI] [PubMed] [Google Scholar]
- Papakonstantinou E, Klangas I, Kouri F, Karakiulakis G, Eickelberg O. Lung fibrosis is associated with a significant increase of hyaluronic acid in the lung. Eur Resp J. 2006;28:604s. [Google Scholar]
- Papakonstantinou E, Kouri FM, Karakiulakis G, Klangas I, Eickelberg O. Increased hyaluronic acid content in idiopathic pulmonary arterial hypertension. Eur Resp J. 2008;32:1504–1512. doi: 10.1183/09031936.00159507. [DOI] [PubMed] [Google Scholar]
- Parthasarathy N, Gotow LF, Bottoms JD, Kute TE, Wagner WD, Mulloy B. Oligosaccharide sequence of human breast cancer cell heparan sulfate with high affinity for laminin. J Biol Chem. 1998;273:21111–21114. doi: 10.1074/jbc.273.33.21111. [DOI] [PubMed] [Google Scholar]
- Penc SF, Pomahac B, Eriksson E, Detmar M, Gallo RL. Dermatan sulfate activates nuclear factor-kappa b and induces endothelial and circulating intercellular adhesion molecule-1. J Clin Invest. 1999;103:1329–1335. doi: 10.1172/JCI4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper MS. Transforming growth factor-β: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- Peter H, Seeberger PH, Werz DB. Synthesis and medical applications of oligosaccharides. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- Petrigni G, Allegra L. Aerosolised hyaluronic acid prevents exercise-induced bronchoconstriction, suggesting novel hypotheses on the correction of matrix defects in asthma. Pulm Pharmacol Ther. 2006;19:166–171. doi: 10.1016/j.pupt.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Poole AR. Proteoglycans in health and disease: structures and functions. Biochem J. 1986;236:1–14. doi: 10.1042/bj2360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porru D, Cervigni M, Nasta L, Natale F, Lo Voi R, Tinelli C, et al. Results of endovesical hyaluronic acid/chondroitin sulfate in the treatment of interstitial cystitis/painful bladder syndrome. Rev Recent Clin Trials. 2008;3:126–129. doi: 10.2174/157488708784223817. [DOI] [PubMed] [Google Scholar]
- Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:434–439. doi: 10.1513/pats.200601-006AW. [DOI] [PubMed] [Google Scholar]
- Praus R, Brettschneider I. Glycosaminoglycans in embryonic and postnatal human cornea. Ophthal Res. 1975;7:452–458. [Google Scholar]
- Praveen MR, Koul A, Vasavada AR, Pandita D, Dixit NV, Dahodwala FF. DisCoVisc versus the soft-shell technique using Viscoat and Provisc in phacoemulsification: randomized clinical trial. J Cataract Refract Surg. 2008;34:1145–1151. doi: 10.1016/j.jcrs.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Pyda M, Korybalska K, Ksiazek K, Grajek S, Lanocha M, Lesiak M, et al. Effect of heparin on blood vascular endothelial growth factor levels in patients with ST-elevation acute myocardial infarction undergoing primary percutaneous coronary intervention. Am J Cardiol. 2006;98:902–905. doi: 10.1016/j.amjcard.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M. Pathobiology of pulmonary hypertension. Extracellular matrix. Clin Chest Med. 2001;22:433–449. doi: 10.1016/s0272-5231(05)70282-3. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2008;118:2372–2379. doi: 10.1172/JCI33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman R, Sasisekharan V, Sasisekharan R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem Biol. 2005;12:267–277. doi: 10.1016/j.chembiol.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- Rodén L, Campbell P, Fraser JR, Laurent TC, Pertoft H, Thompson JN. Enzymic pathways of hyaluronan catabolism. Ciba Found Symp. 1989;143:60–76. doi: 10.1002/9780470513774.ch5. Discussion 76–86, 281–285. Review. [DOI] [PubMed] [Google Scholar]
- Rodriguez E, Roland SK, Plaas A, Roughley PJ. The glycosaminoglycan attachment regions of human aggrecan. J Biol Chem. 2006;28127:18444–18450. doi: 10.1074/jbc.M512531200. [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Segev Y, Jacob-Hirsch J, Amariglio N, et al. Two faces of chondroitin sulfate proteoglycan in spinal cord repair: a role in microglia/macrophage activation. PLoS Med. 2008;5:e171. doi: 10.1371/journal.pmed.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo V, Scott IU, Stella A, Balducci F, Cosma A, Barone A, et al. Nonpenetrating deep sclerectomy with reticulated hyaluronic acid implant versus punch trabeculectomy: a prospective clinical trial. Eur J Ophthalmol. 2008;18:751–757. doi: 10.1177/112067210801800515. [DOI] [PubMed] [Google Scholar]
- Saito A, Munakata H. Analysis of plasma proteins that bind to glycosaminoglycans. Biochim Biophys Acta. 2007;1770:241–624. doi: 10.1016/j.bbagen.2006.10.015. [DOI] [PubMed] [Google Scholar]
- Savani RC, Wang C, Yang B, Zhang S, Kinsella MG, Wight TN, et al. Migration of bovine aortic smooth muscle cells after wounding injury. The role of hyaluronan and RHAMM. J Clin Invest. 1995;95:1158–1168. doi: 10.1172/JCI117764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid KL, Grundboek-Jusko Kimura JA, Tschopp FA, Zollinger R, Binette JP, et al. The distribution of the glycosaminoglycans in the anatomic components of the lung and the changes in the concentration of these macromolecules during development and ageing. Biochim Biophys Acta. 1982;716:178–187. doi: 10.1016/0304-4165(82)90266-5. [DOI] [PubMed] [Google Scholar]
- Scott JE. Supramolecular organization of extracellular matrix glycosaminoglycans in vitro and in the tissues. FASEB J. 1992;6:2639–2645. [PubMed] [Google Scholar]
- Scuri M, Forteza R, Lauredo I, Sabater JR, Botvinnikova Y, Allegra L, et al. Inhaled porcine pancreatic elastase causes bronchoconstriction via a bradykinin-mediated mechanism. J Appl Physiol. 2000;89:1397–1402. doi: 10.1152/jappl.2000.89.4.1397. [DOI] [PubMed] [Google Scholar]
- Scuri M, Abraham WM, Botvinnikova Y, Forteza R. Hyaluronic acid blocks porcine pancreatic elastase (PPE)-induced bronchoconstriction in sheep. Am J Respir Crit Care Med. 2001;164:1855–1859. doi: 10.1164/ajrccm.164.10.2011115. [DOI] [PubMed] [Google Scholar]
- Seeberger PH, Werz DB. Synthesis and medical applications of oligosaccharides. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- Sherman L, Sleeman J, Herrlich P, Ponta H. Hyaluronate receptors: key players in growth, differentiation, migration and tumor progression. Curr Opin Cell Biol. 1994;6:726–733. doi: 10.1016/0955-0674(94)90100-7. [DOI] [PubMed] [Google Scholar]
- Shields LB, Zhang YP, Burke DA, Gray R, Shields CB. Benefit of chondroitinase ABC on sensory axon regeneration in a laceration model of spinal cord injury in the rat. Surg Neurol. 2008;69:568–577. doi: 10.1016/j.surneu.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slevin M, Krupinski J, Gaffney J, Matou S, West D, Delisser H, et al. Hyaluronan-mediated angiogenesis in vascular disease: uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol. 2007;26:58–68. doi: 10.1016/j.matbio.2006.08.261. [DOI] [PubMed] [Google Scholar]
- Snider GL, Celli BR, Goldstein RH, O'Brien JJ, Lucey EC. Chronic interstitial pulmonary fibrosis produced in hamsters by endotracheal bleomycin. Lung volumes, volume-pressure relations, carbon monoxide uptake, and arterial blood gas studied. Am Rev Respir Dis. 1978;117:289–297. doi: 10.1164/arrd.1978.117.2.289. [DOI] [PubMed] [Google Scholar]
- Soker S, Goldstaub D, Svahn CM, Vlodavsky I, Levi BZ, Neufeld G. Variations in the size and sulfation of heparin modulate the effect of heparin on the binding of VEGF165 to its receptors. Biochem Biophys Res Commun. 1994;203:1339–1347. doi: 10.1006/bbrc.1994.2329. [DOI] [PubMed] [Google Scholar]
- Somerville RP, Longpre JM, Apel ED, Lewis RM, Wang LW, Sanes JR, et al. ADAMTS7B, the full-length product of the ADAMTS7 gene, is a chondroitin sulfate proteoglycan containing a mucin domain. J Biol Chem. 2004;279:35159–35175. doi: 10.1074/jbc.M402380200. [DOI] [PubMed] [Google Scholar]
- Souza-Fernandes AB, Pelosi P, Rocco PR. Bench-to-bedside review: the role of glycosaminoglycans in respiratory disease. Crit Care. 2006;10 doi: 10.1186/cc5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer ML, Banfi A, Ye J, von Degenfeld G, Kraft PE, Saini SA, et al. Localization of vascular response to VEGF is not dependent on heparin binding. FASEB J. 2007;21:2074–2085. doi: 10.1096/fj.06-7700com. [DOI] [PubMed] [Google Scholar]
- Srinoulprasert Y, Kongtawelert P, Chaiyaroj SC. Chondroitin sulfate B and heparin mediate adhesion of Penicillium marneffei conidia to host extracellular matrices. Microb Pathog. 2006;40:126–132. doi: 10.1016/j.micpath.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Stern R. Devising a pathway for hyaluronan catabolism: are we there yet? Glycobiology. 2003;13:105–115. doi: 10.1093/glycob/cwg112. [DOI] [PubMed] [Google Scholar]
- Stern R. Hyaluronan metabolism: a major paradox in cancer biology. Pathol Biol (Paris) 2005;53:372–382. doi: 10.1016/j.patbio.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol. 1993;9:541–573. doi: 10.1146/annurev.cb.09.110193.002545. [DOI] [PubMed] [Google Scholar]
- Sugahara K, Yamada S, Yoshida K, de Waard P, Vliegenthart JF. A novel sulfated structure in the carbohydrate-protein linkage region isolated from porcine intestinal heparin. J Biol Chem. 1992;267:1528–1533. [PubMed] [Google Scholar]
- Sugahara K, Mikami T, Uyama T, Mizuguchi S, Nomura K, Kitagawa H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol. 2003;13:612–620. doi: 10.1016/j.sbi.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Suki B, Ito S, Stamenovic D, Lutchen KR, Ingenito EP. Biomechanics of the lung parenchyma: critical roles of collagen and mechanical forces. J Appl Physiol. 2005;98:1892–1899. doi: 10.1152/japplphysiol.01087.2004. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Gabazza EC, Hayashi T, Kamada H, Adachi Y, Taguchi O. Protective role of activated protein C in lung and airway remodeling. Crit Care Med. 2004;32:S262–S265. doi: 10.1097/01.ccm.0000129668.96935.a8. [DOI] [PubMed] [Google Scholar]
- Tadjkarimi S, O'Neil GS, Luu TN, Allen SP, Schyns CJ, Chester AH, et al. Comparison of cyclic GMP in human internal mammary artery and saphenous vein: implications for coronary artery bypass graft patency. Cardiovasc Res. 1992;26:297–300. doi: 10.1093/cvr/26.3.297. [DOI] [PubMed] [Google Scholar]
- Tammi MI, Day AJ, Turley EA. Hyaluronan and homeostasis: a balancing act. J Biol Chem. 2002;277:4581–4584. doi: 10.1074/jbc.R100037200. [DOI] [PubMed] [Google Scholar]
- Tang T, Muneta T, Ju YJ, Nimura A, Miyazaki K, Masuda H, et al. Serum keratan sulfate transiently increases in the early stage of osteoarthritis during strenuous running of rats: protective effect of intraarticular hyaluronan injection. Arthritis Res Ther. 2008;10:R13.. doi: 10.1186/ar2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, et al. Resolution of lung inflammation by CD44. Science. 2002;296:155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theoharides TC, Whitmore K, Stanford E, Moldwin R, O'Leary MP. Interstitial cystitis: bladder pain and beyond. Expert Opin Pharmacother. 2008;9:2979–2994. doi: 10.1517/14656560802519845. [DOI] [PubMed] [Google Scholar]
- Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-β family. J Med Genet. 2000;37:741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnaer EL, Hafmans TG, Van Kuppevelt TH, Sanders EA, Verweij PE, Curfs JH. Involvement of glycosaminoglycans in the attachment of pneumococci to nasopharyngeal epithelial cells. Microbes Infect. 2006;8:316–322. doi: 10.1016/j.micinf.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–539. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- Toole BP, Slomiany MG. Hyaluronan: a constitutive regulator of chemoresistance and malignancy in cancer cells. Semi Cancer Biol. 2008;18:244–250. doi: 10.1016/j.semcancer.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper JN. TGF-β in the cardiovascular system: molecular mechanisms of a context-specific growth factor. Trends Cardiovasc Med. 2000;10:132–137. doi: 10.1016/s1050-1738(00)00061-x. [DOI] [PubMed] [Google Scholar]
- Tu G, Xu W, Huang H, Li S. Progress in the development of matrix metalloproteinase inhibitors. Curr Med Chem. 2008;15:1388–1395. doi: 10.2174/092986708784567680. [DOI] [PubMed] [Google Scholar]
- Turino GM, Cantor JO. Hyaluronan in respiratory injury and repair. Am J Respir Crit Care Med. 2003;167:1169–1175. doi: 10.1164/rccm.200205-449PP. [DOI] [PubMed] [Google Scholar]
- Turley AE, Noble WP, Bourguignon YWL. Signaling properties of hyaluronan receptors. J Biol Chem. 2002;277:4589–4592. doi: 10.1074/jbc.R100038200. [DOI] [PubMed] [Google Scholar]
- Turley EA. The control of adrenocortical cytodifferentiation by extracellular matrix. Differentiation. 1980;17:93–103. doi: 10.1111/j.1432-0436.1980.tb01085.x. [DOI] [PubMed] [Google Scholar]
- Tyrell DJ, Kilfeather S, Page CP. Therapeutic uses of heparin beyond its traditional role as an anticoagulant. Trends Pharmacol Sci. 1995;16:198–204. doi: 10.1016/s0165-6147(00)89022-7. [DOI] [PubMed] [Google Scholar]
- Uchimura K, Muramatsu H, Kadomatsu K, Fan QW, Kurosawa N, Mitsuoka C, et al. Molecular cloning and characterization of an N-acetylglucosamine-6-O-sulfotransferase. J Biol Chem. 1998;273:22577–22583. doi: 10.1074/jbc.273.35.22577. [DOI] [PubMed] [Google Scholar]
- Uchiyama H, Dobashi Y, Ohkouchi K, Nagasawa K. Chemical change involved in the oxidative reductive depolymerization of hyaluronic acid. J Biol Chem. 1990;265:7753–7759. [PubMed] [Google Scholar]
- Uebelhart D. Clinical review of chondroitin sulfate in osteoarthritis. Osteoarthritis Cartilage. 2008;16:S19–S21. doi: 10.1016/j.joca.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Venge P, Pedersen B, Hakansson L, Hallgren R, Lindblad G, Dahl R. Subcutaneous administration of hyaluronan reduces the number of infectious exacerbations in patients with chronic bronchitis. Am J Respir Crit Care Med. 1996;153:312–316. doi: 10.1164/ajrccm.153.1.8542136. [DOI] [PubMed] [Google Scholar]
- Verez-Bencomo V, Fernandez-Santana V, Hardy E, Toledo ME, Rodriguez MC, Heynngnezz L, et al. A synthetic conjugate polysaccharide vaccine against haemophilus influenzae type b. Science. 2004;305:522–525. doi: 10.1126/science.1095209. [DOI] [PubMed] [Google Scholar]
- Villena J, Brandan E. Dermatan sulfate exerts an enhanced growth factor response on skeletal muscle satellite cell proliferation and migration. J Cell Physiol. 2004;198:169–178. doi: 10.1002/jcp.10422. [DOI] [PubMed] [Google Scholar]
- Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases-structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- Vitale C, Verdecchia C, Bagnis C, Ganzaroli M, Giorcelli G, Marangella M. Effects of dermatan sulfate for anticoagulation in continuous renal replacement therapy. J Nephrol. 2008;21:205–212. [PubMed] [Google Scholar]
- Vitanzo PC, Jr, Sennett BJ. Hyaluronans: is clinical effectiveness dependent on molecular weight? Am J Orthop. 2006;35:421–428. [PubMed] [Google Scholar]
- Wang F, Garza LA, Kang S, Varani J, Orringer JS, Fisher GJ, et al. In vivo stimulation of de novo collagen production caused by cross-linked hyaluronic acid dermal filler injections in photodamaged human skin. Arch Dermatol. 2007;143:155–163. doi: 10.1001/archderm.143.2.155. [DOI] [PubMed] [Google Scholar]
- Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Bourguignon LY. Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer. Arch Otolaryngol Head Neck Surg. 2006;132:771–778. doi: 10.1001/archotol.132.7.771. [DOI] [PubMed] [Google Scholar]
- Weigel PH, Hascall VC, Tammi M. Hyaluronan synthases. J Biol Chem. 1997;272:13997–14000. doi: 10.1074/jbc.272.22.13997. [DOI] [PubMed] [Google Scholar]
- West JB, Mathieu-Costello O. Structure, strength, failure, and remodeling of the pulmonary blood-gas barrier. Annu Rev Physiol. 1999;61:543–572. doi: 10.1146/annurev.physiol.61.1.543. [DOI] [PubMed] [Google Scholar]
- Whitelock JM, Iozzo RV. Heparan sulfate: a complex polymer charged with biological activity. Chem Rev. 2005;105:2745–2764. doi: 10.1021/cr010213m. [DOI] [PubMed] [Google Scholar]
- Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z, et al. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ Res. 2006;99:853–860. doi: 10.1161/01.RES.0000246849.17887.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]